
Abstract
Thrombocytopenia, anasarca, fever, reticulin fibrosis, organomegaly (TAFRO) syndrome is a unique clinicopathologic subtype of multicentric Castleman’s disease that has recently been identified in Japan. However, little is known about its renal histological changes and the optimal treatment for TAFRO syndrome. An 80-year-old Japanese woman was admitted to our hospital for evaluation of severe anasarca and weight gain (10 kg in a month). She had polyneuropathy, monoclonal plasma cell proliferative disorder with positive kappa M-protein, a sclerotic bone lesion, elevation of vascular endothelial growth factor (VEGF), skin changes, and extravascular volume overload, which fulfilled the diagnostic criteria for POEMS (polyneuropathy, organomegaly, endocrinopathy, and monoclonal protein, skin changes) syndrome. However, kappa-type M-protein and thrombocytopenia with positivity of platelet-associated immunoglobulin G antibody were unusual, and fitted the diagnostic criteria for TAFRO syndrome. Renal biopsy showed diffuse endocapillary proliferative glomerulonephritis with endothelial swelling and the infiltration of monocytes and neutrophils without specific immunoglobulin deposits. Her systemic symptoms were refractory to initial treatment with high-dose melphalan and glucocorticoids. Alternative therapy with an anti-interleukin-6 (IL-6) receptor antibody (tocilizumab) effectively controlled the symptoms, while a thrombopoietin receptor agonist (romiplostim) was effective for her thrombocytopenia. Results suggest that IL-6—VEGF axis and an autoimmune mechanism may be responsible for TAFRO syndrome with clinical features of POEMS and refractory thrombocytopenia, which can be successfully treated with combination of tocilizumab and romiplostim.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multicentric Castleman’s disease (MCD) is a rare lymphoproliferative disorder that presents with systemic inflammatory symptoms and multiple organ impairment due to hypersecretion of interleukin-6 (IL-6) and other proinflammartory cytokines [1]. POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes) syndrome is characterized by the presence of monoclonal plasma cell disorder and peripheral neuropathy in addition to multiple features resembling MCD, and it can be classified as a subtype of MCD. In POEMS syndrome, extravascular volume overload is another critical complication that causes symptoms such as edema, pleural effusion, or ascites, and is considered to be related to the secretion of vascular endothelial growth factor (VEGF) driven by a paraneoplastic mechanism with plasma cell dyscrasia. VEGF targets endothelial cells and induces a rapid increase of vascular permeability, which can lead to renal dysfunction requiring dialysis [2]. TAFRO (thrombocytopenia, anasarca, fever, reticulin fibrosis, and organomegaly) syndrome is considered to be another subtype of MCD, which has recently been identified and is characterized by thrombocytopenia, while POEMS syndrome tends to feature thrombocytosis [3, 4]. Renal dysfunction is another common clinical feature of TAFRO syndrome; however, renal histologic findings have not been well evaluated in patients with this syndrome because of thrombocytopenia.
Several regimens employing chemotherapy and immunosuppressive agents have been reported for the treatment of MCD and POEMS syndrome, such as high-dose melphalan and dexamethasone with autologous hematopoietic cell transplantation to target the underlying paraneoplastic mechanism [5, 6], but the optimal treatment for TAFRO syndrome remains unclear.
We report a unique case of TAFRO syndrome with refractory thrombocytopenia, which responded to treatment with the combination of tocilizumab and romiplostim.
Case report
An 80-year-old Japanese woman was admitted to our hospital for evaluation of severe anasarca. She had a 2-year history of recurrent peripheral edema, mild thrombocytopenia, and deep venous thrombosis. Despite treatment with a corticosteroid and dabigatran (a non-vitamin K antagonist oral anticoagulant), her symptoms persisted.
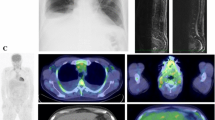
On admission, the patient was 154.0 cm tall and weighed 55.5 kg (weight gain of 10 kg in 1 month), with a blood pressure of 164/80 mmHg, pulse rate of 92/min, and temperature of 37.5 °C. She complained of dyspnea (oxygen saturation was 93% on room air) and dysesthesia of both feet. Multiple raised erythematous lesions 5 mm in size with an angiomatous appearance were noted on her trunk (Fig. 1a), and she had edema of the bilateral lower extremities (Fig. 1b).

a Multiple raised erythematous lesions 5 mm in size on the trunk. b Edema of the bilateral lower extremities. c Computed tomography shows an enlarged mediastinal lymph node measuring 15 mm in size (arrow). d Computed tomography showed bilateral pleural effusions (large arrow) and slight accumulation of pericardial fluid (small arrow). e Computed tomography displays a small sclerotic lesion measuring 10 mm (arrow)
Laboratory data were as follows (Table 1). The white blood cell count was 4.1 × 103/L with 56.0% neutrophils, hemoglobin was 11.1 g/dL with fragmented red blood cells of less than 0.05% of the total red blood cell population, and the platelet count was 3.6 × 104/L with 9.8% immature platelets. Haptoglobin level was 249 mg/dL (normal range 71–160). In addition, total protein was 6.4 g/dL, serum albumin was 2.8 g/dL, lactic dehydrogenase was 179 IU/L, alkaline phosphatase was 1,438 IU/L, and urea nitrogen was 32 mg/dL. Serum creatinine was 1.17 mg/dL with an estimated glomerular filtration rate of 34.5 mL/min, while serum creatinine had been 0.59 mg/dL 1 month earlier. C-reactive protein was 7.3 mg/dL. Urinalysis showed proteinuria of 0.41 g daily and slight hematuria (5–10 dysmorphic red blood cells per high-power field in the sediment). Levels of pituitary, thyroid, and adrenal hormones were all normal. Serum immunoglobulin (Ig) G, IgA, IgM, and CH50 were normal, and various autoantibodies were negative, including antinuclear antibody, myeloperoxidase anti-neutrophil cytoplasmic antibody, lupus anticoagulant, and anti-cardiolipin–β2 glycoprotein I complex antibodies. Platelet-associated IgG was positive at 75.0 ng/107 cells (normal < 46), and the thrombopoietin level was 2.09 Fmol/mL (normal < 3.0); however, anti-platelet autoantibody-producing B cells were not detected. Serum M-protein was not much elevated, but IgM-κ was positive by immunofixation (Supplemental Fig. 1), while the serum free light chain κ/γ ratio was 1.00 (normal range 0.26–1.65). Coagulation tests showed that the prothrombin time was 95.1% (normal: >75%), activated partial thromboplastin time was 28.9 s (normal range 27.0–40.0), fibrinogen was 337.8 mg/dL (normal range 120–300), fibrinogen degradation products was 37.3 µg/mL (normal < 5), and D-dimer was 25.0 µg/L (normal < 1). The plasma level of VEGF was 454.0 pg/mL (normal < 38.3 pg/mL), while the serum levels of IL-6 and tumor necrosis factor-α were 21.3 ng/L (normal < 4.0 ng/L) and 3.1 pg/mL (normal < 2.8 pg/mL), respectively. Cultures of blood, urine, and sputum were all negative for bacteria, acid-fast bacilli, and fungi. Serum tests for HIV, HHV-8, HCV, and HBV were also negative. The medical history and clinical examination ruled out the possibility of infection with rickettsial disease, lyme disease, severe fever with thrombocytopenia syndrome, and EB virus.
Computed tomography (CT) showed an enlarged mediastinal lymph node 15 mm in diameter (Fig. 1c), as well as bilateral pleural effusions and slight accumulation of pericardial fluid and ascites (Fig. 1d). While hepatomegaly was not seen, slight splenomegaly was noted. The skull X-ray film and head CT also displayed a small sclerotic lesion 10 mm in diameter (Fig. 1e). The results of a nerve conduction study were consistent with peripheral axonal neuropathy affecting the tibial and sural nerves. Carotid ultrasound detected thrombus in her right internal jugular vein. Bone-marrow biopsy showed normocellular marrow, with no changes of megakaryocytes and plasma cells, and no evidence of lymphoma or metastasis of malignancy. Ophthalmologic examination showed no signs of papilledema in the fundus.
Renal biopsy findings
The renal biopsy specimen contained eight glomeruli and one showed global sclerosis. There were diffuse lobular endocapillary proliferative changes with endothelial swelling and the infiltration of monocytes and neutrophils (Fig. 2a). Mesangial proliferation was not evident. Neither thrombus formation nor mesangiolysis were observed. The glomerular basement membrane (GBM) showed thickening with a double contour, but spike formation was not detected (Fig. 2a). Immunofluorescence studies indicated positive staining of the glomerular capillaries and mesangium for IgM and κ light chain, with IgA and C3 also being weakly positive, while the patient was negative for IgG, C1q, C4, and λ light chain (Fig. 2b). Immunohistochemical staining demonstrated that podocytes were weakly positive for IL-6, but negative for VEGF (Fig. 2c). Electron microscopy revealed endothelial cell swelling and edematous expansion of the subendothelial space in the GBM with no electron-dense deposits (Fig. 2d). Tubular atrophy and interstitial fibrosis occupied approximately 20% of the total renal cortical area. Sclerosis of interlobular arteries was mild, and arteriolar hyalinosis was not seen. Diffuse endocapillary proliferative glomerulonephritis was diagnosed from these findings, but a close association with deposition of any immunoglobulin was unclear.


Renal biopsy specimen. a There are diffuse lobular endocapillary proliferative changes with endothelial cell swelling, as well as infiltration of monocytes and neutrophils. b Immunofluorescence demonstrates positive staining of the glomerular capillaries and mesangium for IgM and κ light chain. c Immunohistochemical staining shows that podocytes are weakly positive for IL-6 (arrow head), but VEGF is negative. IL interleukin, VEGF vascular endothelial growth factor. d Electron microscopy reveals endothelial cell swelling and edematous expansion of the subendothelial space (arrow) in the GBM with no electron-dense deposits. GBM glomerular basement membrane
Diagnosis
This patient fulfilled the mandatory criteria for diagnosis of POEMS syndrome (polyneuropathy and monoclonal plasma cell proliferative disorder with positive M-protein), as well as 2 other major criteria (sclerotic bone lesion and elevation of VEGF) and 2 minor criteria (skin changes and extravascular volume overload) [2]. However, she also had fairly unusual clinical manifestations (i.e., a low level of IgM-κ type M-protein with no apparent monoclonal plasma cell proliferation, minimal symptoms of polyneuropathy, a small sclerotic bone lesion, anemia, and thrombocytopenia), so we were unable to make a definitive diagnosis of POEMS syndrome. Since biopsy of the affected lymph node was not performed due to its location, our patient could not fit one of the two newly proposed sets of diagnostic criteria for TAFRO syndrome [3, 4], namely, Iwaki’s criteria that require histopathological confirmation by lymph node biopsy [3]. On the other hand, when we considered Masaki’s diagnostic criteria for TAFRO syndrome [4], this patient fulfilled all three major criteria (anasarca, thrombocytopenia, and systemic inflammation) and two minor criteria (mild organomegaly and progressive renal insufficiency). Although Masaki’s criteria require exclusion of POEMS syndrome, the overall clinical picture of this patient with atypical features for POEMS syndrome led us to conclude that TAFRO syndrome was the most likely diagnosis.
Clinical course
According to the standard regimen for POEMS syndrome, high-dose melphalan (4 mg daily) and dexamethasone (40 mg daily) were initiated [5, 6], but these medications were not effective (Supplemental Fig. 2). Intravenous immunoglobulin and oral prednisolone (1 mg/kg) were added [7, 8], but the patient still did not respond. Next, treatment with an anti-IL-6 receptor antibody (tocilizumab) was started at a dose of 8 mg/kg biweekly [9]. Edema, pleural effusion, polyneuropathy, and renal function were markedly improved after 4 weeks of tocilizumab administration, along with disappearance of serum M-protein, but her severe thrombocytopenia persisted and required platelet transfusion two or three times per week. Although hypermegakaryocytosis was not detected in the bone marrow, considering the possible involvement of platelet surface IgG in the mechanism of immune-mediated thrombocytopenia, she was treated with a thrombopoietin receptor agonist (romiplostim) at a dose of 45 µg weekly combined with an anticoagulant as prophylaxis against exacerbation of her hypercoagulable state associated with this agent [10]. Within 1 month after initiation of romiplostim therapy, the platelet count increased dramatically and returned to normal along with improvement of her coagulopathy, and she was discharged from hospital. The plasma level of VEGF decreased to 203.0 pg/mL at discharge, which remained roughly unchanged thereafter. At present (1 year after discharge), prednisolone has been tapered to 3 mg/day and the patient is doing well.
Discussion
The etiology of MCD involves viral, paraneoplastic, or autoimmune mechanisms [1]. Clinical features of POEMS syndrome are associated with hypersecretion of VEGF by malignant plasma cells [2]. In contrast, it has been proposed that TAFRO syndrome represents a systemic inflammatory disorder characterized by thrombocytopenia, anasarca, fever, reticulin fibrosis, and organomegaly including lymphadenopathy [3, 4]. As aforementioned, this patient had both TAFRO and POEMS-like features and fulfilled the respective diagnostic criteria (i.e., Masaki’s diagnostic criteria for TAFRO syndrome [4] and the diagnostic criteria for POEMS syndrome [2]); hence it was difficult, and may be impossible, to make a definitive diagnosis as having TAFRO syndrome.
In patients with MCD, renal involvement has been reported to cover a wide spectrum, including secondary amyloidosis, minimal change disease, mesangial proliferative glomerulonephritis, membranous glomerulonephritis, membranoproliferative glomerulonephritis, interstitial nephritis, nephrosclerosis, and thrombotic microangiopathy (TMA)-like lesions [11, 12]. Karoui et al. reported that TMA-like lesions were the most common feature (55–60%) [13]. Overproduction of IL-6 may be responsible for renal involvement in patients with MCD. On the other hand, some reports have discussed renal histology of patients with POEMS syndrome. Navis et al. reported that the majority of these patients had membranoproliferative glomerulonephritis-like glomerulopathy or glomerular microangiopathy with evidence of endothelial damage, but did not show characteristic immunofluorescent staining for immunoglobulin or complement [14]. Nakamoto et al. reviewed the renal pathology of 52 Japanese patients with POEMS syndrome, and reported that the glomerular lesions included glomerular enlargement, cellular proliferation, mesangiolysis, and marked swelling of endothelial–mesangial cells. Immunofluorescence showed no specific deposition pattern of immunoglobulin heavy and light chains or mediators like complement components and fibrin. Electron microscopy displayed variable lucent widening of the subendothelial space, while the glomerular basement membrane was well preserved. A serum monoclonal protein (IgA-λ or IgG-λ) was identified in 81% of these patients. In addition, 96% had minor proteinuria of less than 1.0 g/ day, while renal dysfunction and extravascular fluid retention were characteristic findings [15]. A study of transgenic mice with overexpression of VEGF showed similar histologic findings to those in the present case [16], suggesting that excessive secretion of VEGF may be responsible for renal involvement in patients with POEMS syndrome. In patients with TAFRO syndrome, however, renal pathology has not been well evaluated because of its accompanying thrombocytopenia; hence, it remains unknown.
Some combined regimens have been reported for the treatment of TAFRO syndrome refractory to corticosteroids. Kawabata et al. reported a 47-year-old Japanese woman with TAFRO syndrome in whom prednisolone and methylprednisolone pulse therapy were initiated, but were not effective. On day 28, tocilizumab was started. The urine volume suddenly increased on day 30, and the platelet count returned to more than 10.0 × 104/µL after 4 months [9]. In addition, Yamaga et al. reported a 61-year-old man with TAFRO syndrome that was refractory to initial treatment with methylprednisolone pulse therapy [17]. Tocilizumab was initiated on day 42, but it was also ineffective. 1 month after the administration of cyclosporin A (4 mg/kg /day) on day 51, the platelet count had increased to the normal range and the patient’s ascites and pleural effusion had completely resolved. Furthermore, Tatekawa et al. reported a 56-year-old man who had TAFRO syndrome [18]. Combined administration of tocilizumab and prednisolone improved lymphadenopathy, pleural effusion, anemia, thrombocytopenia, and hypoalbuminemia, but elevation of C-reactive protein and massive ascites persisted. Therefore, thalidomide was initiated at 100 mg/day, leading to complete resolution of ascites after 2 months.
Romiplostim is a thrombopoietin receptor agonist that binds competitively to the c-MPL receptor, a thrombopoietin receptor expressed on haemopoietic stem cells, and activates the signaling pathway that stimulates platelet production. It has been reported to improve the platelet count in patients with immune-mediated thrombocytopenic purpura refractory to conventional therapy [10].
In summary, an anti-IL-6 receptor antibody (tocilizumab) was effective for the systemic symptoms of our patient, including edema, renal dysfunction, and elevation of inflammatory markers, but severe thrombocytopenia persisted. Addition of a thrombopoietin receptor agonist (romiplostim) subsequently controlled her thrombocytopenia. These findings suggest the underlying contribution of IL-6 to systemic inflammation and increased vascular permeability via stimulation of VEGF and the potential involvement of an autoimmune mechanism in her persistent thrombocytopenia, and may provide additional insights into the pathophysiologic mechanisms and treatment of TAFRO syndrome.
References
Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123:2924–33.
Dispenzieri A. POEMS syndrome: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:814–29.
Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicenteric Castleman disease. Am J Hematol. 2016;91:220–6.
Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103:686–92.
Li J, Zhang W, Jiao L, et al. Combination of melphalan and dexamethasone for patients with newly diagnosed POEMS syndrome. Blood. 2011;117:6445–9.
Dispenzieri A. How I treat POEMS syndrome. Blood. 2012;119:5650–8.
Wei Y, Ji XB, Wang YW, et al.: High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. Blood 2016;127(3):296–302.
Godeau B, Caulier MT, Decuypere L, Rose C, Schaeffer A, Bierling P. Intravenous immunoglobulin for adults with autoimmune thrombocytopenic purpura: results of a randomized trial comparing 0.5 and 1 g/kg b.w. Br J Haematol. 1999;107:716–9.
Kawabata H, Kotani S, Matsumura Y, et al. Successful treatment of a patient with multicentric castleman’s disease who presented with thrombocytopenia, ascites, renal failure and myelofibrosis using tocilizumab, an anti-interleukin-6 receptor antibody. Intern Med. 2013;52:1503–7.
Kuter DJ, Bussel JB, Newland A, et al. Long-term treatment with romiplostim in patients with chronic immune thrombocytopenia: safety and efficacy. Br J Haematol. 2013;161:411–23.
Leung KT, Wong KM, Choi KS, Chau KF, Li CS. Multicentric Castleman’s disease complicated by secondary renal amyloidosis. Nephrology (Carlton). 2004;9(6):392–3.
Imafuku A, Suwabe T, Hasegawa E, et al. Castleman’s disease accompanied by hypolipidemic cerebral hemorrhage and nephrosclerosis. Intern Med. 2013;52(14):1611–6.
El Karoui K, Vuiblet V, Dion D, et al. Renal involvement in Castleman disease. Nephrol Dial Transplant. 2011;26(2):599–609.
Navis GJ, Dullaart RP, Vellenga E, Elema JD, de Jong PE. Renal disease in POEMS syndrome: report on a case and review of the literature. Nephrol Dial Transplant. 1994;9:1477–81.
Nakamoto Y, Imai H, Yasuda T, Wakui H, Miura AB. A spectrum of clinicopathological features of nephropathy associated with POEMS syndrome. Nephrol Dial Transplant. 1999;14:2370–8.
Sison K, Eremina V, Baelde H, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol. 2010;21:1691–701.
Yamaga Y, Tokuyama K, Kato T, et al. Successful Treatment with Cyclosporin A in Tocilizumab-resistant TAFRO Syndrome. Intern Med. 2016;55(2):185–90.
Tatekawa S, Umemura K, Fukuyama R, et al. Thalidomide for tocilizumab-resistant ascites with TAFRO syndrome. Clin Case Rep. 2015;3(6):472–8.
Acknowledgements
This study was funded by the Okinaka Memorial Institute for Medical Research. We wish to thank Akira Shimizu (Department of Pathology, Nihon University School of Medicine, Tokyo, Japan) and Noriaki Hashiguchi (Department of Pathology, Keio University School of Medicine, Tokyo, Japan) for valuable advice regarding the renal pathology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Research involving human participants and/or animals
This article does not contain studies with human or animal participants.
Informed consent
Written informed consent was obtained from the patient for publication of this case report.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Noda-Narita, S., Sumida, K., Sekine, A. et al. TAFRO syndrome with refractory thrombocytopenia responding to tocilizumab and romiplostim: a case report. CEN Case Rep 7, 162–168 (2018). https://doi.org/10.1007/s13730-018-0319-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-018-0319-0