
Abstract
Glomerulopathy associated with shunt infection is commonly membranoproliferative glomerulonephritis, whereas the causative organisms of secondary membranous nephropathy are usually viruses. We report a case of membranous nephropathy associated with shunt infection. The patient was born at 29-week gestation with a birth weight of 1178 g. Ventriculoperitoneal shunt surgery had been performed for congenital hydrocephalus. Thereafter, she had experienced seven shunt infections. At the age 13 years, proteinuria was detected in a school urinary screening. Urinalysis at our hospital demonstrated 3 + protein and 3 + blood. Laboratory testing demonstrated a serum creatinine 0.5 m/dl, albumin 2.5 g/dl, C-reactive protein (CRP) 13.7 mg/dl, and C3 182 mg/dl. Prior to repeat urinalysis, the patient developed vomiting and was admitted with suspected shunt infection. On admission, her body temperature was 36.0 ºC. Physical examination was unremarkable other than small stature and a palpable mass in the left upper quadrant. Urinalysis demonstrated 2 + protein and 1 + blood with no cells or casts. The urinary protein excretion was 3 g/day. Abnormal laboratory tests included erythrocyte sedimentation rate 102 mm/hr, CRP 11.67 mg/dl, IgG 2442 mg/dl, C3 177 mg/dl, and C4 44 mg/dl. Antibiotic therapy was initiated for a presumptive diagnosis of shunt infection and the shunt catheter was removed. Cultures obtained after antibiotic administration were negative. Proteinuria persisted after control of the shunt infection. Histology of a renal biopsy demonstrated membranous nephropathy with diffuse granular IgG staining and subepithelial deposits. Three possible pathomechanisms for her membranous nephropathy were considered.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pathological findings in shunt nephritis are commonly membranoproliferative glomerulonephritis, diffuse proliferative glomerulonephritis, or mesangial proliferative glomerulonephritis. Membranous nephropathy (MN) associated with shunt nephritis has not previously been described except in cases of tuberculosis. Even in non-tuberculosis bacterial infections other than shunt infection, only two reports of MN were found. Herein, we report a case of MN associated with ventriculoperitoneal shunt infection.
Case report
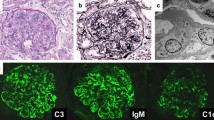
The patient is a 13-year-old girl with mental retardation and congenital hydrocephalus. She was found to have hydrocephalus and was delivered by C-section at 29 weeks gestation with a birth weight of 1178 g. Ventriculoperitoneal shunt placement was performed on day 18 of life. Thereafter, she experienced seven shunt infections. She had been treated with phenobarbital for seizures. At the age 13 years and one month, she was found to have proteinuria on school urinary screening. Repeated urinalysis at our hospital demonstrated 3 + protein, 3 + blood, 5–9 red blood cells per high-powered field (HPF), 1–4 white blood cells/HPF, and no casts. Laboratory testing revealed hemoglobin 13.6 g/dl, serum creatinine 0.5 mg/dl (eGFR 97 ml/min/1.73 m2), urea nitrogen 13.1 mg/dl, total protein 8.0 g/dl, albumin 2.5 g/dl, uric acid 4.5 mg/dl, C3 182 (60–111) mg/dl, CH50 88 (30–50) U/ml, antinuclear antibody titer < 40, anti-DNA antibody 6.9 (< 6.0) U/ml, and C-reactive protein (CRP) 13.7 mg/dl. Elevated levels of C3 and CRP were thought to be due to otitis externa being treated by another clinic. Repeat urinalysis was planned as the patient was menstruating at the time of presentation. Three months later, she presented with vomiting and reduced activity. On admission, height was 138.0 cm (less than third percentile), weight 29 kg (less than third percentile), body temperature 36.0 ºC, heart rate 72 beats per minute, respirations 18 per minute, and blood pressure 108/72 mmHg. Physical examination revealed a wheelchair-bound girl with dull appearance. Her consciousness was clear, and the lungs and heart were normal. The abdomen was soft and flat with no organomegaly. A mass was palpable in the left upper quadrant. There was no skin eruption or edema. Laboratory testing demonstrated white blood cell count 7,800/µl, red cell count 5,540,000/µl, hematocrit 43.6%, hemoglobin 13.2 g/l, platelet count 433,000/µl, serum creatinine 0.5 mg/dl, sodium 136 mmol/l, potassium 4.3 mmol/l, chloride 101 mmol/l, calcium 9.3 mg/dl, phosphorus 4.2 mg/dl, uric acid 4.3 mg/dl, total protein, 8.7 g/dl, albumin 2.5 g/dl. alkaline phosphatase 138 IU/l, total cholesterol 173 mg/dl, IgG 2442 (861–1747) mg/dl, IgA 457 (93–393) mg/dl, IgM 290 (50–269) mg/dl, C3 177 mg/dl, C4, 44 (11–31) mg/dl, antinuclear antibody titer < 40, anti-DNA antibody 7.4 U/ml, anti-dsDNA antibody 4.5 (0–20) U/ml, anti-ssDNA antibody 8.6 (0.1–40) U/ml, CRP 11.67 mg/dl, erythrocyte sedimentation rate 102 mm/hr; and normal venous blood gas analysis. Rheumatoid factor, LE cell, anti-Sm, anti-RNA, anti-cardiolipin, anti-RNP antibodies, HBs antigen, HCV antibody, anti-GBM antibody, circulating immune complex c1q, MPO-ANCA, PR3-ANCA, cryoglogulin, TPLA, and RPR were negative. The anti-streptolysin O (ASO) titer was 640 (< 209) IU/ml with a serum anti-streptokinase (ASK) level of 167 (< 2560) IU/ml. Urinalysis demonstrated 1 + protein and trace blood with no cells or casts. Twenty-four-hour urinary protein excretion was 3.0 g/day. The urinary ß2 microglobulin level was 629 (< 230) μg/l. Ultrasonography of the abdomen revealed a 10 cm × 30 cm cystic mass in the left upper to left lower quadrant. The appearance of the kidneys was unremarkable except for a difference in kidney length with the right kidney 82.7 mm (− 1.2 SD) and the left kidney 107.4 mm (+ 1.3 SD). Computed tomography demonstrated a cystic mass between the stomach and the spleen, with the distal tip of the ventriculoperitoneal shunt catheter located within the cyst. Right kidney length was 84 mm and width 43 mm, and left kidney length was 100 mm and width 48 mm. A presumptive diagnosis of shunt infection was made and antibiotic therapy with ampicillin/sulbactam was initiated. Cultures of blood, spinal fluid, the shunt catheter, and cystic fluid taken after the initiation of antibiotic treatment were negative. The catheter was removed and cyst drainage was performed. After the operation and a 6 week course of antibiotic treatment, CRP, immunoglobulin, and anti-DNA antibody levels normalized. Urinary protein excretion decreased to 1 g/day with intermittent hematuria. Percutaneous biopsy of the left kidney was performed 3 months later. On light microscopy, 2 out of 10 glomeruli were globally sclerosed (Fig. 1a, left). A slight increase in mesangial matrix and occasional arteriolar hialynosis were observed (Fig. 1a, right). Maximal glomerular tuft diameters was 192 µm. PASM staining revealed a characteristic “bubble wrap” appearance with spike formation in the glomerular basement membrane (Fig. 1b). Tubular atrophy and interstitial fibrosis affected approximately 10% of the parenchyma. Diffuse granular IgG deposits 2 + were observed along the capillary loops (Fig. 1c). IgM was 1 + , IgA ± , C3 ± , C4-, C1q-, fibrinogen ± , κ light chain + , λ light chain + , IgG1 1 + , IgG2 -, IgG3 -, IgG4 -, and phospholipase A2 receptor (PLA2R) -. On electron microscopy, subepithelial deposits with spikes were observed (Fig. 1d). A pathological diagnosis of stage III diffuse membranous glomerulonephritis was made. Losartan potassium was administered with complete resolution of proteinuria after 9 months.

Renal biopsy findings. A PAS staining. Left, lower magnification; right, higher magnification. B PAMS staining. C Immunofluorescence with IgG. D Electron microscopy. See text for figure descriptions
Discussion
We report the case of a girl with MN associated with ventriculoperitoneal shunt infection. The causative agent was not identified but was thought to be a bacterium as treatment with ampicillin/sulbactam was effective. Other than for tuberculosis, bacterial infection associated with MN has rarely been reported. We found two reports of MN associated with non-tuberculosis bacterial infection. Iida et al. reported the case of a 78-year-old male with enterococcal endocarditis and MN [1]. In this patient, light microscopic examination of the kidney demonstrated diffuse capillary loop wall thickening with mild mesangial proliferation. Electron microscopy showed diffuse thickening of the capillary basement membrane with intramembranous and subepithelial electron-dense deposits. Immunofluorescence studies showed granular deposits of IgG, IgA, IgM, C1q, C3, IgG1, and fibrinogen along capillary loops. The patient had hypocomplementemia and circulating immune complexes suggesting an antigen–antibody complex-mediated mechanism. Bulucu et al. reported the case of a 21-year-old male with MN associated with meningococcal meningitis and septicemia [2]. In this patient, deep venous thrombosis in the legs and proteinuria developed during the recovery period. Light microscopy of the kidney revealed marked thickening of the glomerular membrane with subepithelial spikes and mild expansion of the mesangial matrix. Immunofluorescence studies demonstrated a diffuse granular pattern of C3 along with moderate fibrinogen and IgM deposits. The serum C3 level was low and anti-cardiolipin antibodies were elevated. Other tests for secondary MN, including HBs antigen, anti-HCV antibody, and antinuclear antibody, were negative. An association between MN and antiphospholipid syndrome without lupus has previously been reported [3]. However, MN and antiphospholipid syndrome developed simultaneously during recovery from meningococcal disease in Bulucu et al.’s case, suggesting the infection had induced both conditions.
Transformation from diffuse proliferative glomerulonephritis has also been described as a mechanism underlying the development of infection-related MN [4, 5]. In these studies, development of membranous changes was demonstrated on repeated biopsies performed 5–19 months after the initial biopsy. Among seven of these patients, four had streptococcal infection. Similar cases of transformation from severe acute poststreptococcal glomerulonephritis to MN have been reported [6, 7]. The contribution of streptococcus infection to the transformation of glomerulonephritis to MN is currently unknown. In our case, the serum ASO titer was elevated raising a possibility of streptococcus as the causative bacterium of shunt infection. Common causative bacteria in shunt nephritis are Staphylococcus epidermidis, Corynebacterium, Staphylococcus aureus, and bacilli. Common causative agents in shunt infection include coagulase-negative Staphylococcus epidermidis, Staphylococcus aureus, Propionobacterium acnes, Escherichia coli, and Enterococcus faecalis, whereas group A Streptococcus has been reported as a cause of shunt infection [8]. In our case, CRP levels were elevated at the time of presentation. School urinary screening performed one year before had been normal. Shunt infection and consequent glomerulopathy may have developed during this period. Proliferative glomerulonephritis and low serum complement level may have been present earlier in the clinical course prior to transformation into MN.
An alternative mechanism other than infection may have contributed to the present case, although this alone is unlikely to explain the overall clinical course. Our patient was born at the gestational age of 29 weeks with a birth weight of 1178 g. Preterm and low birth weight are associated with reduced nephron number [9]. Individuals born preterm or with a low birth weight often present with proteinuria with or without hypertension during adolescence. The underlying pathology is typically glomerular hypertrophy and focal glomerulosclerosis. The present patient was found to have sclerosed glomeruli, which may be a feature of chronic glomerulonephritis. This finding, however, may be a consequence of reduced nephron number. The absence of glomerular hypertrophy in the present case supports the former possibility, while hyalinosis of glomerular arterioles may be an early sign of the changes related to reduced nephron number. Of interest, a recent study reported MN-like lesions in patients with renal hypoplasia [10]. Among five of the reported patients, two were born preterm with extremely low birth weight. Four patients had focal glomerulosclerosis or global sclerosis. All patients had segmental IgG staining, spikes, subepithelial deposits, and mild mesangial hypercellularity. Of interest, MN has previously been reported in a patient with unilateral renal agenesis, which is characterized by reduced nephron number [11]. The patient was a 13-year-old male with hematuria and proteinuria. Serological tests for glomerular diseases were negative and renal biopsy showed glomerular hypertrophy, diffuse and segmental spike formation, and thickening of the capillary walls with a segmental increase in mesangial cells and extracellular matrix. Electron microscopy revealed segmental deposition of subepithelial, intramembranous, and mesangial electron-dense deposits. Immunofluorescence studies demonstrated diffuse and granular staining for IgG, fibrinogen, C3, and C1q along capillary walls. The author of this report referred to a paper written in Japanese that described a similar patient with unilateral renal agenesis and MN. In our patient, the right kidney length was − 1.2 SD with a slightly hypertrophied left kidney at + 1.3 SD. These findings may indicate a hypoplastic right kidney with compensatory hypertrophy of the left kidney. Glomerular hypertrophy, however, was absent on renal biopsy. Unlike previously reported patients with reduced nephron number, membranous changes were diffuse in our case. A recent cohort study of pediatric non-lupus MN included one patient with a solitary kidney and non-segmental MN. The patient was an 11-year-old female and was PLA2R positive. The predominant IgG was IgG3 [12]. Her Ehrenreich and Churg stage was III. She had a relapse of MN after kidney transplantation.
In the pediatric population, secondary MN is far more prevalent than autoantibody-associated (primary) or idiopathic MN [13]. Primary MN, including PLA2R-associated MN, is uncommon in childhood. Unlike in adults, findings such as mesangial or subendothelial deposits, tubuloreticular inclusions, and IgG4 dominance are not helpful in differentiating primary MN from secondary MN in pediatric populations [12], all of which were absent in the present case. Of note, a case with positive serum HBsAg and positive biopsy staining for PLA2R was reported in the cohort of Miller et al. [12]. A previous study demonstrated that many cases of HB virus-associated MN are in fact tissue PLA2R-positive [14]. Other newly discovered autoantigens have also been shown to be associated with “secondary” MN [15, 16], blurring the distinction between autoantibody-associated and secondary MN. It is possible that secondary causes elicit podocyte injury that exposes PLA2R, thereby increasing the risk of autoantibody formation.
In conclusion, we report a case of MN associated with ventriculoperitoneal shunt infection and discuss three possible pathomechanisms underlying the development of MN in the present case.
References
Iida H, Mizumura Y, Uraoka T, et al. Membranous glomerulonephritis associated with enterococcal endocarditis. Nephron. 1985;40:88. https://doi.org/10.1159/000183435.
Bulucu F, Can C, Oktenli C, et al. Membranous glomerulonephritis, antiphospholipid syndrome, and persistent low C3 levels associated with meningococcal disease. Nephron. 2002;91:336–8. https://doi.org/10.1159/000058415.
Levy Y, George J, Ziporen L, et al. Massive proteinuria as a main manifestation of primary antiphospholipid syndrome. Pathobiology. 1998;66:49–52. https://doi.org/10.1159/000027995.
Richet G, Fillastre JP, Morel-Maroger L, et al. Change from diffuse proliferative to membranous glomerulonephritis: serial biopsies in four cases. Kidney Int. 1974;5:57–71. https://doi.org/10.1038/ki.1974.57-71.
Kapur S, Salcedo J, Chandra R, et al. Evolution of membranous nephropathy from a proliferative and exudative glomerulonephritis–a report of three cases studied by serial biopsies. Int J Pediatr Nephrol. 1985;6:105–10.
Sotsiou F, Dimitriadis G, Liapis H. Diagnostic dilemmas in atypical postinfectious glomerulonephritis. Semin Diagn Pathol. 2002;19:146–59.
Sotsiou F. Postinfectious glomerulonephritis. Nephrol Dial Transplant. 2001;16:68. https://doi.org/10.1093/ndt/16.suppl_6.68.
Patel C, Chaudhuri NR, Gaur S. Group A streptococcus ventriculoperitoneal shunt infection in a child. Pediatr Infect Dis J. 2012;31:660. https://doi.org/10.1097/INF.0b013e31824c04a5.
Luyckx VA, Brenner BM. The clinical importance of nephron mass [Review]. J Am Soc Nephrol. 2010;21:898–910. https://doi.org/10.1681/ASN.2009121248.
Takizawa K, Miura K, Kaneko N, et al. Renal hypoplasia can be the cause of membranous nephropathy-like lesions. Clin Exp Nephrol. 2020;24:813–20. https://doi.org/10.1007/s10157-020-01902-y.
Watanabe T. Membranous glomerulonephritis in a patient with unilateral renal agenesis. Nephron. 2002;91:159–61. https://doi.org/10.1159/000057619.
Miller P, Lei L, Charu V, et al. Clinicopathologic features of non-lupus membranous nephropathy in a pediatric population. Pediatr Nephrol. 2022;25:1–1.
Menon S, Valentini RP. Membranous nephropathy in children: clinical presentation and therapeutic approach. Pediatr Nephrol. 2010;25:1419–28. https://doi.org/10.1007/s00467-009-1324-5.
Xie Q, Li Y, Xue J, et al. Renal phospholipase A2 receptor in hepatitis B virus-associated membranous nephropathy. Am J Nephrol. 2015;41:345–53. https://doi.org/10.1159/000431331.
Sethi S, Madden BJ, Debiec H, et al. Exostosin 1/Exostosin 2-associated membranous nephropathy. J Am Soc Nephrol. 2019;30:1123–36. https://doi.org/10.1681/ASN.2018080852.
Caza TN, Hassen SI, Dvanajscak Z, et al. NELL1 is a target antigen in malignancy-associated membranous nephropathy. Kidney Int. 2021;99:967–76. https://doi.org/10.1016/j.kint.2020.07.039.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Consent for publication
Informed consent was obtained from the parents.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Awazu, M., Miyahara, M., Chiga, M. et al. A girl with membranous nephropathy associated with ventriculoperitoneal shunt infection. CEN Case Rep 12, 130–134 (2023). https://doi.org/10.1007/s13730-022-00732-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-022-00732-z