
Abstract
In this work, a novel chitosan/P(2-acrylamido-2-methyl-1-propanesulfonic acid-co-acrylic amide) (CS/P(AMPS-co-AM)) hydrogel was successfully prepared by a simple one-step method using glow-discharge-electrolysis plasma (GDEP) initiated copolymerization, in which N,N′-methylenebisacrylamide was used as a cross-linking agent. A copolymerization mechanism of AMPS and AM onto CS initiated by GDEP was proposed. The structure, thermal stability and morphology of CS/P(AMPS-co-AM) hydrogel were characterized by Fourier transform infrared (FTIR), X-ray diffraction (XRD), TG/DTG, and scanning electron microscope (SEM). This hydrogel was employed as an absorbent for the removal of methylene blue (MB) and malachite green (MG) from aqueous solutions. The effects of pH, contact time and equilibrium concentration on the dye adsorption were investigated batchwise. FTIR and XRD indicated that AM and AMPS were grafted onto the CS backbone successfully, forming copolymer. TG/DTG suggested that grafted AMPS and AM onto CS could change the thermal stability of the CS. SEM showed a unique three-dimensional porous structure for the CS/P(AMPS-co-AM) hydrogel. The optimum pH for the removal of cationic dyes was 5.8, and time of adsorption equilibrium was achieved in 90 min. The CS/P(AMPS-co-AM) hydrogel exhibited a very high adsorption potential, and its adsorption capacities calculated based on the Langmuir isotherm for MB and MG were 1,538.5 and 917.4 mg g−1, respectively. The dye adsorption data fitted well to the pseudo-second-order model and Langmuir model at 25 °C with pH 5.8.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dyes are extensively used in chemical industries, such as textile, printing, food, paper, cosmetics and plastics industries. However, the release of dyes into the environment is harmful, not only because of their color, but also due to the fact that many dyes and their degradation products are mutagenic and/or toxic to living organisms [1]. Therefore, dye removal from the contaminated water has been considered a worldwide problem and has attracted the attention of academia and industry. At present, various treatment techniques, such as photocatalysis [2], electrochemical oxidation [3], advanced oxidation [4], adsorption [5], and biological methods [6], have been developed to remove dyes from aqueous solutions. Among all the techniques for wastewater treatment, adsorption has been recognized as one of the most convenient and effective methods for the removal of pollutants because of its low initial investments, simple design, easy operation, high efficiencies, and eco-friendly nature [5, 7]. Although activated carbon has been widely used in adsorption, its microporous nature has limited the pore utility and the adsorption capacity for large molecules [8].
In recent years, hydrogels with three-dimensional network structure as well as functional groups have received great attention for wastewater purification because of their high adsorption capacity and economic advantages [9, 10]. Hydrogels can absorb a lot of water without dissolving. Due to the unique swelling property and the three-dimensional network structure, such as honeycomb, layer and ribbon-like morphology, hydrogels have been investigated as promising adsorbents for the removal of dyes from aqueous solutions [9–11]. However, most of the hydrogels have been synthesized using petroleum chemical products. As we all know, these hydrogels are toxic and non-biodegradable, therefore, they can produce long-term and serious environmental issues. Recently, the development and application of biopolymer hydrogels have increased owing to their unique properties, such as non-toxicity, biodegradability and biocompatibility [12, 13]. Consequently, natural polymer-based hydrogels are gaining great attention.
Chitosan (CS), a natural cationic amino polysaccharide copolymer, obtained by the deacetylation of chitin, is a linear polysaccharide and is chemically defined as a copolymer consisting of two residues: glucosamine(2-amino-2-deoxy-b-glucopyranose) and N-acetylglucosamine(2-acetamido-2-deoxy-b-d-glucopyranose) [14, 15]. CS is the first most abundant natural alkaline polysaccharide and the second most abundant natural polymer. In addition, this polysaccharose has gained immense interest due to its well non-toxicity, biodegradability, biocompatibility, and mucoadhesive properties [16]. However, the protonation of amino groups results in dissolution of CS in acidic solution, which limits CS to be used as an adsorbent in acidic media [17]. Therefore, improving the acid resistance is one of the hot topics for studying the modified CS recently.
For improving the performance, many methods have been used to prepare chitosan-based hydrogels, such as chemical initiation [13], UV-curing technique [17], and radiation [18]. Glow-discharge-electrolysis is a non-conventional electrolysis process in which plasma is sustained by direct current glow discharges between the electrode and the liquid electrolyte around it [19, 20]. In fact, this technique has been widely used in the field of synthetic chemistry [21], wastewater treatment [22], oxidative degradation [23], and surface modification [24]. However, application of glow-discharge-electrolysis plasma (GDEP) to initiate synthesis of hydrogel in aqueous solution is rarely reported. Recently, our works showed that hydrogels could be efficiently prepared with GDEP to initiate copolymerization that would allow improvements in the adsorption [9, 25] and water absorbency [26] properties. Results indicated that GDEP-initiated copolymerization is a promising method which has advantages over the others, such as simple steps, mild reaction conditions, lower cost of setup, and temporal control over polymerization [9, 25, 26].
Vinyl monomers acrylic amide (AM) and 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS) were chosen because of the following reasons [27, 28]: the toxicities of AM and AMPS are lower than that of the acrylic acid. AM, a non-ionic and cheap common monomer for preparing hydrogel materials, can be used to improve the strength of hydrogels [9, 27]. AMPS is a hydrophilic monomer containing anionic and non-ionic groups. Increasing number of ionic groups in a hydrogel increases its swelling capacity, and the non-ionic groups can improve its salt tolerance [28].
In this work, a chitosan/P(2-acrylamido-2-methyl-1-propanesulfonic acid-co-acrylic amide) (CS/P(AMPS-co-AM)) hydrogel was successfully prepared through GDEP technique, and then employed as an absorbent for the removal of cationic dyes such as methylene blue (MB) and malachite green (MG) from aqueous solutions. The structure, thermal stability and morphology of hydrogel were characterized by Fourier transform infrared (FTIR), TG/DTG, X-ray diffraction (XRD), and scanning electron microscope (SEM), respectively. The effects of pH, contact time and initial concentration on the dye adsorption were investigated by batchwise experiments. In addition, a mechanism of initiated copolymerization by GDEP was also proposed.
Experimental
Reagents
Chitosan (CS, the average molecular weight M w = 1 × 105 to 3 × 105 g mol−1, and the degree of deacetylation >85 %) was purchased from Zhejiang Golden-Shell Biochemical Co., Ltd (Zhejiang, China). AMPS (analytical reagent grade) was obtained from Shandong Shouguang Runde Chemical Co., Ltd (Shandong, China). Acrylamide (AM, analytical reagent grade) was received from Tianjin Guangfu Fine Chemical Industry Research Institute (Tianjin, China). N,N′-methylenebisacrylamide (MBA, chemical pure) was supplied by Shanghai Chemical Reagent Corporation (Shanghai, China). MB and MG were analytical grade and supplied by Jiangsu Sopo Chemical Share-holding Co., Ltd (Jiangsu, China). All chemicals were used without further purification. Other chemicals, such as sodium hydroxide (NaOH) and hydrochloric acid (HCl), were all of analytical grades. The pH was adjusted with 0.1 mol L−1 solutions of NaOH and HCl.
Preparation of CS/P(AMPS-co-AM) hydrogel
The experimental apparatus was similar to the one described in our previous work [9]. The CS/P(AMPS-co-AM) hydrogel was prepared as the following procedure. In a 250 mL three-neck flask, 0.5 g CS was added in 28 mL distilled water and stirred for 10 min at 25 °C. Subsequently, 2.0 g AMPS was placed into the flask. To dissolve completely, the mixture was stirred for additional 20 min. The amounts of 4.0 g AM and 0.1 g MBA were simultaneously added into the above solution and stirred for 10 min. After that, two electrodes were immersed into the solution to start the glow discharge for 54 s under a 540 V and 77 mA. At this stage, the reaction mixture was raised to 75 °C and stirred for additional 3–6 h. The final product was cooled to 25 °C and cut into small pieces about 2–5 mm. Finally, the resulting product was washed several times with distilled water, dried in a vacuum oven at 60 °C, and milled through a 100-mesh sieve.
Characterization of the CS/P(AMPS-co-AM) hydrogel
Fourier transform infrared spectra of the samples were carried out on a DIGILAB FTS 3000 FTIR spectrophotometer (USA) in the range of 400–4,000 cm−1 with a KBr pellet. XRD of the samples was recorded by a Rigaku D/max-2400 X-ray power diffractometer with Cu-Kα radiation (λ = 0.15406 nm), running at 40 kV and 150 mA, scanning from 5° to 80°. Thermogravimetric analysis (TG/DTG) of the samples was obtained on a PE TG/DTA 6300 instrument with a nitrogen flow rate of 50 mL min−1 and a heating rate of 10 °C min−1, over a temperature range of 20–800 °C. Surface morphology of the samples was observed on a Zeiss Ultra Plus field emission (FE)-SEM. Before SEM observation, the samples were freeze-dried to remove the water completely, and then coated with gold. Absorbance measurement of dyes was recorded on an UV757CRT Ver 2.00 spectrophotometer (Shanghai, China) in the range from 200 to 800 nm.
Adsorption experiments
Effect of pH on adsorption capacity
Effect of pH on adsorption capacity was tested as follows. The pHs ranged from 2 to 10 were adjusted by 0.1 mol L−1 HCl and NaOH, and 1,000 mg L−1 dyes solutions were prepared at several pH. In 40 mL of each pH dye solution 0.025 g dried hydrogel was placed and shaken at 25 °C for 5 h. The mixture was filtered, and the absorbance was determined by UV spectrophotometer. The equilibrium adsorption capacity (q e) was calculated as the following equation [6, 9]:
where q e (mg g−1) is the equilibrium adsorption capacity of dyes, C 0 and C e (mg L−1) the concentrations of dyes in the aqueous phase before and after the adsorption equilibrium, respectively; V (L) the volume of the aqueous phase and m (g) is the amount of dry hydrogel.
Adsorption kinetics
Dried hydrogel of 0.06 g was added in 200 mL of each dye solution (1,000 mg L−1, pH 5.8) and shaken at 25 °C. The sample was removed at the desired time-interval and filtered, and determined the absorbance by the UV spectrophotometer. The adsorption capacity was calculated according to Eq. (1).
Adsorption isotherms
Adsorption isotherms of the hydrogel for the two dyes were measured at different concentrations. 40 mL MB (from 700 to 1,500 mg L−1) and MG (from 400 to 1,400 mg L−1) were added into 0.03 g hydrogels, respectively. The solutions were shaken for 5 h, then filtered, and measured the absorbance. The adsorption capacity was calculated according to Eq. (1). Each adsorption study was repeated three times, and then the average was calculated.
Results and discussion
Copolymerization mechanism of AMPS and AM onto GDEP-initiated CS
Generally speaking, in the process of glow discharge, energetic electrons produced by GDEP are about 5–20 eV [23], which can transfer energy to gas water molecules in the gas sheath layer to form a variety of active free radicals such as HO· and H· [20]. HO· and H· within plasma distributed in solution can provide a reactive intermediate source for solution chemical reaction. The results proved that HO· played an important role in the degradation of organic compounds because it was the most powerful oxidizing species after fluorine [23]. Apart from hydrogen abstraction on labile H atoms of hydrocarbon chains (e.g., RH + HO· → H2O + R·), HO· is able to add to double bonds [23]:

Thus, in the initiated graft copolymerization by GDEP, the produced HO· radicals can be added onto polymer chains, so the hydrogels have promising properties [25, 26]. Previous studies showed that the mechanism of GDEP-initiated copolymerization was similar to that of the chemical initiation and γ-ray irradiation in hydrogels’ synthetic process [26, 29], which was concluded by the free radical process. A possible copolymerization mechanism of AMPS and AM onto CS initiated by GDEP is shown in Scheme 1. Initially, the gas water molecules absorb enough energy from plasma in the gas sheath layer to form HO· and H· radicals, which is termed “free radical formation process” (reaction 1). Apart from hydrogen abstraction on the labile H atoms from reactive hydroxyl (–OH) groups at C-3, C-6 and amino (–NH2) groups at C-2 position of CS, the HO· radicals can react with the double bonds of AM and AMPS, forming a new CS (a and b), AM and AMPS radicals (c), which is termed “chain initiation process” (reaction 2). Thereafter, random reactions of radicals with AM and AMPS monomers lead to the chain propagation and growth (reaction 3), so that monomers are grafted onto CS backbone successfully forming d and e radicals. The degrading reactions caused by radicals (HO·) and polymer chains are ignored at this stage, owing to the formation of hydrogel to cease from further reaction if discharge conditions (i.e., discharge time and discharge voltage) are controlled appropriately [25, 26]. Meanwhile, radicals (f) of homopolymer and copolymer of AMPS and/or AM can also be formed in reaction 3, but they do not cause appreciable unwanted effects on the properties of the final products. Because the cross-linker MBA can crosslink the polymer by reaction of its two double bonds [25, 26]. In the above stages, the copolymer radicals, homopolymer radicals, and cross-linker are co-existing which can form a three-dimensional network of copolymer by radical–radical and termination reactions (reaction 4).

The copolymerization mechanism of AMPS and AM onto GDEP-initiated CS. (1) Radical formation, (2) chain initiation, (3) chain propagation, (4) chain termination
Characterization of the CS/P(AMPS-co-AM) hydrogel
FTIR analysis
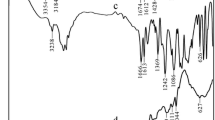
The FTIR spectra of CS (a), AM (b), AMPS (c) and CS/P(AMPS-co-AM) (d) are shown in Fig. 1. As can be seen from Fig. 1a, the overlapped peak at around 3,433 cm−1 corresponds to the stretching vibration of –OH and –NH. The peak at 1,599 cm−1 is assigned to the N–H bending vibration. The peak at 1,382 cm−1 is attributed to the deformation vibration for the amide III of CS [27]. The peaks at 1,157, 1,030 and 1,082 cm−1 are attributed to the C–O–C (glucosidic bond), C6–OH and C3–OH, respectively [30].

FTIR spectra of a CS, b AM, c AMPS, and d CS/P(AMPS-co-AM)
In Fig. 1b, the peaks at 3,354 and 3,184 cm−1 are assigned to the stretching vibration of N–H. The peaks at 1,674 and 1,612 cm−1 are attributed to the stretching vibration of C=O and C=C, respectively [9]. The peaks at 1,428 and 1,136 cm−1 are assigned to the bending vibration and stretching vibration of C–N in AM.
In Fig. 1c, the peak at 3,238 cm−1 is attributed to the stretching vibration of N–H. The peaks at 1,666 and 1,613 cm−1 are assigned to the stretching vibration of C=O and C=C, respectively. The peaks at 1,369 and 1,242 cm−1 are assigned to the bending vibration and stretching vibration of C–N. The peaks at 1,086 and 626 cm−1 are attributed to the stretching vibration of S=O and S–O, respectively [26].
Comparison with CS, AM, and AMPS, the peak at 3,433 cm−1 of CS, corresponding to the stretching vibrations of –OH and –NH2 groups, shifted to lower wavenumber (3,427 cm−1) in the FTIR spectrum of CS/P(AMPS-co-AM) hydrogel (Fig. 1d). The peaks at 1,157 and 1,082 cm−1, corresponding to the C–O–C, and C3–OH of CS, are greatly weaken and shifted to 1,113 and 1,091 cm−1. However, the peak at 1,030 cm−1, corresponding to the C6–OH of CS, has disappeared completely. The C=C peaks at 1,612 cm−1 of AM and at 1,613 cm−1 of AMPS have disappeared completely, and the C=O peaks of AM (1,674 cm−1) and AMPS (1,666 cm−1) are strengthened and shifted to lower wavenumber (1,660 cm−1). The peak at 1,086 cm−1 which corresponds to the stretching vibration of S=O is weakened and shifted to lower wavenumber (1,042 cm−1) [18]. The peaks at 1,405 and 1,207 cm−1 are assigned to the C–N bending and stretching vibration. The above information indicates that AM and AMPS are grafted onto the CS backbone successfully and the copolymer is formed.
XRD analysis
X-ray diffraction is used to study the crystal lattice arrangements and provides very useful information on degree of sample’s crystallinity [31]. The XRD patterns of CS (a) and CS/P(AMPS-co-AM) (b) are shown in Fig. 2. It can be seen that CS shows two strong crystal diffraction peaks at 2θ = 12.14° and 2θ = 20.10°, respectively. This is because strong intermolecular and intramolecular hydrogen bonds make CS form crystalline regions easily [32, 33]. However, the peak at 12.14° disappears, and the peak at 20.10° is obviously weakened and shifted to 21.18° after grafting copolymerization with AM, AMPS and CS. What is more, the intensity of diffraction peak at 21.18° is decreased and its shape is widened, which indicate that grafting copolymerization changes the crystallinity of CS and the structure of copolymer becomes very disorder [17]. This indicates that grafting copolymerization destroys the intermolecular hydrogen bonds between amine groups and hydroxyl groups of the native CS, and forms a new hydrogen bond. Therefore, it is reasonable to draw a conclusion from the XRD results that AM and AMPS monomers are grafted onto CS successfully by GDEP to form a totally amorphous composite, and that the graft copolymerization destroys the ordered crystal structure of the native CS [17].

XRD analysis of a CS and b CS/P(AMPS-co-AM)
Thermal analysis
The grafting copolymerization influenced the thermal stability of CS. The thermal stability of CS (a) and CS/P(AMPS-co-AM) (b) are shown in Fig. 3. We can see from the thermogravimetry (TG) curve in Fig. 3a, there are two weight loss stages for the thermal decomposition of CS. Weight loss of 6.2 wt% from 25 to 148 °C of CS is ascribed to the removal of free water, bound water and a little constitutive water. The second weight loss of 59.2 wt% from 148 to 787 °C can be attributed to the decomposition of C–C bond in the main chain of CS [34]. However, the CS/P(AMPS-co-AM) in Fig. 3b exhibits a four-step continuous thermal decomposition. Weight loss of 3.0 wt% from 25 to 145 °C is assigned to the removal of bonded and absorbed water. The second weight loss of 12.6 wt% from 145 to 211 °C can be attributed to the decomposition of a low molecular weight polymer [26, 35]. The third weight loss by 30.4 wt% from 211 to 350 °C is ascribed to the loss of hydroxide groups, amine groups, sulfonic groups, the destruction of cross-linked network structure and decomposition of CS main chain [28]. The fourth weight loss by 29.2 wt% from 350 to 785 °C is ascribed to the breakage of main chains in the polymeric backbone. The total weight losses of CS and CS/P(AMPS-co-AM) are, respectively, 65.4 and 75.2 wt% at about 800 °C.

TG curves of a CS and b CS/P(AMPS-co-AM)
To further prove the result that graft copolymerization of AMPS and AM onto CS backbone can change the thermal stability of CS, the DTG curves of CS and CS/P(AMPS-co-AM) are shown in Fig. 4. It can be seen from Fig. 4a that the maximum degradation temperatures (T max) of CS appear at 61.6 and 301.5 °C. However, the maximum degradation temperatures (T max) of CS/P(AMPS-co-AM) from Fig. 4b are at 74.7, 165.6, 301.5 and 404.8 °C. All results show that grafting AMPS and AM onto CS changes the thermal stability of the CS.

DTG curves of a CS and b CS/P(AMPS-co-AM)
SEM analysis
In general, hydrogels, as adsorbents, need to have a high surface area and porous structure networks to allow diffusion of the dye [18]. SEM images of the CS/P(AMPS-co-AM) hydrogel are shown in Fig. 5. It is clearly observed that the CS/P(AMPS-co-AM) hydrogel exhibits a three-dimensional porous structure, which forms hydrogel particles with a larger surface area compared to smoother particles. The sizes of the pores are about 2–10 μm. In addition, the CS is well distributed in the hydrogel, and there is no phase separation. The interface among AMPS, AM and CS is not very clear. This indicates that AM and AMPS are grafted onto the CS backbone successfully to form a three-dimensional network structure. This unique structure can increase the contact area of the dyes, facilitate water molecules to diffuse into the porous structure network, accelerate the adsorption rate, and improve the adsorption capacity effectively [12, 13, 34].

SEM images of CS/P(AMPS-co-AM) hydrogel (×5,000)
Adsorption study
Effect of pH on cationic dyes adsorption
The pH is one of the key parameters affecting adsorption process. Since the adsorption of “anionic” hydrogels was strongly affected by ionic strength, no buffer solutions were used [9, 25]. As can be seen from Fig. 6, the adsorption capacities of CS/P(AMPS-co-AM) hydrogels are higher at pH 5–10 than those of at pH 2–5. This is because the CS/P(AMPS-co-AM) hydrogels contain –NH2, SO3H and –OH, and the structure of hydrogel can change in different pH solutions.

The effect of pH on adsorption capacity
At higher pH range (pH 5–10 solution), the cationic dyes (D–Cl) are first dissolved, dissociated, and then converted to ionic dyes:
Meanwhile, CS/P(AMPS-co-AM) hydrogel is dissociated:
A substantial number of –SO3H groups in hydrogel are deprotonated and ionized to a negatively charged –SO3 −, which cause high anion–anion repelling and enhance the osmotic pressure difference between the internal and external particles [25]. As a result, the hydrogel gives rise to the swelling [26]. Water molecules and dye cation (D+) diffuse into the three-dimensional network structure effectively. The adsorption process occurs because of the electrostatic attractions between these two counter ions:
In addition, hydrogen-bonding interaction between the –NH2 and –OH groups of the CS/P(AMPS-co-AM) hydrogel and the amino groups (–NR2) of the dyes can also occur [9]. So the hydrogel has higher adsorption capacity at pH 5–10 solution.
Under acidic pH (pH <5), most of sulfonic groups (–SO3 −) in hydrogel are protonated, forming –SO3H [18]:
The hydrogen-bonding interaction is strengthened and the anion–anion electrostatic repulsion is restricted, consequently, the osmotic pressure of inside the hydrogel particles is decreased [9]. Therefore, the swelling of hydrogel is decreased and the network tends to shrink and collapse. Water molecules and dye molecules do not effectively diffuse into the polymeric network, and consequently, adsorption capacity is decreased.
Adsorption kinetics
To investigate the adsorption kinetics, the effects of the adsorption capacities of CS/P(AMPS-co-AM) hydrogels for two dyes on contact times at pH 5.8 are shown in Fig. 7. It shows that the adsorption capacities increase sharply within 30 min, then continue to increase with a slower rate between 30 and 60 min, and finally level off after 90 min, implying that CS/P(AMPS-co-AM) hydrogel has quite fast adsorption rate.

The effect of time on adsorption capacity at pH 5.8 and 25 °C
For practical applications of adsorption such as process control and design, it is significant to understand the adsorption rate and the dynamic behavior of the system. Adsorption kinetics describes how fast the adsorption occurs and also gives information on the factors affecting the rate of reaction.
To investigate the mechanism and rate-controlling step, three kinetic models, such as the pseudo-first-order, the pseudo-second-order and the intra-particle diffusion, were used to evaluate the experimental data [25].
The pseudo-first-order kinetics model assumes that the adsorption is originated from physical process [36]. It is given as [9]:
where k 1 (min−1) is the rate constant of the pseudo-first-order model, q e and q t (mg g−1) are the amounts of dyes adsorbed on the adsorbent at equilibrium and at any time t (min). The value of the k 1 and q e can be obtained from the slope and intercept of the straight-line plots of log(q e − q t ) against t [25].
The pseudo-second-order kinetics model can predict the behavior over the whole adsorption processes and is in agreement with the chemisorption mechanism being the rate-controlling step [37]. It is expressed as:
where k 2 (g mg−1 min−1) is the rate constant of the pseudo-second-order model. The values of k 2 and q e can be obtained from the plot of t/q t against t.
Figure 8a and b show the linear plots of the two models, and the correlation coefficients (R 2), k 1, k 2, the calculated q e values and experimental q e(exp) values are all shown in Table 1. By comparing Fig. 8a and b, it is obvious that the pseudo-second-order model agrees with the experimental data better than the pseudo-first-order model. It was observed from Table 1 that the q e values (MB 1,061.9 mg g−1 and MG 746.3 mg g−1) obtained from the pseudo-second-order model are closer to the experimental q e(exp) values (MB 1,019.1 mg g−1 and MG 734.5 mg g−1) than those from the pseudo-first-order model (MB 588.7 mg g−1 and MG 289.8 mg g−1). In addition, the R 2 values (MB 0.9997 and MG 0.9999) of the pseudo-second-order model are much closer to unity 1, and higher than those of the pseudo-first-order model (MB 0.8996 and MG 0.9982). These results suggested that the adsorption kinetics fit well to the pseudo-second-order model. The rate-limiting chemisorption process involves the valence forces through sharing or exchange of electrons between the dyes and the hydrogel [25].

The linear plots of kinetic models
The Weber’s intra-particle diffusion model is one of the most commonly used methods for identifying the adsorption mechanism of porous materials. It can be given as [38]:
where k int (mg g−1 min−1/2) is the intra-particle diffusion rate constant, and I (mg g−1) is the intercept which gives information about the thickness of the boundary layer. When the adsorption mechanism follows the intra-particle diffusion model, a plot of q t against t 1/2 should be given a linear line. Generally, if the intra-particle diffusion is the only rate-controlling step, then the plot passes through the origin, if not, the boundary layer diffusion affects the adsorption to some extent [37, 38].
Figure 9 shows that the adsorption plots of q t versus t 1/2 are not linear over the whole time range, and can be split into multilinear plots of intra-particle diffusion process with different slopes, which indicates that two or more steps occur in the adsorption process. The first portion can be assigned to the film diffusion of dye through the solution towards the external surface of hydrogel or the boundary layer diffusion of dyes molecules, where the adsorption rate is very high. The second portion is intra-particle diffusion of dye molecules through the pores of hydrogel and illustrates the gradual adsorption stage, where intra-particle diffusion rate is rate-controlling. The third portion involves the final equilibrium stage, where the intra-particle diffusion starts to slow down and level out [39]. It can be also observed from Fig. 9 that the plots do not pass through the origin (I ≠ 0), suggesting that the intra-particle diffusion is not the only rate-limiting step and some other adsorption mechanisms may also play an important role [40].

Intra-particle diffusion kinetics for adsorption of MB and MG on CS/(AMPS-co-AM) hydrogel
The parameters k int and R 2 of all three linear segments of q t versus t 1/2 are shown in Table 2. It can be easily observed that k int values are increased as k 1 > k 2 > k 3; indicating that film diffusion is a rapid process while intra-particle diffusion is a gradual process. Besides, all R 2 values reflect the specific applicability of Weber’s intra-particle diffusion model in various stages of dye adsorption onto the CS/P(AMPS-co-AM) hydrogel. Similar results were reported on the adsorption of Congo Red on coal-based mesoporous activated carbon and MB onto poly(cyclotriphosphazene-co-4,4′-sulfonyldiphenol) nanotubes [39, 40].
Effect of concentration on adsorption capacity
An ideal adsorbent for the removal of dyes should not only have a fast rate of adsorption but also a large adsorption capacity [40]. Adsorption isotherms give a description of how adsorbates interact with adsorbents and can provide information about the relationship between the amount of dye adsorbed on the solid phase and the concentration of dye in solution. Figure 10 shows the effect of the adsorption capacity on the equilibrium concentration at pH 5.8 and 25 °C. It was observed that the adsorption capacity increased with the increase of dyes equilibrium concentration and reached saturation adsorption at higher equilibrium concentrations. This is because at lower concentrations almost all dyes molecules can contact with the active sites on the surface of hydrogel [40, 41]. Nevertheless, the adsorption sites will reach saturation at high equilibrium concentrations, indicating that the hydrogel is an effective adsorbent for high concentration dyes.

The effect of equilibrium concentration on adsorption capacity at pH 5.8 and 25 °C
Adsorption isotherms
To investigate an interaction of the adsorbent surface and adsorbate molecules, the Freundlich and Langmuir isotherms models were selected to elucidate dye–hydrogel interaction in this work. In general, the linear regression is the most commonly used method to study the isotherm parameters.
Freundlich isotherm is an empirical model that is the earliest known relationship describing the adsorption equation and it is based on adsorption on a heterogeneous surface. It is given by the equation as follows [42]:
where K F (mg g−1) is the Freundlich characteristic constant, and n is the adsorption intensity. If a value for n is above unity, adsorption is a favorable and physical process. If a value for n is below unity, this implies that adsorption process is governed by a chemical mechanism [42, 43].
The Langmuir isotherm model assumes monolayer adsorption on a surface with a finite number of identical sites. It is given as:
where b (L mg−1) is the Langmuir adsorption equilibrium constant, and q m (mg g−1) is the maximum amount of adsorption. The essential characteristic of the Langmuir isotherm model can be expressed as the dimensionless separation factor R L:
where C 0 (mg L−1) is the initial dyes concentration. The R L indicates the type of isotherm whether: unfavorable (R L > 1), linear (R L = 1), favorable (0 < R L < 1) or irreversible (R L = 0) [44, 45].
If the adsorption isotherm is closely fitted with the Langmuir model, the values of free energy change (∆G 0) may be calculated according to the following equation:
The ∆G 0 for the chemisorption is at a range of −80 to −400 kJ mol−1, the physisorption together with chemisorption is at the range of −20 to −80 kJ mol−1, and the physisorption is generally between −20 and 0 kJ mol−1 [42].
Figure 11 shows the experimental data and predicted equilibrium curve of Freundlich (a) and Langmuir (b) isotherms. The relative parameters are listed in Table 3. The plots suggest that the adsorption processes can be described by Langmuir and Freundlich isotherms, though Langmuir isotherm provides a better fit to the experimental data because Langmuir isotherm agrees with the experimental data better than that of Freundlich. In addition, it is found that R 2 obtained from the Langmuir isotherm model is 0.9987 and 0.9943, which is higher than those of Freundlich isotherm (0.9658 and 0.8983) as listed in Table 3. The result suggests that the experimental data is not fitted well with the Freundlich isotherm.

a Freundlich isotherm and b Langmuir isotherm
It is found from Table 3 that the values of n in Freundlich isotherms are 8.32 and 3.21, representing favorable adsorption, and therefore, this suggests that a physical mechanism, which is referred to the weak adsorption bond and conducted with Van der Waals forces and hydrophobic interaction [42], is dominant rather than a chemical adsorption.
Langmuir isotherm indicates the monolayer adsorption of dye onto the hydrogel. The R L values of adsorption dye onto hydrogels are 0.01–0.04 and 0.08–0.25 for MB (700–1,500 mg L−1) and MG (400–1,400 mg L−1), respectively, which lie in the range of 0–1, a favorable adsorption of MB and MG on hydrogel. In addition, the ∆G 0 values are –23.03 and –19.64 kJ mol−1 for MB and MG, which lie between –80 and 0 kJ mol−1. Thus, the adsorption of dyes on hydrogel is a physisorption together with chemisorption process due to electrostatic interactions, ion exchange, hydrophobic interaction, and Van der Waals forces. Negative ∆G 0 values suggest that the adsorption process takes place spontaneously.
Many works have reported the use of modified chitosan to remove dyes [46, 47]. To evaluate the hydrogel synthesized by GEDP among the adsorbents prepared from other techniques for the removal of two dyes from aqueous solutions, a comparison based on the Langmuir adsorption capacity, q m, is listed in Table 4. The results show that the CS/P(AMPS-co-AM) hydrogel prepared by GDEP has higher adsorption capacity for the two dyes, and this novel composite has a great potential as an alternative promising adsorbent for the removal of dyes from aqueous solutions.
Conclusion
The CS/P(AMPS-co-AM) hydrogels were prepared by GDEP-initiated graft copolymerization in aqueous solution, in which MBA was used as a cross-linking agent. FTIR and XRD indicated that AM and AMPS were grafted onto the CS backbone successfully, forming copolymer. TG/DTG suggested that grafted AMPS and AM onto CS could change the thermal stability of the CS. SEM showed a unique three-dimensional porous structure for the CS/P(AMPS-co-AM). The hydrogel was applied to the removal of MB and MG from aqueous solutions. The adsorption capacities were affected significantly by pH, contact time and dye initial concentration. The results showed that the adsorption capacities were much higher in neutral solutions than those in acidic conditions. Adsorption kinetic indicated that adsorption behavior of dyes on CS/P(AMPS-co-AM) hydrogel followed the pseudo-second-order model with multi-step diffusion process. The equilibrium experimental data fitted perfectly with the Langmuir isotherm. The CS/P(AMPS-co-AM) hydrogel showed very large adsorption capacities at pH 5.8 and 25 °C, whose maximum adsorption capacity for MB and MG based on Langmuir isotherm were 1,538.5 and 917.4 mg g−1, respectively. Negative ∆G 0 values revealed the spontaneous nature of the adsorption process. All results indicated that GDEP may provide a new method for the preparation of CS/P(AMPS-co-AM) hydrogels. The hydrogel is believed to be a very promising candidate for highly efficient removal of cationic dyes from aqueous solutions.
References
Singh RL, Singh PK, Singh RP (2015) Enzymatic decolorization and degradation of azo dyes—a review. Int Biodeterior Biodegrad 104:21–31
Wang SM, Guan Y, Wang LP, Zhao W, He H, Xiao J, Yang SG, Sun C (2015) Fabrication of a novel bifunctional material of BiOI/Ag3VO4 with high adsorption–photocatalysis for efficient treatment of dye wastewater. Appl Catal B 168–169:448–457
Raghu S, Lee CW, Chellammal S, Palanichamy S, Basha CA (2009) Evaluation of electrochemical oxidation techniques for degradation of dye effluents—a comparative approach. J Hazard Mater 171:748–754
Muthukumar M, Sargunamani D, Selvakumar N (2005) Statistical analysis of the effect of aromatic, azo and sulphonic acid groups on decolouration of acid dye effluents using advanced oxidation processes. Dyes Pigments 65:151–158
Reddy DHK, Lee SM (2013) Application of magnetic chitosan composites for the removal of toxic metal and dyes from aqueous solutions. Adv Colloid Interface Sci 201–202:68–93
Punzi M, Anbalagan A, Börner RA, Svensson BM, Jonstrup M, Mattiasson B (2015) Degradation of a textile azo dye using biological treatment followed by photo-Fenton oxidation: evaluation of toxicity and microbial community structure. Chem Eng J 270:290–299
Ungureanu G, Santos S, Boaventura R, Botelho C (2015) Arsenic and antimony in water and wastewater: overview of removal techniques with special reference to latest advances in adsorption. J Environ Manag 151:326–342
Crini G (2006) Non-conventional low-cost adsorbents for dye removal: a review. Bioresour Technol 97:1061–1085
Yu J, Yang GG, Pan YP, Lu QF, Yang W, Gao JZ (2014) Poly(acrylamide-co-acrylic acid) hydrogel induced by glow-discharge electrolysis plasma and its adsorption properties for cationic dyes. Plasma Sci Technol 16:767–776
Wang L, Zhang JP, Wang AQ (2011) Fast removal of methylene blue from aqueous solution by adsorption onto chitosan-g-poly(acrylic acid)/attapulgite composite. Desalination 266:33–39
Liu PF, Gao WJ, Zhang QS, Chen K, Zhang J, Chen L, Zhang XY, Wang K (2015) Temperature-sensitive hydrogel modified by polymerizable liquid crystal AAc-Brij-58: optical and protein adsorption/desorption behaviors. React Funct Polym 89:1–8
Wang L, Zhang JP, Wang AQ (2008) Removal of methylene blue from aqueous solution using chitosan-g-poly(acrylic acid)/montmorillonite superadsorbent nanocomposite. Colloids Surf A 322:47–53
Liu Y, Zheng Y, Wang AQ (2010) Enhanced adsorption of methylene blue from aqueous solution by chitosan-g-poly(acrylic acid)/vermiculite hydrogel composites. J Environ Sci 22:486–493
Luo JW, Han GC, Xie MJ, Cai ZR, Wang XY (2015) Quaternized chitosan/montmorillonite nanocomposite resin and its adsorption behavior. Iran Polym J 24:531–539
Bekçi Z, Özveri C, Seki Y, Yurdakoç K (2008) Sorption of malachite green on chitosan bead. J Hazard Mater 154:254–261
Xing Y, Sun XM, Li BH (2009) Poly(methacrylic acid)-modified chitosan for enhancement adsorption of water-soluble cationic dyes. Polym Eng Sci 49:272–280
Nguyen NT, Liu JH (2013) Fabrication and characterization of poly(vinyl alcohol)/chitosan hydrogel thin films via UV irradiation. Eur Polym J 49:4201–4211
Gad YH (2008) Preparation and characterization of poly(2-acrylamido-2-methylpropanesulfonic acid)/chitosan hydrogel using gamma irradiation and its application in wastewater treatment. Radiat Phys Chem 77:1101–1107
Wang XY, Zhou MH, Jin XL (2012) Application of glow discharge plasma for wastewater treatment. Electrochim Acta 83:501–512
Joshi AA, Locke BR, Arce P, Finney WC (1995) Formation of hydroxyl radicals, hydrogen peroxide and aqueous electrons by pulsed streamer corona discharge in aqueous solution. J Hazard Mater 41:3–30
Harada K, Iwaski T (1974) Syntheses of amino acids from aliphatic carboxylic acid by glow discharge electrolysis. Nature 250:426–428
Malik MA, Ghaffar A, Malik SA (2001) Water purification by electrical discharges. Plasma Sources Sci Technol 10:82–91
Brisset JL, Moussa D, Doubla A, Hnatiuc E, Hnatiuc B, Youbi GK, Herry JM, Naïtali M, Bellon-Fontaine MN (2008) Chemical reactivity of discharges and temporal post-discharges in plasma treatment of aqueous media: examples of gliding discharge treated solutions. Ind Eng Chem Res 47:5761–5781
Friedrich JF, Mix R, Schulze RD, Meyer-Plath A, Joshi R, Wettmarshausen S (2008) New plasma techniques for polymer surface modification with monotype functional groups. Plasma Process Polym 5:407–423
Lu QF, Yu J, Gao JZ, Yang W, Li Y (2011) Glow-discharge electrolysis plasma induced synthesis of polyvinylpyrrolidone/acrylic acid hydrogel and its adsorption properties for heavy-metal ions. Plasma Process Polym 8:803–814
Yu J, Pan YP, Lu QF, Yang W, Gao JZ, Li Y (2012) Synthesis and swelling behaviors of P(AMPS-co-AAc) superabsorbent hydrogel produced by glow-discharge electrolysis plasma. Plasma Chem Plasma Process 33:219–235
Mukhopadhyay P, Sarkar K, Bhattacharya S, Bhattacharyya A, Mishra R, Kundu PP (2014) pH sensitive N-succinyl chitosan grafted polyacrylamide hydrogel for oral insulin delivery. Carbohydr Polym 112:627–637
Bao Y, Ma JZ, Li N (2011) Synthesis and swelling behaviors of sodium carboxymethyl cellulose-g-poly(AA-co-AM-co-AMPS)/MMT superabsorbent hydrogel. Carbohydr Polym 84:76–82
Malik MA, Ahmed M, Rehman E, Naheed R, Ghaffar A (2003) Synthesis of superabsorbent copolymers by pulsed corona discharges in water. Plasmas Polym 8:271–279
Mukhopadhyay P, Sarkar K, Soam S, Kundu PP (2013) Formulation of pH-responsive carboxymethyl chitosan and alginate beads for the oral delivery of insulin. J Appl Polym Sci 129:835–845
Bhattacharyya R, Ray SK (2014) Micro- and nano-sized bentonite filled composite superabsorbents of chitosan and acrylic copolymer for removal of synthetic dyes from water. Appl Clay Sci 101:510–520
Ahn JS, Choi HK, Cho CS (2001) A novel mucoadhesive polymer prepared by template polymerization of acrylic acid in the presence of chitosan. Biomaterials 22:923–928
Zhou JQ, Wang JW (2009) Immobilization of alliinase with a water soluble–insoluble reversible N-succinyl-chitosan for allicin production. Enzyme Microb Technol 45:299–304
Zhang JP, Wang Q, Wang AQ (2007) Synthesis and characterization of chitosan-g-poly(acrylic acid)/attapulgite superabsorbent composites. Carbohydr Polym 68:367–374
Limparyoon N, Seetapan N, Kiatkamjornwong S (2011) Acrylamide/2-acrylamido-2-methylpropane sulfonic acid and associated sodium salt superabsorbent copolymer nanocomposites with mica as fire retardants. Polym Degrad Stab 96:1054–1063
Pourjavadi A, Tehrani ZM, Salimi H, Banazadeh A, Abedini N (2015) Hydrogel nanocomposite based on chitosan-g-acrylic acid and modified nanosilica with high adsorption capacity for heavy metal ion removal. Iran Polym J 24:725–734
Wang Y, Wang W, Wang A (2013) Efficient adsorption of methylene blue on an alginate-based nanocomposite hydrogel enhanced by organo-illite/smectite clay. Chem Eng J 228:132–139
Vimonses V, Lei S, Jin B, Chow CWK, Saint C (2009) Adsorption of Congo red by three Australian kaolins. Appl Clay Sci 43:465–472
Lorenc-Grabowska E, Gryglewicz G (2007) Adsorption characteristics of Congo red on coal-based mesoporous activated carbon. Dyes Pigments 74:34–40
Chen ZH, Zhang JA, Fu JW, Wang MH, Wang XZ, Han RP, Xu Q (2014) Adsorption of methylene blue onto poly(cyclotriphosphazene-co-4,4′-sulfonyldiphenol) nanotubes: kinetics, isotherm and thermodynamics analysis. J Hazard Mater 273:263–271
Li XL, Li YF, Zhang SD, Ye ZF (2012) Preparation and characterization of new foam adsorbents of poly(vinyl alcohol)/chitosan composites and their removal for dye and heavy metal from aqueous solution. Chem Eng J 183:88–97
Şölener M, Tunali S, Özcan AS, Özcan A, Gedikbey T (2008) Adsorption characteristics of lead(II) ions onto the clay/poly(methoxyethyl)acrylamide (PMEA) composite from aqueous solutions. Desalination 223:308–322
Kumar PS, Ramalingam S, Senthamarai C, Niranjanaa M, Vijayalakshmi P, Sivanesan S (2010) Adsorption of dye from aqueous solution by cashew nut shell: studies on equilibrium isotherm, kinetics and thermodynamics of interactions. Desalination 261:52–60
Mahdavinia GR, Aghaie H, Sheykhloie H, Vardini MT, Etemadi H (2013) Synthesis of CarAlg/MMt nanocomposite hydrogels and adsorption of cationic crystal violet. Carbohydr Polym 98:358–365
Panic VV, Madzarevic ZP, Volkov-Husovic T, Velickovic SJ (2013) Poly(methacrylic acid) based hydrogels as sorbents for removal of cationic dye basic yellow 28: kinetics, equilibrium study and image analysis. Chem Eng J 217:192–204
Ngah WSW, Ariff NFM, Hashim A, Hanafiah MAKM (2010) Malachite green adsorption onto chitosan coated bentonite beads: isotherms, kinetics and mechanism. Clean Soil Air Water 38:394–400
Peng Q, Liu MX, Zheng JW, Zhou CR (2015) Adsorption of dyes in aqueous solutions by chitosan–halloysite nanotubes composite hydrogel beads. Microporous Mesoporous Mater 201:190–201
Acknowledgments
This work was supported in part by National Natural Science Foundation of China (Nos. 21367023 and 21567025), Natural Science Foundation of Gansu Province (Nos. 1308RJZA144 and 1208RJZA161), Scientific Research Project in Higher Education Institutions of Gansu Province (No. 2013-019), and Key Project of Young Teachers’ Scientific Research Promotion of Northwest Normal University (No. NWNU-LKQN-12-9), China.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Yu, J., Li, Y., Lu, Q. et al. Synthesis, characterization and adsorption of cationic dyes by CS/P(AMPS-co-AM) hydrogel initiated by glow-discharge-electrolysis plasma. Iran Polym J 25, 423–435 (2016). https://doi.org/10.1007/s13726-016-0434-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13726-016-0434-8