
Abstract
The regiochemistry of 1,3-dipolar cycloaddition reactions of C-phenyl carbamoyl-N-phenyl nitrone with some dialkyl-substituted 2-benzylidenecyclopropane-1,1-dicarboxylates as dipolarophile was investigated using density functional theory-based reactivity indexes and activation energy calculations at B3LYP/6-31G(d) level of theory. Analysis of the geometries and bond orders at the TS structures associated with the different reaction pathways shows that these 1,3-dipolar cycloaddition reactions occur via an asynchronous concerted mechanism. Analysis of the local electrophilicity and nucleophilicity indexes based on Parr functions only for reaction between 1 + 2a and based on Fukui functions only for 1 + 2b gives correct regioselectivity. The theoretical results obtained in the work clearly predict the regiochemistry of the isolated cycloadducts and agree to experimental results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring [1]. These reactions are ones of the most important processes with both synthetic and mechanistic interest in organic chemistry. Current understanding of the underlying principle in the Diels–Alder reactions and the 1, 3-dipolar cycloadditions (1,3-DC) has grown from worthwhile interaction between theory and experiment [2–4].
The 1,3-DC reactions possess several interesting characteristics, in particular, regioselectivity. Although transition state theory remains the most widely used and the most exact approach for the study of the mechanism and the regiochemistry of these reactions, the localization of transition states is not always easier. Furthermore, transition-state calculations are often much time consuming when bulky substituents are present in reactive systems.
Recently, reactivity descriptors based on the density functional theory (DFT), such as Fukui indexes, local softness and local electrophilicity, have been extensively used for the prediction of the regiochemistry. For instance, several treatments of 1, 3-DC reactions of nitrones with various dipolarophiles can be found in the literature [5, 6]. The 1,3-DC reactions of nitrones with alkenes is an important method for preparing isoxazolidines in a regioselective and stereoselective manner [1]. Isoxazolidines have been successfully transformed into alkaloids, some important natural products and other bioactive molecules [7]. Experimentally, it has been found that the cycloaddition reaction of C-phenyl carbamoyl-N-phenyl nitrone 1 with dimethyl 2-(4-methylbenzylidene)cyclopropane-1,1-dicarboxylate 2a and dimethyl 2-(4-chlorobenzylidene)cyclopropane-1,1-dicarboxylate 2b gives preferentially the cycloadduct 3a and 3b, respectively, shown in Fig. 1 [8].

The regioisomeric pathways for 1,3-dipolar cycloadditions
Computational details
All calculations were carried out with GAUSSIAN03 program suite [9]. Geometry optimization of the reactants was carried out using DFT methods at the B3LYP/6-31G (d) level of theory [10]. The transition states (TSs) for the 1,3-DC reactions have been localized at the B3LYP/6-31G(d) level of theory. Frequency calculations characterized the stationary points to verify that the TSs had one and only one imaginary frequency. In a stable molecule, the potential energy curve for all the vibrational modes slopes upward, meaning that the energy increases as the atoms vibrate away from their equilibrium positions. The upward potential energy curve means that the force constant is positive, so the vibrational frequency obtained is real. This is true for all the vibrational frequencies in the transition state, except for the one and only one vibrational normal mode that corresponds to the reaction coordinate. As we can see when we look at a reaction coordinate diagram, the potential energy curve for the reaction coordinate slopes downward. The downward slope causes the force constant for the reaction coordinate to be negative, which in turn gives an imaginary vibrational frequency [11]. The intrinsic reaction coordinate (IRC) [12] calculation was performed in forward and backward path to identify that each saddle point connects to the two associated minima using the second-order González–Schlegel integration method [13, 14]. The atomic electronic populations were evaluated according to Merz–Kollman scheme (MK option) [15, 16]. The electronic chemical potential μ was evaluated in terms of the one electron energies of the HOMO and LUMO, using Eq. (1) [17]:
The global electrophilicity ω for dipoles and dipolarophile was evaluated using Eq. (2) [18]:
As usual, local indexes are computed in atomic condensed form [19]. The well-known Fukui function [20, 21] for electrophilic \(f_{k}^{ - }\) and nucleophilic attack \(f_{k}^{ + }\) can be written as
where ρ k (N), ρ k (N − 1) and ρ k (N + 1) are the electronic populations of the site k in neutral, cationic, and anionic systems, respectively. The local electrophilicity index, ω k , condensed to atom k is easily obtained by projecting the global quantity onto any atomic center k in the molecule using the electrophilic Fukui function (e.g. the Fukui function for nucleophilic attack, \(f_{k}^{ + }\) [22] )
Domingo et al. has introduced an empirical (relative) nucleophilicity index, N, [23] based on the HOMO energies obtained within the Kohn–Sham scheme [17], and defined as:
This nucleophilicity scale is referred to tetracyanoethylene (TCE) taken as a reference. Local nucleophilicity index, N k , [24] was evaluated using the following equation:
where, \(f_{k}^{ - }\) is the Fukui function for an electrophilic attack [20, 21].
Very recently, Domingo proposed two new electrophilic, \(P_{k}^{ + }\), and nucleophilic, \(P_{k}^{ - }\), Parr functions based on the atomic spin density distribution at the radical anion and at the radical cation of a neutral molecule [25]. The electrophilic, \(P_{k}^{ + }\), and nucleophilic, \(P_{k}^{ - }\), Parr functions, were obtained through the analysis of the Mulliken atomic spin density of the radical anion and the radical cation by single-point energy calculations over the optimized neutral geometries using the unrestricted UB3LYP formalism for radical species.
Results and discussion
The polar cycloaddition of nitrones can take place through high asynchronous TSs associated with two-center interactions. In these cases, analysis of the most electrophilic center of the electrophile and the most nucleophilic center of the nucleophile accounts for the regioselectivity in these polar processes [26]. To put in evidence the preferential cyclization mode and consequently the major cycloadducts of 1, 3-dipolar cycloaddition reactions under investigation, we will consider two-center reaction mechanism. Our theoretical predictions of the regiochemistry will be based on the activation energy calculations and DFT-based reactivity indexes.
Activation energy calculations
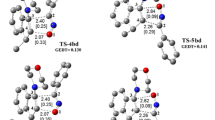
The transition states have been localized for both cyclization modes. The corresponding activation energies and TS structures are given in Table 1 and Fig. 2, respectively. An analysis of the geometries at the TS structures given in Fig. 2 shows that they correspond to an asynchronous bond formation processes. The extent of bond formation along a reaction pathway is provided by the concept of bond order (BO) [27]. The BO (Wiberg indexes) values of the O–C and C–C forming bonds at TSs are shown in brackets in Fig. 2. These values are within the range of 0.24–0.36. The BO analysis shows that these TSs correspond to asynchronous concerted processes. The BO values of the O–C and C–C forming σ bonds at the TS structures are 0.24 (O1–C3) and 0.36 (C2–C4) at TS3a, 0.33 (O1–C4) and 0.36 (C2–C3) at TS4a, 0.36 (O1–C3) and 0.24 (C2–C4) at TS3b, 0.34 (O1–C4) and 0.36 (C2–C3) at TS4b. These results show that the TS3a is more asynchronous than the TS4a and the TS3b is more asynchronous than the TS4b. In general, the asynchronicity shown by the geometrical data is accounted for by the BO values. All the reactions progressed exothermically with large ΔE r (relative energies between products and reactants) energy values. According to Hammond’s postulate, the TSs should then be closer to the reactants. Hammond’s postulate can also be interpreted in terms of the position of the transition state along the reaction coordinate, n T , as defined by Agmon [28]:

Optimized geometries for transition state structures at the B3LYP/6-31G(d) level of theory. Hydrogen atoms have been omitted for clarity. Distances of forming bonds are given in angstroms. The bond orders are given in brackets
where ∆G ≠ and ∆G r are the relative Gibbs free energy change between the reactants and their corresponding transition states and relative Gibbs free energy change between products and reactants, respectively.
The extent of n T shows the degree of similarity between the transition state and the product. According to this equation, the situation of the transition state along the reaction coordinate is determined exclusively by ∆G r (a thermodynamic quantity) and ∆G ≠ (a kinetic quantity). If n T < 0.5 the transition state is similar to reactants (early TS) and if n T > 0.5 the transition state is similar to products (late TS) [28]. The values of n T for these 1,3-DCs are 0.3296 (for 1 + 2a→3a), 0.5321 (for 1 + 2a→4a), 0.3271 (for 1 + 2b→3b) and 0.5318 (for 1 + 2b→4b). Therefore, we can conclude that TSs should then be closer to the reactants. The activation energy values, ΔE a, also favor the formation of the cycloadducts 3a and 3b against their regioisomers 4a and 4b, respectively. The presence of the chloro group in dipolarophile 2b reduces the barrier.
A comparison of the results presented in Table 1 with above-mentioned BO values shows a relationship between the activation energy of the TS structures and the asynchronicity of the reactions. In the reaction between 1 and 2a, the less energetic TS3a is more asynchronous than the TS4a. In the reaction between 1 and 2b, the less energetic TS3b is more asynchronous than the TS4b. These findings state the empirical rule that holds for a variety of [4 + 2] cycloaddition reactions that ‘‘for asymmetrically substituted dienophiles, the more asynchronous TS has the lower energy’’ [29–31].
The polar nature of the two cyclization modes can be estimated by a charge transfer (CT) analysis at the TSs. The CT from dipolarophile 2a to dipole 1 is 0.09 e at TS3a and 0.03 e at TS4a. Therefore, the CT calculations show an IED (inverse electron demand) character for this reaction. For 1 + 2b, The CT from dipole 1 to dipolarophile 2b is 0.1 e at TS3b and 0.03 e at TS4b. Thus, the CT calculations show a normal electron demand (NED) character for this process.
IRC calculations were carried out for all studied reactions, and presented only for the reaction between 1 and 2a at the pathway due to 3a (Fig. 3). This figure shows saddle point clearly and demonstrates that the TS connect to the associated minima of the concerted mechanism.

B3LYP/6-31G(d) IRC plot for the pathway of 1,3-DC reaction between 1 and 2a due to 3a
DFT-based reactivity indexes
The HOMO and LUMO energies, electronic chemical potential μ, chemical hardness η, global electrophilicity ω and global nucleophilicity N of the nitrone and dipolarophiles are given in Table 2.
As it can be seen in Table 2, the electronic chemical potential (μ) of dipolarophile 2a (−0.1383) is greater than that of dipole 1 (−0.1477) which shows that the charge transfer is taking place from dipolarophile 2a to dipole 1. The electronic chemical potential (μ) of dipole 1 is greater than that of dipolarophile 2b which indicates that the charge transfer is taking place from dipole 1 to dipolarophile 2b. It is important to note here that even though dipole 1 shows a larger electrophilicity value than dipolarophile 2b, the latter has a lower chemical potential, which is the index that determines the direction of electron flux along the cycloaddition [32]. Consequently, the dipole 1 can act as electrophile in reaction between 1 and 2a and in reaction between 1 and 2b acts as nucleophile. These results are in agreement with CT calculations at the TSs.
The difference in electrophilicity for the dipole/dipolarophile pair, ∆ω, was found to be a measure of the high- or low-polar character of the cycloaddition [33]. The small ∆ω between 1 and 2a, 0.82 eV, and 0.57 eV for 1 + 2b, shows a low-polar character for these 1,3-DC reactions.
The Fukui and Parr indexes for the atoms O1 and C2 of the dipole (nitrone 1) and for the atoms C3 and C4 of the dipolarophiles (2a and 2b) are given in Table 3 (see Fig. 5 for atom numbering). Fukui and Parr functions were computed based on the MK and the Mulliken atomic spin density analysis, respectively.
In Fig. 4, we have reported the values of local electrophilicities ω k for atoms O1 and C2 of the dipole 1 (nitrone) and the local electrophilicities N k for atoms C3 and C4 of dipolarophile 2a in (3). For (4) the values of N k for atoms O1 and C2 of the dipole 1 and ω k for atoms C3 and C4 of dipolarophile 2b were reported. According to the Domingo’s model [22, 23], in a polar cycloaddition reaction between unsymmetrical reagents, the more favorable two-center interaction will take place between the more electrophile center characterized by the highest value of the local electrophilicity index ω k at the electrophile, and the more nucleophile center characterized by the highest value of the local nucleophilicity index N k at the nucleophile. For better visualization we have illustrated these interactions in Fig. 4. According to Parr functions prediction, in the reaction between 1 and 2a, the most favorable two-center interaction takes place between C2 of dipole 1 and C4 of the dipolarophile 2a leading to the formation of the 3a regioisomer. However, according to Fukui functions prediction in this reaction, the most favorable two-center interaction takes place between C2 of the dipole 1 and C3 of the dipolarophile 2b leading to the formation of the 4a regioisomer which is in disagreement with experimental observations [8]. In (4) based on Parr functions the most favorable two-center interaction takes place between C2 of the dipole and C4 of the dipolarophile leading to the formation of the 4b regioisomer which is in disagreement with experimental findings. Based on Fukui functions, the most favorable two-center interaction takes place between O1 of the dipole and C3 of the dipolarophile leading to the formation of the 3b regioisomer which agrees to experimental outcomes.

Illustration of the favorable interactions using local nucleophilicities, N k and local electrophilicities, ω k
These results show that, in (3), the two-center polar model based on Fukui functions failed to predict experimental regiochemistry. On the other hand, in (4), the two-center polar model based on Parr functions failed to predict experimental regiochemistry.
Conclusion
Mechanism and regiochemistry for the 1, 3-dipolar cycloaddition reactions of nitrone 1 with dipolarophiles 2a and 2b have been investigated using activation energy calculations and DFT-based reactivity indexes at the B3LYP/6-31G (d) level of theory. The results obtained in this work allow us to conclude that activation energy calculations clearly predict the regiochemistry of the isolated cycloadducts, but DFT-based reactivity indexes based on Parr functions for reaction between 1 + 2a and based on Fukui functions for 1 + 2b give correct regioselectivity.
References
K.V. Gothelf, K.A. Jorgensen, Chem. Rev. 98, 863 (1998)
A. Padwa, 1,3-Dipolar cycloaddition chemistry, 1st edn. (Wiley-Interscience, New York, 1984)
L.R. Domingo, M.J. Aurell, J.A. Saez, J. Org. Chem. 72, 4220 (2007)
G.O. Jones, K.N. Houk, J. Org. Chem. 73, 1333 (2008)
O. Bortolini, M. D’Agostini, A. De Nino, L. Maiuolo, M. Nardi, G. Sindona, Tetrahedron 64, 8078 (2008)
A. Banerji, P. Sengupta, J. Indian Inst. Sci. 81, 313 (2001)
W. Carruthers, Cycloaddition reactions in organic synthesis (Pergamon, Oxford, 1990)
T.Q. Tran, R.S. Savinkov, V.V. Diev, G.L. Starova, A.P. Molchanov, Tetrahedron 69, 5173 (2013)
J.R. Cheeseman, J.A. Montgomery, T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, H.P. Hratchian, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K. Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S. Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K. Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J. Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, J.A. Pople, GAUSSIAN 03 (Revision B.03) (Gaussian Inc, Pittsburgh, 2003)
H.B. Schlegel, J. Comput. Chem. 3, 214 (1982)
E.G. Lewars, Computational chemistry: introduction to the theory and applications of molecular and quantum mechanics (Springer, Berlin, 2011)
M.H. Gordon, J.A. Pople, J. Chem. Phys. 89, 5777 (1988)
C. González, H.B. Schlegel, J. Phys. Chem. 94, 5523 (1990)
C. González, H.B. Schlegel, J. Chem. Phys. 95, 5853 (1991)
U.C. Singh, P.A. Kollman, J. Comput. Chem. 5, 129 (1984)
B.H. Besler, K.M. Merz, P.A. Kollman, J. Comput. Chem. 11, 431 (1990)
R.G. Parr, R.G. Pearson, J. Am. Chem. Soc. 105, 7512 (1983)
R.G. Parr, L. Von Szentpaly, S. Liu, J. Am. Chem. Soc. 121, 1922 (1999)
W. Yang, W.J. Mortier, J. Am. Chem. Soc. 108, 5708 (1986)
K. Fukui, Science 218, 747 (1982)
R.G. Parr, W. Yang, J. Am. Chem. Soc. 106, 4049 (1984)
L.R. Domingo, M.J. Aurell, P. Pérez, J. Phys. Chem. A 106, 6871 (2002)
L.R. Domingo, P. Pérez, J. Org. Chem. 73, 4615 (2008)
P. Pérez, L.R. Domingo, M. Duque-Noreña, E. Chamorro, J. Mol. Struc. (Theochem) 895, 86 (2009)
L.R. Domingo, P. Pérez, J.A. Saez, RSC Adv. 3, 1486 (2013)
R. Herrera, A. Nagarajan, M.A. Morales, F. Me′ndez, H.A. Jime′nez-Va′zquez, L.G. Zepeda, J. Tamariz, J. Org. Chem. 66, 1252 (2001)
K.B. Wiberg, Tetrahedron 24, 1083 (1968)
N. Agmon, R.D. Levine, Chem. Phys. Lett. 52, 197 (1977)
J.I. Garcı′a, V. Martı′nez-Merino, J.A. Mayoral, L. Salvatella, J. Am. Chem. Soc. 120, 2415 (1998)
L.R. Domingo, M. Arno′, J. Andre′s, J. Org. Chem. 64, 5867 (1999)
L. R. Domingo, Eur. J. Org. Chem. 12, 2265 (2000)
M.J. Aurell, L.R. Domingo, P. Pérez, R. Contreras, Tetrahedron 60, 11503 (2004)
H. Chemouri, S.M. Mekelleche, Int. J. Quantum Chem. 112, 2294 (2012)
Acknowledgments
The authors are grateful to Islamic Azad University, Bandar Abbas Branch for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moeinpour, F., Khojastehnezhad, A. 1, 3-dipolar cycloaddition of C-phenyl carbamoyl-N-phenyl nitrone with some dialkyl-substituted 2-benzylidenecyclopropane-1,1-dicarboxylates: theoretical analysis of mechanism and regioselectivity. J IRAN CHEM SOC 11, 1459–1465 (2014). https://doi.org/10.1007/s13738-014-0414-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-014-0414-x