
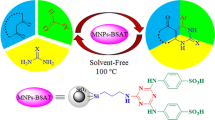
Abstract
Novel urea-functionalized silica-coated magnetic core–shell Fe3−x Ti x O4 nanoparticles were prepared and fully characterized by different methods such as Fourier transform infrared spectroscopy, energy-dispersive X-ray spectroscopy, field emission scanning electron microscopy, high-resolution transmission electron microscopy, vibrating sample magnetometer, X-ray diffraction, and thermogravimetric analyses. These magnetic nanoparticles have been explored as highly efficient and recoverable heterogeneous nanocatalyst for one-pot three-component reactions under mild conditions for the synthesis of diverse range of 5-oxo-dihydropyrano[3,2-c]chromenes known as coumarins and 1H-pyrazolo[1,2-b] phthalazine-5,10-diones in high yields. Simple isolation of the products, use of green solvent, and magnetically easy isolation and reusability of the catalyst with no significant loss of activity are the main advantages of the present method.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The last few years have witnessed much advancement in developments and applications of nanoparticles (NPs) as heterogeneous nanocatalysts or supports for various homogeneous catalysts in different fields of synthetic organic and bioorganic chemistry. Their importance stems from their amazing level of catalytic performance and improved selectivity [1–3]. On the other hand, the issue of green chemistry has emerged as major concern for the scientists in organic chemistry who have taken considerable efforts for development and application of green approaches, more selective, and environmentally benign heterogeneous nanocatalysts in industrial and synthetic organic chemistry [4–8]. In this context, various nanoparticles (NPs), especially nanometal oxides such as Fe2O3, Fe3O4, TiO2, Al2O3, ZnO, have recently been developed and applied as nanocatalysts in different research and technological areas [9–13]. These nanoparticles offer many advantages due to their unique size and extraordinary physical and chemical properties such as high surface area, oxidizing, reducing, and acid–base characters on their surfaces [14, 15]. Moreover, nanometal oxides serve as excellent adsorbents or supports for immobilization of a wide variety of homogeneous organic and inorganic catalysts [16, 17]. Usually, immobilization of homogeneous catalysts on nanosupports enhances their stability and catalytic power in chemical transformations due to their interesting properties like high ratio of surface area to volume, high selectivity, long-term stability, and reusability [18–21]. However, many of these nanocatalysts are subject to tedious process of isolation and recyclization via filtration or centrifugation [22]. Such problem has prompted the application of magnetically separable and reusable magnetic nanoparticles (MNPs) as excellent supports for various catalysts [2, 23–28]. Nevertheless, these magnetic nanoparticles suffer from quick aggregation into large bunch and thereby loss of their unique properties. In order to prevent the aggregation of magnetic nanoparticles and preserve their nanoscale properties, their surface is coated with organic or inorganic components such as silica to form core–shell structures [29]. The surface-modified magnetic nanoparticles contain reactive sites with high mobility which can be grafted with different functional groups with oxidizing, acidic, basic, and ionic liquid properties and widely employed as selective heterogeneous catalysts in various organic transformations [30–33]. In recent years, magnetic nanoparticles (MNPs) have shown a rapid growth in their developments and applications in various industrial and biological areas such as drug delivery, hyperthermia, bioseparations, magnetic resonance imaging (MRI), and catalytic processes [23, 25, 28, 34].
The presence of chromene scaffold in the framework of various pharmacologically active compounds possessing spasmolytic [35], diuretic [36], anti-HIV [37], antitumor [38], antimalarial [39], anti-Alzheimer [40], and antileukemic [41, 42] properties continues to spur synthetic efforts regarding their acquisition. In addition, different derivatives of chromenes are known as valuable synthones used for potential biodegradable agrochemicals [43] and as cosmetics and pigments [35]. Chromenes are also used as antimicrobial and antituberculosis agents [44].
A number of methods have been reported in the literature for the preparation of chromenes which utilize different catalytic systems including diammonium hydrogen phosphate (DAHP) [45], H6P2W18O62.18H2O [46], MgO [47], ionic liquids [48], hexadecyltrimethyl ammonium bromide [49], SiO2PrSO3H [50], Mg/La mixed metal oxides [51], nanosilica [52], α-Fe2O3 nanoparticles [53], and silica-grafted ionic liquid [54].
In addition, pyrazolo-fused phthalazine derivatives, constituting a bridgehead hydrazine, are reported to possessing multiplicity of pharmacological properties including anticonvulsant [55], vasorelaxant [56], and cardiotonic [57] activities. Albeit, there are several methods available for the synthesis of different phthalazine derivatives [58, 59]. Raghuvanshi and Singh [60] have recently reported a highly efficient green synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-diones of photophysical importance.
The broad range utility of chromenes and phthalazine derivatives as useful precursors in synthetic organic and medicinal chemistry has accentuated the need to develop more improved synthetic approaches for scaffold manipulation of heterocycles containing chromene and phthalazine moieties.
In continuation of our efforts for the development of versatile and efficient catalysts for the synthesis of various heterocyclic compounds including the chromenes and phthalazines and other organic transformations [61–64], we were encouraged to initiate for the first time the synthesis of hitherto unreported urea-functionalized silica-modified titanomagnetite nanoparticles and explore its catalytic activity as a novel and highly efficient heterogeneous basic nanocatalyst toward the synthesis of dihydropyranochromenes 2a-j and 1H-pyrazolo[1,2-b]phthalazine-5,10-diones 3a-j (Scheme 1). It is to mention that the catalytic activity of urea-grafted nanoparticles such as Fe3O4/SiO2 magnetic NPs has been previously explored for one-pot synthesis of other heterocyclic compounds [65].

Synthesis of dihydropyrano[2,3-c]chromenes 2a-j and 1H-pyrazolo[1,2-b]phthalazine-5,10-diones 3a-j catalyzed by Fe3−x Ti x O4@SiO2@urea MNPs
Experimental setup
Materials and instrumentation
Chemicals were purchased from Merck Chemical Company. 1H NMR and 13C NMR spectra were measured for samples in CDCl3 or DSMO-d6 using a 400 MHz Bruker AVANCE instrument at 400 and 100 MHz, respectively, using Me4Si as internal standard. Fourier transform infrared (FT-IR) spectra were recorded on a Shimadzu 435-U-04 FT spectrophotometer from KBr pellets. Melting points were measured on a BUCHI 510 apparatus in open capillary tubes. Qualitative analysis of Fe3−x Ti x O4@SiO2@urea MNPs was performed by field emission scanning electron microscopy (FE-SEM) on a MIRA3 FE-SEM instrument (TESCAN company) operated at 15 kV accelerating voltage. High-resolution transmission electron microscopy (HRTEM) images of the catalyst were provided by a Philips CM30 TEM (300 kV) instrument. The energy-dispersive X-ray (EDX) analysis was performed using an SAMX model instrument. The X-ray diffraction measurements of the synthesized Fe3−x Ti x O4, Fe3−x Ti x O4@SiO2 and Fe3−x Ti x O4@SiO2@urea samples were carried out using an XRD Model D500 Ziemence instrument from 2θ 10° to 120° at room temperature. The vibrating sample magnetometry (VSM) measurement was performed on a MDKFT instrument. Thermal stability of the nanocatalyst was studied by thermogravimetric analysis (TGA) performed on a PerkinElmer, Pay Rise Diamond, USA, TG/DTG at the heating rate of 5 °C/min in the range of 25–700 °C under N2 flow. Ultrasonication was performed in a 2200 ETH-SONICA ultrasound cleaner with a frequency of 45 kHz. The pH measurement of the catalyst was performed using a pH meter Model Metrohm 827 (Herisan, Switzerland) with a combined glass electrode.
Preparation of the Fe3−x Ti x O4 MNPs
Titanomagnetite nanoparticles were prepared according to our previously reported procedure [64] based on almost a similar procedure as reported by Yang et al. [66]. FeSO4.7H2O (1.903 g, 6.8 mmol) was dissolved in deionised water (10 mL). The pH of the solution was reduced to <1 by addition of HCl solution. Then, TiCl4 (1.3 g, 6.8 mmol) and hydrazine monohydrate (2 mL) were successively added to the reaction mixture. The resulted mixture was refluxed at 90 °C under N2 atmosphere for 30 min followed by dropwise addition of aqueous solutions of NaOH (1.6 g) and NaNO3 (0.77 g) in deionised water (10 mL) under vigorous stirring at a rate of 500 rpm for 1 h. Finally, the resulting reaction mixture was cooled to room temperature. The precipitated titanomagnetite nanoparticles were isolated simply by using a magnet bar, washed with water several times, and dried in air.
Synthesis of silica-coated magnetite Fe3−x Ti x O4 @SiO2
The prepared magnetic Fe3−x Ti x O4 nanoparticles (1 g) were placed in the mixture of EtOH (10 mL) and deionized water (30 mL), and the mixture was sonicated for 30 min. Then, a 25 % aqueous solution of NH4OH (20 mL) and tetraethyl orthosilicate (TEOS) (1.2 mL) was successfully added dropwise to the resulted mixture followed by sonication for an additional 15 min. Finally, the mixture was vigorously stirred at room temperature for 24 h. The resulted nanoparticles were magnetically separated by a magnet bar, successively washed with deionized water and ethanol, and dried in air.
Preparation of the urea-functionalized Fe3−x Ti x O4@SiO2@urea nanoparticles
A dispersion of Fe3−x Ti x O4@SiO2 MNPs (1 g) in dry xylene (30 mL) was ultrasonicated for 1 h. Then, 3-(trimethoxysilyl)propyl urea (1.93 mL, 10 mmol) was added dropwise to the resulted dispersion under N2 atmosphere and refluxed for 24 h with vigorous stirring. The resulted black powders were then isolated by an external magnetic field, successively washed with xylene and ethanol, and dried in an oven to obtain the Fe3−x Ti x O4@SiO2@urea NPs (3.1 g).
Typical procedure for the synthesis of dihydropyrano[3,2-c]chromen-2-ones
To a mixture of aldehyde 1 (1 mmol), malononitrile (0.08 g, 1.2 mmol), and 4-hydroxycoumarin (0.162 g, 1 mmol) in H2O (1 mL) and EtOH (4 mL) was added the catalyst Fe3−x Ti x O4@SiO2@urea (0.01 g), and the mixture was refluxed for an appropriate time (Table 2). After completion of the reaction as monitored by TLC, the resulted reaction mixture was diluted with hot ethanol (5 mL) and stirred for 10 min. Then, the catalyst was isolated by an external magnetic field, and the remaining supernatant was diluted with water (50 mL) and let to stir for 20 min. The precipitated crude product was filtered, washed with water, dried in oven, and recrystallized from ethanol to obtain the pure product. All the synthesized products 2a-j are known compounds which are characterized by their melting points and spectral (FT-IR, 1H NMR, and 13C NMR) analysis and compared with the corresponding reported data (Table 2).
Typical procedure for the synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-dione derivatives
To the mixture of aryl aldehyde (1 mmol), malononitrile (1 mmol), and phthalhydrazide (1 mmol) was added the catalyst Fe3−x Ti x O4@SiO2@urea (0.03 g), and the resulted mixture was stirred at 100 °C for an appropriate time (Table 4). After the completion of the reaction as monitored by thin-layer chromatography (TLC), the reaction mixture was cooled to room temperature, diluted with ethanol (10 mL), and stirred for 10 min. The catalyst was isolated in an external magnetic field. The remaining insoluble phthalhydrazide was removed by filtration, and the supernatant liquid was evaporated under reduced pressure to leave the crude product which was washed with acetone and 10 % NaHCO3 solution and finally recrystallized from ethyl acetate/n-hexane (5:7) to yield the reasonably pure product. All the synthesized products 3a-j are known compounds which are characterized by their melting points and spectral (FT-IR, 1H NMR, and 13C NMR) analyses and compared with the corresponding reported data (Table 4).
Selected data
2-Amino-4-(phenyl)-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile ( 2a )
Cream solid, m.p. 254–256 °C; FT-IR (KBr) ν (cm−1): 3380, 3285, 3179 (NH2), 3096 (CAr –H), 2198 (C≡N), 1709 (C=O), 1674, 1604,1568 (C=C), 1211, 1056 (C–O), 1493, 1380 (C–N); 1H NMR (400 MHz, DMSO-d6) δ: 4.45 (s, 1H, CH), 7.5–8.2 (m, 11H, Ar–H and NH2) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 13C NMR (125 MHz, CDCl3): δ 39.1(C–H), 57.9 (=C–CN), 103.9, 112.7 (C=C), 116.4 (C–HAr), 120.2 (C≡N), 122.3, 125.6, 128.0, 127.5, 128.4, 133.8 (C–HAr), 143.2 (C=C), 153.1 (=C–O),, 154.3 (=C–NH2), 157.9 (=C–O), 159.4 (O=C–O) ppm.
2-Amino-4-(2,4-dichlorophenyl)-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile ( 2e )
Cream solid, m.p. 255–258 °C; FT-IR (KBr) ν (cm−1): 3463, 3297,3164 (NH2), 3069 (CAr–H), 2200 (C≡N), 1716 (C=O), 1674, 1631, 1590 (C=C), 1376 (C–N), 1062 (C–O); 1H NMR (400 MHz, DMSO-d6): δ 4.98 (1H, s, CH), 7.35–7.92 (m, 9H, Ar–H and NH2) ppm; 13C NMR (100 MHz, DMSO-d6): δ 34.3 (C–H), 56.4 (=C–CN), 102.9, 113.3 (C=C), 117.1 (C–HAr), 119.1 (C≡N), 123.0, 125.2, 128.3, 129.3, 132.5, 132.9 (C–HAr), 133.6, 133.8 (C–Cl), 139.9 (C=C), 152.7 (=C–O), 158.5 (=C–NH2), 158.6 (=C–O), 159.9 (O=C–O) ppm.
2-Amino-4-(3-nitrophenyl)-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile ( 2 h )
Cream solid, m.p. 260–263 °C; FT-IR (KBr) ν (cm−1): 3398, 3323, 3190 (NH2), 3087 (CAr –H), 2194 (C≡N), 1712 (C=O), 1674, 1603 (C=C), 1532, 1379 (C–N), 1212, 1063 (C–O); 1H NMR (400 MHz, DMSO-d6) δ: 4.70 (s, 1H, CH), 7.2–7.9 (m, 10H, Ar–H and NH2) ppm; 13C NMR (100 MHz, CDCl3): δ 38.0 (C–H), 59.8 (=C–CN), 102.7, 112.8 (C=C), 116.4 (C–HAr), 119.8 (C≡N), 122.1, 124.3, 123.5, 125.5, 130.0, 133.0, 134.6 (C–HAr), 145.4 (C=C), 148.7 (=C–NO2), 152.2 (=C–NH2), 153.7, 159.1 (=C–O), 159.5 (O=C–O) ppm.
2-Amino-5-oxo-4-(pyridin-3-yl)-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile ( 2i )
Cream solid, m.p. 257–260 °C; FT-IR (KBr) ν (cm−1): 3365, 3282, 3177 (NH2), 3029 (CAr–H), 2202 (C≡N), 1707 (C=O), 1673, 1605 (C=C), 1377 (C–N), 1060 (C–O); 1H NMR (400 MHz, DMSO-d6) δ: 4.57 (s, 1H, CH), 7.4–8.5 (m, 10H, Ar–H and NH2) ppm; 13C NMR (100 MHz, CDCl3): δ 37.8 (C–H), 56.9 (=C–CN), 102.9, 112.8 (C=C), 116.4 (C–HAr), 119.0 (C≡N), 122.5, 123.7, 124.5, 132.9, 135.4, 138.7, 146.2 (C–HAr), 149.0 (C=N), 152.1 (=C–NH2), 153.7, 158.0 (=C–O), 159.5 (O=C–O) ppm.
3-Amino-1-phenyl-5,10-dioxo-5,10-dihydro-1H-pyrazolo[1,2-b]phthalazine-2-carbonitrile ( 3a )
Yellow solid, m.p. 274–276 °C; FT-IR (KBr) ν (cm−1): 3362, 3261, 3192 (NH2), 3035 (CAr–H), 2198 (C≡N), 1681 (C=O), 1660, 1605 1568 (C=C), 1439, 1383 (C–N); 1H NMR (90 MHz, DMSO-d6): δ 6.12 (s, 1H, CH), 7.37–8.05 (m, 11H, Ar–H and NH2). 13C NMR (75 MHz, DMSO-d6,): δ 61.4 (HC–N), 62.9 (=C–CN), 116.0 (C≡N), 126.6, 126.7, 127.2, 128.2, 128.5, 128.6, 128.7 (CHAr), 129.1, (=C–CO), 133.7, 134.6 (CHAr), 138.4 (=C–CH), 150.6 (C=O), 153.6 (=C–NH2), 156.6 (C=O) ppm.
3-Amino-1-(2,3-dichlorophenyl)-5,10-dioxo-5,10-dihydro-1H-pyrazolo[1,2-b]phthalazine-2-carbonitrile ( 3e )
Yellow solid, m.p. 262–264 °C; FT-IR (KBr) ν (cm−1): 3373, 3238, 3178 (NH2), 2211 (C≡N), 1683 (C=O), 1659, 1603, 1566 (C=C), 1415, 1380 (C–N); 1H NMR (400 MHz, DMSO-d6): 6.30 (1H, s, CH), 7.63–8.37 (m, 9H, Ar–H and NH2) ppm. 13C NMR (100 MHz, DMSO-d6): δ 60.6 (HC–N), 62.5 (=C–CN), 116.3 (C≡N), 122.2, 123.8, 127.1 (CHAr), 127.7 (=C–Cl), 128.7, 129.3 (CHAr), 130.7 (=C–CO), 134.2, 134.3 (CHAr), 135.1 (=C–Cl), 148.3 (=C–CH), 151.5 (C=O), 154.3 (=C–NH2), 157.2 (C=O) ppm.
3-Amino-1-(4-methoxyphenyl)-5,10-dioxo-5,10-dihydro-1H-pyrazolo[1,2-b]phthalazine-2-carbonitrile ( 3g )
Yellow solid, m.p. 238–240 °C; FT-IR (KBr) ν (cm−1): 3373, 3267 (NH2), 2192 (C≡N), 1665 (C=O), 1603, 1584 (C=C), 1417,1382 (C–N); 1H NMR (400 MHz, DMSO-d6): δ 3.80 (s, 3H, OCH3), 6.15 (s, 1H, CH), 6.95–8.31 (m, 10H, Ar–H and NH2) ppm. 13C NMR (100 MHz, DMSO-d6): δ 55.1 (CH3), 61.3 (HC–N), 62.6 (=C–CN), 113.8 (CHAr), 116.1 (C≡N), 125.1, 126.6, 127.2, 128.5 128.7 (CHAr), 130.1 (=C–CO), 132.5, 133.7 (CHAr), 134.6 (=C–CH), 150.6 (C=O), 153.6 (=C–NH2), 156.6 (=C–OMe), 159.2 (C=O) ppm.
3-Amino-5,10-dioxo-1-(pyridin-4-yl)-5,10-dihydro-1H-pyrazolo[1,2-b]phthalazine-2-carbonitrile ( 3j )
Yellow solid, m.p. 264–267 °C; FT-IR (KBr) ν (cm−1): 3388, 3285 (NH2), 2198 (C≡N), 1681 (C=O), 1664, 1604, 1579 (C=C), 1441, 1384 (C–N); 1H NMR (400 MHz, DMSO-d6): δ 6.21 (s, 1H, CH), 7.57–8.64 (m, 10H, Ar–H and NH2) ppm. 13C NMR (100 MHz, DMSO-d6): δ 60.1 (HC–N), 61.7 (=C–CN), 115.7 (C≡N), 121.3, 126.7, 127.2, (CHAr), 128.3, 128.9 (=C–CO), 133.8, 134.6 (CHAr), 147.1 (=C–CH), 149.9 (CHAr), 150.9 (C=O), 153.7 (=C–NH2), 156.7 (C=O) ppm.
Result and discussion
Characterization of the catalyst Fe3−x Ti x O4@SiO2@urea MNPs
In continuation of our efforts to develop green and recoverable heterogeneous catalysts, herein, we are encouraged to synthesize the hitherto unreported urea-functionalized silica-modified Fe3−x Ti x O4 nanoparticles (Fe3−x Ti x O4@SiO2@urea) as a new basic nanocatalyst and examine its catalytic activity in the synthesis of the titled products. It is has been shown that the presence of Ti+4 cations in the structure of nanoparticles can increase the number of superficial hydroxyl groups [67]. Such a structural quality can improve the loading capacity of sulfonic acid groups on the surface of the titanomagnetite nanoparticles (4.0–5.5 mmol/g) compared with the magnetite Fe3O4 NPs (1.76 mmol/g) [68] and magnetite silica-coated Fe3O4 NPs (0.32 mmol/g) [69].
As illustrated in Scheme 2, this heterogeneous nanocatalyst is synthesized following our previously reported procedure [64]. Initially, a mixture of equimolar amounts of FeSO4.7H2O and TiCl4 in acidic solution is treated with hydrazine hydrochloride in deionized water while refluxing under nitrogen atmosphere. The resulting Fe3−x Ti x O4 MNPs are separated from the reaction mixture simply by using a magnetic bar. In the second step, in order to protect the Fe3−x Ti x O4 MNPs from possible oxidation or aggregation, the nanoparticles are modified on their external surface by treating with tetraethyl orthosilicate (TEOS) in aqueous NH4OH solution under sonication. Then, the hydroxyl groups on the resulted Fe3−x Ti x O4@SiO2 MNPs are reacted with 3-(trimethoxysilyl)propylurea in dry xylene under sonication and nitrogen atmosphere to produce the urea-functionalized magnetic nanoparticles Fe3−x Ti x O4@SiO2@urea.

Steps for the synthesis of the catalyst Fe3−x Ti x O4@SiO2@urea MNPs
The structure of the prepared catalyst was established by performing different analytical methods such as Fourier transform infrared (FT-IR) spectroscopy, X-ray diffraction (XRD) analysis, field emission scanning electron microscopy (FE-SEM), high-resolution transmission electron microscopy (HRTEM), thermogravimetric analysis (TGA), energy-dispersive X-ray (EDX) spectroscopy, and vibrating sample magnetometric (VSM) analysis as described below.
The FT-IR spectra of the Fe3−x Ti x O4, Fe3−x Ti x O4@SiO2 and Fe3−x Ti x O4@SiO2@urea MNPs are presented in Fig. 1. As shown in Fig. 1a, the FT-IR spectrum of the Fe3−x Ti x O4 MNPs as prepared in the first step (Scheme 2) performs the characteristic broad absorption bands centered at 3418 and 1630 cm−1 which are attributed to the symmetrical and asymmetrical vibrational modes of the O–H bonds attached to the surface metal atoms. In addition, the absorption band at 470 cm−1 is due to the introduction of titanium into magnetite structure and also the bands appearing at 735 and 587 cm−1 are assigned to the symmetric stretching vibrations of the Ti–O and Fe–O bonds, respectively. The appearance of the band at 1095 cm−1 in the infrared spectrum of Fe3−x Ti x O4@SiO2 MNPs as shown in Fig. 1b underlines the covalent bond between silane and titanomagnetite surface characteristic for Si–O groups. In addition, the absorption bands at 951 and 715 cm−1 are likely associated with the stretching vibrations of Si–O–Si groups. These results confirm the successful formation of SiO2 layer on the surface of Fe3−x Ti x O4. The bands appeared at 2841–2943 cm−1 (C–H stretching vibrations) in the FT-IR spectrum of Fe3−x Ti x O4@SiO2 (Fig. 1c) substantiate the presence of the propyl groups which are successfully anchored on the Fe3−x Ti x O4@SiO2 MNPs. Moreover, the signals appeared at 3307 and 1549 cm−1 regions are attributed to the stretching and bending vibrations of N–H groups, respectively. Also, appearance of the absorption band at 1689 cm−1 is assigned to the amidyl C=O stretching of urea moiety grafted on the surface of Fe3−x Ti x O4@SiO2 MNPs.

FT-IR spectra of synthesized Fe3−x Ti x O4 (a), Fe3−x Ti x O4@SiO2 (b), and Fe3−x Ti x O4@SiO2@urea NPs (c)
The spectra obtained from the energy-dispersive X-ray (EDX) analysis of Fe3−x Ti x O4 and Fe3−x Ti x O4@SiO2@urea MNPs are illustrated in Fig. 2a, b. As shown in this figure, the expected elemental compositions of these nanoparticles are clearly demonstrated by the spectra 2a and 2b, respectively. Based on these spectra, the elemental analysis performed on the Fe3−x Ti x O4 and Fe3−x Ti x O4@SiO2@urea MNPs clearly indicates the presence of Fe, Ti, O and Fe, Ti, Si, C, O, N in these nanoparticles, respectively. The EDX pattern shown in Fig. 2 obviously approves the well dispersion of Fe3−x Ti x O4 nanoparticles.

EDX spectra of Fe3−x Ti x O4 (a) and Fe3O4@SiO2@urea (b) NPs
As illustrated in Figs. 3 and 4, the morphology and particle sizes of the Fe3−x Ti x O4 and Fe3−x Ti x O4@SiO2@urea MNPs are studied by field emission scanning electron microscopy (FE-SEM) and high-resolution transmission electron microscopy (HRTEM). As can be seen in the SEM images (Fig. 3), the Fe3−x Ti x O4 and Fe3−x Ti x O4@SiO2@urea nanoparticles exhibit a regularly spherical morphology which are made up of nanometer-sized particles with an average diameter of 23 and 31 nm, respectively. In addition, based on the HRTEM analysis (Fig. 4), the core–shell structure and the nanometer-sized particles (~70 nm) of Fe3−x Ti x O4@SiO2@urea nanoparticles are verified.

FE-SEM images of Fe3−x Ti x O4 (a) and Fe3−x Ti x O4@SiO2@urea (b) NPs

HRTEM image of Fe3−x Ti x O4@SiO2@urea NPs
Figure 5a exhibits the crystalline structure of Fe3−x Ti x O4 nanoparticles identified by X-ray diffraction (XRD) technique. The diffraction peaks are appeared at 2θ° values of 18.5, 30.2, 35.6, 37.2, 43.2, 53.6, 57.1, 62.7, 74.1, 89.9, 106. The broad peak observed at 2θ° region between 20° and 28° in the XRD pattern of the Fe3−x Ti x O4@SiO2 nanoparticles as shown in Fig. 5b confirms the successful coating of Fe3−x Ti x O4 nanoparticles with amorphous SiO2 layers. Also, as seen in the XRD pattern obtained for the Fe3−x Ti x O4@SiO2@urea nanoparticles (Fig. 5c), the positions and relative intensities of the corresponding peaks are almost in accord with those shown for the non-grafted Fe3−x Ti x O4@SiO2 nanoparticles and no significant changes are observed.

X-ray diffraction (XRD) patterns of Fe3−x Ti x O4 (a), Fe3−x Ti x O4@SiO2 (b), and Fe3−x Ti x O4@SiO2@ urea NPs (c)
The magnetic measurements of the Fe3−x Ti x O4, Fe3−x Ti x O4@SiO2, and Fe3−x Ti x O4@SiO2@urea nanoparticles were registered by using vibrating sample magnetometer (VSM) at 300 k. The magnetization curves determined for these nanoparticles are compared and shown in Fig. 6. As shown in this figure, the values of the saturation magnetization were found to be 33.85, 22.92, and 13.22 emu/g for Fe3−x Ti x O4 nanoparticles, Fe3−x Ti x O4@SiO2 core–shell, and Fe3−x Ti x O4@SiO2@urea, respectively, in 20,000 Oe. The reduction in the saturation magnetization values (Ms) of the Fe3−x Ti x O4@SiO2 and Fe3−x Ti x O4@SiO2@urea in comparison to the Fe3−x Ti x O4 NPs proposes the successful coating of SiO2 and grafting the trimethoxysilylpropyl urea moiety for the formation of Fe3−x Ti x O4@SiO2@urea. However, despite the considerable decrease in the magnetization value, the catalyst could still be separated from the solution by using an external magnetic field.

VSM curve of the Fe3−x Ti x O4 (A), Fe3−x Ti x O4@SiO2 (B), and Fe3−x Ti x O4@SiO2@urea NPs (C)
Thermogravimetric (TG) and derivative thermogravimetric (DTG) analyses were performed on the prepared Fe3−x Ti x O4@SiO2@urea MNPs to monitor its thermal decomposition profile (Fig. 7). According to the TG thermogram, the weight loss process of the catalyst could be divided into three stages. The first weight loss of 10.70 mg around 100 °C was due to surface physisorbed water. The second weight loss of 9.80 mg centered at about 240 °C can be attributed to the immobilized urea moiety followed by third decomposition weight loss of 8.85 mg centered at about 390 °C likely due to the complete removal of the organic residue. Accordingly, as shown in Fig. 7b, the derivative thermogravimetric (D-TG) thermogram performs three major peaks centered at about 100, 246, and 393 °C which correspond to the aforementioned major decomposition steps shown by the TG thermogram.

TG (a) and DTG (b) curves of Fe3−x Ti x O4@SiO2@urea NPs
In order to determine the pH value of the catalyst, we used a simple and rapid procedure in this paper. First, 0.05 g of the catalyst (Fe3−x Ti x O4@SiO2@urea NPs) in distilled water (10 mL) was stirred for 5 min. Subsequently, the pH measurement of the mixture was performed at room temperature using a pH meter instrument. As expected, the measured pH value of 9.24 confirms that the synthesized catalyst is basic in nature. This result could be explained by the presence of the grafted urea moiety and the remaining hydroxyl groups on the surface of the catalyst.
Catalytic activity of the Fe3−x Ti x O4@SiO2@urea MNPs for the synthesis of dihydropyranochromen-2-ones
In order to evaluate the catalytic potential of the newly prepared Fe3−x Ti x O4@SiO2@urea MNPs as heterogeneous nanocatalyst in organic transformations, we decided to examine its activity in one-pot synthesis of dihydropyrano[3,2-c]chromen-2-ones from three-component reaction of aromatic aldehydes, malononitrile, 4-hydroxycoumarin, and one-pot synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-diones from three-component reaction between aromatic aldehydes, malononitrile, and phthalhydrazide. Preliminarily, we carried out the reaction of benzaldehyde with malononitrile and 4-hydroxycoumarin as model reaction for optimization of the reaction conditions. The effects of different reaction parameters like the catalyst loading, solvent, and temperature were screened on the model reaction. According to the experimental results summarized in Table 1, the best results in terms of the reaction rate and yield of the product 2-amino-5-oxo-4-phenyl-4H,5H-dihydropyrano[3,2-c]chromene-3-carbonitrile were obtained when the reaction was carried out in the mixture of EtOH and H2O (4:1) as the solvent of choice with using the catalyst loading of 0.01 g under reflux conditions (entry 9). In addition, the important role of the catalyst in the reaction was substantiated by conducting the reaction in the absence of the catalyst under optimized conditions which resulted in a very low yield of the expected product after a prolonged reaction time (entry 15). Finally, it was noted that when the reaction is conducted under ultrasonication at 35 and 50 °C no improvement of the reaction rates and yields is observed (entries 13 and 14).
Similarly, to establish the conditions for the synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-diones, we chose the one-pot condensation reaction between benzaldehyde, malononitrile, and phthalhydrazide as model reaction. The effects of the reaction parameters like solvents, catalyst loading, and reaction temperature on the reaction were screened. Based on the experimental results summarized in Table 2, the optimal results were obtained for the reaction under solvent-free condition using the catalyst loading of 0.03 g at 100 °C (entry 13). The necessary involvement of the catalyst in the reaction was substantiated by conducting the reaction in the absence of the catalyst for a prolonged reaction time that resulted in only a trace amount of the expected product with almost full recovery of the starting materials (entry 15).
To establish the scope and general applicability of these reactions, a series of variously substituted aldehydes were reacted under the aforementioned optimized conditions to furnish the corresponding chromenes 2a-j and 1H-pyrazolo[1,2-b] phthalazine-5,10-diones 3a-j. According to the experimental results summarized in Tables 3 and 4, all the reactions proceeded smoothly to yield the respected products in relatively low reaction times and high yields. Generally, as seen in these tables, the aldehydes bearing electron-withdrawing substituent groups react more rapidly in comparison with those carrying electron-donating groups. This can be attributed to the inductive and resonance electronic effects of the electron-withdrawing groups which activate the carbonyl group toward the nucleophilic addition reaction with malononitrile. All the products are known compounds which are characterized by their melting points and spectral (FT-IR, 1H NMR, 13C NMR) analysis and compared with the reported data. The characteristic data for some selected products are presented in the experimental section.
The superiority and efficiency of the present method to a number of other reported works are compared for the synthesis of 4H,5H-dihydropyrano[3,2-c]chromene-3-carbonitriles and 1H-pyrazolo[1,2-b]phthalazine-2-carbonitriles as summarized in Table 5. Although each of these methods has their own advantages, they suffer from some drawbacks such as the use of organic solvent, long reaction time, low yield, and use of non-recyclable catalyst. As seen in Table 5, the ungrafted Fe3−x Ti x O4 MNPs exhibited a very low catalytic activity in comparison with the catalyst Fe3−x Ti x O4@SiO2@urea and the products were produced in lower yields and higher reaction times (entries 2 and 10).
Proposed catalytic reaction mechanisms
A plausible mechanism to explain the three-component condensation reactions between aldehydes, malononitrile, and 4-hydroxycoumarin is depicted in Scheme 3. Likely, the initial step involves the condensation reaction between the aldehyde and malononitrile under the catalytic effect of Fe3−x Ti x O4@SiO2@urea as a basic nanocatalyst via its NH2 group to produce the intermediate arylidenemalononitrile (I). Subsequently, the catalyst-activated 4-hydroxycoumarin undergoes a Michael-type addition reaction with the intermediate (I) to produce the adduct (II). Finally, under the catalytic acceleration, intramolecular nucleophilic cyclization of the intermediate (II) followed by a 1,3-proton rearrangement to furnish the expected 1,4-dihydropyranochromen-2-ones.

Possible mechanism for the synthesis of 4H-chromenes catalyzed by Fe3−x Ti x O4@SiO2@urea
A similar pathway can be suggested for the synthesis of 1H-pyrazolo[1,2-b]phthalazine-5,10-diones as illustrated in Scheme 4. Likewise, the initial step in this reaction involves the condensation of aromatic aldehydes with malononitrile under the catalytic effect of the nanocatalyst Fe3−x Ti x O4@SiO2@urea to produce the intermediate arylidenemalononitrile (I). Subsequently, the catalyst-activated phthalhydrazide undergoes the Michael-type addition to the intermediate (I) followed by successive intramolecular cyclization and tautomerization steps to afford the respective 1H-pyrazolo[1,2-b]phthalazine-5,10-dione products.

Possible mechanism for the synthesis of 1H-pyrazolo[1,2-b] phthalazine-5,10-dione catalyzed by Fe3−x Ti x O4@SiO2@urea
Catalyst recyclability
Recycling experiments were performed on the model reaction between 3-nitrobenzaldehyde, malononitrile, and 4-hydroxycoumarin under the optimized conditions. After completion of the reaction, the catalyst was magnetically separated from the reaction mixture, washed with hot ethanol, and reused for at least five fresh runs without significant loss of its catalytic activity (Fig. 8). Based on the IR analysis, the integrity of the recovered catalyst was examined and proved to be as active as the originally used sample.

Catalytic reusability of Fe3−x Ti x O4@SiO2@urea NPs for the synthesis of 4H-chromenes
Conclusion
In conclusion, we have successfully prepared the magnetic nanocatalyst Fe3−x Ti x O4@SiO2@urea and fully characterized by FT-IR, SEM, HRTEM, EDX, TGA, VSM, and XRD analytical techniques. This newly prepared nanocatalyst has been explored as an efficient, versatile, and selective heterogeneous nanocatalyst with basic nature for the synthesis of dihydropyranochromen-2-ones and 1H-pyrazolo[1,2-b] phthalazine-5,10-diones. The attractive features of the present method are high yields of the products, low reaction times, simple work-up procedure, use of green solvents, and easy recyclability and reusability of the catalyst. With respect to these advantages, the present method offers improvements on the synthesis of the titled products over many other existing methods.
References
L.M. Rossi, N.J.S. Costa, F.P. Silva, R.V. Goncalves, Nanotechnol. Rev. 2, 597 (2013)
R.B.N. Baig, R.S. Varma, Chem. Commun. 49, 752 (2013). (and references cited therein)
M.B. Gawande, P.S. Branco, R.S. Varma, Chem. Soc. Rev. 42, 3371 (2013)
K. Tanaka, F. Toda, Chem. Rev. 100, 1025 (2000)
P.T. Anastas, T. Williamson, Green Chemistry; Frontiers in Benign Chemical Synthesis and Process (Oxford University Press, Oxford, 1998)
P.T. Anastas, L.B. Bartlett, M.M. Kirchhof, T.C. Williamson, Catal. Today 55, 11 (2000)
A. Maleki, S. Javanshir, M. Naimabadi, RSC Adv. 4, 30229 (2014)
A. Maleki, M. Aghaei, N. Ghamari, Chem. Lett. 44, 259 (2015)
A. Yamashita, F. Uejo, T. Yoda, T. Uchida, Y. Tanamura, T. Yamashita, N. Teramae, Nat. Mater. 3, 337 (2004)
A. Maleki, A.H. Rezayan, Tetrahedron Lett. 55, 1848 (2014)
A. Maleki, R. Paydar, RSC Adv. 5, 33177 (2015)
A. Maleki, R. Rahimi, S. Maleki, Environ. Chem. Lett. 14, 195 (2016)
M. Shakourian-Fard, A.H. Rezayan, S. Kheirjou, A. Bayat, M. Mahmoodi Hashemi, Bull. Chem. Soc. Jpn 87, 982 (2014)
H. Itoh, S. Utamapanya, J.V. Stark, K.J. Klabunde, J.R. Schlup, Chem. Mater. 5, 71 (1993)
A. Maleki, R. Rahimi, S. Maleki, Synth. Met. 194, 11 (2014)
R.J. White, R. Luque, V.L. Budarin, J.H. Clark, D.J. Macquarrie, Chem. Soc. Rev. 38, 481 (2009)
D.J. Cole-Hamilton, Science 299, 1702 (2003)
F. Shi, M.K. Tse, M.M. Pohl, A. Bruckner, S. Zhang, M. Beller, Angew. Chem. Int. Ed. 46, 8866 (2007)
D.H. Zhang, G.D. Li, J.X. Li, J.S. Chen, Chem. Commun. 29, 3414 (2008)
A. Saxena, A. Kumar, S. Mozumdar, J. Mol. Catal. A Chem. 269, 35 (2007)
A.T. Bell, Science 299, 1688 (2003)
D. Guin, B. Baruwati, S.V. Manorama, Org. Lett. 9, 1419 (2007)
S. Shyles, V. Schünemann, W.R. Thiel, Angew. Chem. Int. Ed. 49, 3428 (2010)
M.B. Gawande, A.K. Rathi, P.S. Branco, R.S. Varma, Appl. Sci. 3, 656 (2013). (and references cited therein)
Y. Zhu, L.P. Stubbs, F. Ho, R. Liu, C.P. Ship, J.A. Maguire, Chem. Cat. Chem. 2, 365 (2010)
P. Wang, H. Liu, J. Niu, R. Li, J. Ma, Catal. Sci. Technol. 4, 1333 (2014)
R. Tayebee, M.M. Amini, N. Abdollahi, A. Aliakbari, S. Rabiee, H. Ramshini, Appl. Catal. A Gen. 468, 75 (2013)
V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrera, J.M. Basset, Chem. Rev. 111, 3036 (2011). (and references cited therein)
G. Schmid, Chem. Rev. 92, 1709 (1992)
B. Karimi, M. Khalkhali, J. Mol. Catal. A Chem. 232, 113 (2005)
I.K. Mbaraka, D.R. Radu, V.S. Lin, B.H. Shanks, J. Catal. 219, 329 (2003)
S.R. Jetti, A. Bhatewara, T. Kadre, S. Jain, Chin. Chem. Lett. 25, 469 (2014)
A. Maleki, E. Akhlaghi, R. Paydar, Appl. Organomet. Chem. 30, 382 (2016)
M. Aghayee, M.A. Zolfigol, H. Keypour, M. Yarie, L. Mohammadi, Appl. Organomet. Chem. 30, 612 (2016)
G.P. Ellis, The chemistry of heterocyclic compounds, in Chromenes, Chromenes and Chromenes, vol. 2, ed. by A. Weissberger, E.C. Taylor (Willey, New York, 1977), p. 13
E.A.A. Hafez, M.H. Elnagdi, A.G.A. Elagamey, F.M.A.A. El-Taweel, Heterocycles 26, 903 (1987)
A. Tanabe, H. Nakashima, O. Yoshida, N. Yamamoto, O. Tenmyo, T. Oki, J. Antibiot. 41, 1708 (1988)
G. Shijay, H.T. Cheng, T. Chi, Y. Ching-Fa, Tetrahedron 64, 9143 (2008)
A. Bolognese, G. Correale, M. Manfra, A. Lavecchia, O. Mazzoni, E. Novellino, P. La Colla, G. Sanna, R. Loddo, J. Med. Chem. 47, 849 (2004)
T.A. Bayer, S. Schafer, H. Breyh, O. Breyhan, C. Wirths, G.A. Treiber, A vicious circle. Clin. Neuropathol. 25, 163 (2006)
N. Fokialakis, P. Magiatis, L. Chinou, S. Mitaka, F. Tillequin, Chem. Pharm. Bull. 50, 413 (2002)
P. Beagley, M.A.L. Blackie, K. Chibale, C. Clarkson, R. Meijboom, J.R. Moss, P. Smith, H. Su, Dalton Trans. 15, 3046 (2003)
F.M. Abdel Galil, B.Y. Riad, S.M. Sherif, M.H. Elnagdi, Chem. Lett. 11, 1123 (1982)
D.C. Mungra, M.P. Patel, D.P. Rajani, R.G. Patel, Eur. J. Med. Chem. 46, 4192 (2011)
S. Balalaie, S. Abdolmohammadi, Tetrahedron Lett. 48, 3299 (2007)
M.M. Heravi, B. Alimadadi Jani, F. Derikvand, F.F. Bamoharram, H.A. Oskooie, Catal. Commun. 10, 272 (2008)
M. Seifi, H. Sheibani, Catal. Lett. 126, 275 (2008)
Y. Peng, G. Song, Catal. Commun. 8, 111 (2007)
T.-S. Jin, L.-B. Liu, Y. Zhao, T.-S. Li, Synth. Commun. 35, 1859 (2005)
G. Mohammadi Ziarani, A. Badiei, M. Azizi, P. Zarabadi, Iran. J. Chem. Chem. Eng. 30, 59 (2011)
B.N. Seshu, N. Pasha, R.K.T. Venkateswara, P.P.S. Sai, N. Lingaiah, Tetrahedron Lett. 49, 2730 (2008)
S. Banerjee, A. Horn, H. Khatri, G. Sereda, Tetrahedron Lett. 52, 1878 (2011)
H. Nagabhushana, S.S. Saundalkar, L. Muralidhar, B.M. Nagab-hushana, C.R. Girija, D. Nagaraja, M.A. Pasha, V.P. Jayashankara, Chin. Chem. Lett. 22, 143 (2011)
K. Niknam, A. Piran, Green Sustain. Chem. 3, 1 (2013)
S. Grasso, G. DeSarro, N. Micale, M. Zappala, G. Puia, M. Baraldi, C. Demicheli, J. Med. Chem. 43, 2851 (2000)
N. Watanabe, Y. Kabasawa, Y. Takase, M. Matsukura, K. Miyazaki, H. Ishihara, K. Kodama, H. Adachi, J. Med. Chem. 41, 3367 (1998)
Y. Nomoto, H. Obase, H. Takai, M. Teranishi, J. Nakamura, K. Kubo, Chem. Pharm. Bull. 38, 2179 (1990)
H.W. Heine, M. Baclawski, S.M. Bonser, G.D. Wachob, J. Org. Chem. 41, 3221 (1976)
L.P. Liu, J.M. Lu, M. Shi, Org. Lett. 9, 1303 (2007)
D.S. Raghuvanshi, K.N. Singh, Tetrahedron Lett. 52, 5702 (2011)
A. Azarifar, R. Nejat-Yami, M. AlKobaisi, D. Azarifar, J. Iran. Chem. Soc. 10, 439 (2012)
A. Azarifar, R. Nejat-Yami, D. Azarifar, J. Iran. Chem. Soc. 10, 297 (2013)
D. Azarifar, R. Nejat-Yami, F. Sameri, Z. Akrami, Lett. Org. Chem. 9, 435 (2012)
D. Azarifar, Y. Abbasi, Synth. Commun. 46, 745 (2016)
A. Maleki, Z. Alrezvani, S. Maleki, Catal. Commun. 69, 29 (2015)
S. Yang, H. He, D. Wu, D. Chen, X. Liang, Z. Qin, M. Fan, J. Zhu, P. Yuan, Appl. Catal. B. Environ. 89, 527 (2009)
X. Liang, Y. Zhong, S. Zhu, L. Ma, P. Yuan, J. Zhu, H. He, Z. Jiang, J. Hazard. Mater. 199–200, 247 (2012)
M.B. Gawande, A.K. Rathi, I.D. Nogueira, R.S. Varma, P.S. Branco, Green Chem. 15, 1895 (2013)
F. Nemati, M.M. Heravi, R. SaeediRadi, Chin. J. Catal. 33, 1825 (2012)
K. Tabatabaeian, H. Heidari, M. Mamaghani, N.O. Mahmoodi, Appl. Organomet. Chem. 26, 56 (2012)
F. Khorami, H.R. Shaterian, Chin. J. Catal. 35, 242 (2014)
S.S. Mansoor, K. Logaiya, K. Aswin, P. Nithiya Sudhan, J. Taibah Univ. Sci. 9, 213 (2015)
M. Khoobi, L. Ma’mani, F. Rezazadehb, Z. Zareieb, A. Foroumadia, A. Ramazanib, J. Mol. Catal. A Chem. 359, 74 (2012)
R. Ghahremanzadeh, Gh Imani Shakibaei, A. Bazgir, Synlett 8, 1129 (2008)
M.R. Nabid, S.J. Tabatabaei Rezaei, R. Ghahremanzadeh, A. Bazgir, Ultrason. Sonochem. 17, 159 (2010)
A. Vafaee, A. Davoodnia, M. Pordel, M.R. Bozorgmehr, Orient. J. Chem. 31, 2153 (2015)
A. Mulika, M. Deshmukha, D. Chandama, P. Patilb, S. Jagdalea, D. Patila, S. Sankpal, Der Pharma Chem 5, 19 (2013)
A. Montaghami, N. Montazeri, Orient. J. Chem. 30, 1361 (2014)
H.J. Wang, J. Lu, Z.H. Zhang, Monatsh. Chem. 141, 1107 (2010)
A. Alizadeh, M.M. Khodaei, M. Beygzadeh, D. Kordestani, M. Feyzi, Bull. Korean Chem. Soc. 33, 2546 (2012)
G. Karthikeyana, A. Pandurangan, J. Mol. Catal. A Chem. 361–362, 58 (2012)
M. Kidwai, R. Chauhan, J. Hetercyclic Chem. 51, 1689 (2014)
B. Maleki, S. Barat Nam Chalaki, S. Sedigh Ashrafi, E.R. ezaeiSeresht, F. Moeinpour, A. Khojastehnezhad, R. Tayebee, Appl. Organomet. Chem. 29, 290 (2015)
Acknowledgments
The authors wish to thank the Research Council of Bu-Ali Sina University for financial support to carry out this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Azarifar, D., Badalkhani, O., Abbasi, Y. et al. Urea-functionalized silica-coated Fe3−x Ti x O4 magnetic nanoparticles: as highly efficient and recyclable heterogeneous nanocatalyst for synthesis of 4H-chromene and 1H-pyrazolo[1,2-b]phthalazine-5,10-dione derivatives. J IRAN CHEM SOC 14, 403–418 (2017). https://doi.org/10.1007/s13738-016-0989-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-016-0989-5