
Abstract
Purpose of Review
Since the mid-twentieth century, obesity and its related comorbidities, notably insulin resistance (IR) and type 2 diabetes (T2D), have surged. Nevertheless, their underlying mechanisms remain elusive. Evolutionary medicine (EM) sheds light on these issues by examining how evolutionary processes shape traits and diseases, offering insights for medical practice. This review summarizes the pathogenesis and genetics of obesity-related IR and T2D. Subsequently, delving into their evolutionary connections. Addressing limitations and proposing future research directions aims to enhance our understanding of these conditions, paving the way for improved treatments and prevention strategies.
Recent Findings
Several evolutionary hypotheses have been proposed to unmask the origin of obesity-related IR and T2D, e.g., the "thrifty genotype" hypothesis suggests that certain "thrifty genes" that helped hunter-gatherer populations efficiently store energy as fat during feast-famine cycles are now maladaptive in our modern obesogenic environment. The "drifty genotype" theory suggests that if thrifty genes were advantageous, they would have spread widely, but proposes genetic drift instead. The "behavioral switch" and "carnivore connection" hypotheses propose insulin resistance as an adaptation for a brain-dependent, low-carbohydrate lifestyle. The thrifty phenotype theory suggests various metabolic outcomes shaped by genes and environment during development. However, the majority of these hypotheses lack experimental validation. Understanding why ancestral advantages now predispose us to diseases may aid in drug development and prevention of disease.
Summary
EM helps us to understand the evolutionary relation between obesity-related IR and T2D. But still gaps and contradictions persist. Further interdisciplinary research is required to elucidate complete mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
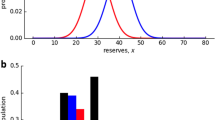
Obesity is a serious global health issue that affects over 500 million individuals across the world [1,2,3]. The incidence of obesity has risen significantly since the mid-twentieth century [4] (Fig. 1a). Even though recent studies have reported that obesity rates in most economically advanced countries have stabilized since 2000, low-income nations continue to experience an uninterrupted rise in obesity rates. Also, an escalating number of high- and middle-income nations are confronting an epidemic of severe obesity. Projections forecast a twofold increase in severe obesity prevalence among high-income populations by 2035, presenting a formidable challenge to healthcare infrastructures. Even if there is a temporary stabilization, global obesity prevalence persists at unacceptably high levels, and there is no assurance of prolonged stability [5]. With the development of obesity in the last decade, it is also observed an increase in the incidence type 2 diabetes (T2D) (Fig. 1b). Apart from T2D, obesity also serves as a significant risk factor for the development of various human diseases, including cardiovascular diseases (CVDs) [6, 7], and cancers [1,2,3, 8] (Fig. 1c). Earlier studies have reported that the occurrence of these diseases is mainly due to obesity-associated insulin resistance (IR) [9]. IR is mainly influenced by insulin hormones secreted by the pancreas. Insulin plays a crucial role in regulating blood sugar (glucose) levels by aiding the absorption of glucose into cells, where it can either be utilized for energy or stored for future use [10]. During IR, cells exhibit reduced responsiveness to insulin's signal to take up glucose. Consequently, the pancreas increases its insulin production to compensate, resulting in elevated insulin levels in the bloodstream (hyperinsulinemia). This in turn could potentially contribute to the development of T2D [11,12,13].

Prevalence and consequences of obesity in different economic regions of the world. a The percentage of adults with obesity, defined as having a Body Mass Index (BMI) equal to or exceeding 30 kg/m2, adjusted for age standardization in specified populations from 1975 to 2019 [27, 28]. b The percentage of individuals within a specified population who have either a fasting glucose level of 126 mg/dl (7.0 mmol/l) or higher, a history of diagnosed diabetes or are currently using insulin or oral hypoglycemic drugs [29, 30] from 1980 to 2014. The survey data didn't differentiate between type 1 and type 2 diabetes, as distinguishing between these conditions in adults can be challenging. However, the majority (85–95%) of diabetes cases in adults are attributed to type 2 diabetes. Hence, the observed increase in diabetes prevalence among adults is highly likely to be driven by the rise in type 2 diabetes cases [29]. c Health risks associated with Obesity [31, 32]. Figure was generated using the rnaturalearth [33] and ggplot2 [34] package of R v. 4.0.5 and https://app.biorender.com/
Earlier studies suggest that a combination of genetic and environmental factors influences the development of T2D [14]. T2D genetics is very complicated [15]. This may be caused by common variants with small effects and rare variants with bigger effects. However, approaches to understanding disease's natural progression based on genetic factors are underdeveloped, and the specific genetic factors underlying T2D development remain largely unknown [16]. This, in turn, may hinder the lack of discovery of effective drugs for T2D. Thus, there is always a quest to understand the key reason for developing obesity-associated IR and T2D and how they affect human health. Recently developed evolutionary medicine (EM), also known as Darwinian medicine, explores how evolutionary forces have shaped human traits and diseases, and how we can use evolutionary concepts in medical practice [17•]. EM and conventional medicine have distinct perspectives on time. In conventional medicine, the focus is primarily on immediate causes, often referred to as "how" questions [18]. Conversely, evolutionary theories try to figure out "why" certain characteristics have changed through time. For instance, when confronted with a cancer diagnosis, a patient may wonder, "Why did I get cancer?" [18]. To this a physician would say, "Because you have a certain mutation(s)". While this addresses the proximate cause, it does not fully answer the patient's underlying question of why cancer arises in the first place. In contrast, EM may hypothesize that cancer is a shared consequence of multicellular life. The same traits that allow complex multicellular organisms to thrive also create vulnerabilities that can lead to the development of cancer. From this viewpoint, cancer is not an aberration, but rather an inevitable byproduct of the evolutionary tradeoffs inherent in the emergence of multicellular life. In these instances, the proverb "timing is everything" becomes particularly relevant in distinguishing the difference in perspective between conventional medicine and EM.
To date, the evolutionary medicine approach has been employed to understand the various human traits and diseases, including mental health [19, 20], the immune system evolution [21, 22], and T2D [23]. For example, in 1962, Neel introduced the thrifty genotype theory to explain the increasing occurrence of diabetes in developed countries. Since then, it has been recognized as a potential explanation for the worldwide rise in obesity and non-insulin-dependent diabetes mellitus (NIDDM) or T2D as societies rapidly adopted Western lifestyles during the twentieth century [23, 24]. Subsequently, several evolutionary hypotheses, like Drifty [25] and Behavioral Switch [26], have been proposed, focusing on the origin and possible causes of obesity-associated IR and T2D. Based on the aforementioned considerations, our current studies initially aimed to summarize the pathogenesis and genetic factors linked to obesity-associated IR and T2D development. Subsequently, we sought to explore the connection between obesity-related IR and T2D from an evolutionary perspective. Later, we also described the limitations associated with these evolutionary theories and how the evolutionary approach can be improvised in the near future to unmask the mechanisms associated with the IR, which in the near future, can be used for treatments and prevention strategies for the disease.
Pathogenesis Associated with Obesity Associated IR and T2D
Adipose tissue plays a crucial role in maintaining the body's energy and glucose homeostasis. Its primary function is to store excess energy (glucose) in the form of lipids, particularly triacylglycerols, when there is an excess of energy intake [35]. When glucose is scarce, adipose tissue plays a crucial adaptive role by increasing lipolysis and free fatty acids (FFA)/glycerol release, modulating adipokine secretion, and regulating thermogenesis. These mechanisms help preserve the limited glucose supply through gluconeogenesis for use by glucose-dependent tissues, particularly the brain, and maintain overall glucose homeostasis [36,37,38,39,40]. However, the adipocytes have a limited capacity for storing calories. In the presence of large amounts of calories, they become physiologically stressed, experiencing profound hypoxia. This triggers the activation of inflammatory signaling cascades, such as those mediated by hypoxia-inducible factor-1 (HIF-1) [41] and c-Jun N-terminal kinase (JNK) [42]. Activating these pathways leads to the upregulation of pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), fostering chronic, low-grade inflammation within the adipose tissue compartment. This inflammatory environment directly hampers insulin signal transmission, resulting in systemic IR affecting vital metabolic tissues like skeletal muscle, liver, and adipose tissue [43] (Fig. 2).

Pathogenesis of obesity associated IR and T2D. Clinically, T2D is characterized by dysfunction of β cells and IR. β cell dysfunction may develop because of numerous environmental as well as genetic predisposition factors. During the initial T2D phase, the β-cell compensates for insulin deficiency through increased insulin secretion. Reduced plasma insulin increases glucose levels. Glucose-sensitive tissues, including the liver, muscle, and adipocytes, are unable to accommodate glucose's enhanced concentration. Persistent glucose release maintains the hyperglycemic environment, which in turn leads to T2D
The worsening of IR in both the liver and muscles is primarily caused by the excessive accumulation of lipids [44]. Fatty deposits in the liver lead to IR, triggering Kupffer cells to release inflammatory cytokines and activate NF-κB. Elevated levels of fatty acids induce IR during euglycemic-hyperinsulinemic conditions and increases DAG levels [44, 45] and ceramide accumulation [44, 46]. Simultaneously, the elevated DAG levels activate protein kinase C epsilon (PKCϵ), which impairs the function of the insulin receptor tyrosine kinase, disrupting insulin signaling and leading to a decrease in insulin-stimulated glycogen synthesis, primarily due to reduced phosphorylation of glycogen synthase kinase 3 (GSK3). Additionally, DAG dampens glycogen synthase activity, further contributing to impaired insulin signaling. Moreover, DAG promotes hepatic gluconeogenesis by reducing the phosphorylation of FoxO, leading to increased translocation of FoxO into the nucleus and enhanced transcription of gluconeogenic genes such as phosphoenolpyruvate carboxykinase (PEP-CK) and glucose-6-phosphatase (G6P) [47]. Likewise, the cytokines and adipocytokines released during systemic inflammation activate NF-κB and JNK, further upregulating the transcription of hepatic gluconeogenic enzymes and contributing to increased glucose production in the liver [48]. Furthermore, ceramide, a potent inflammatory activator, can stimulate JNK and NF-κB/IKK pathways, closely linked to IR [44]. In skeletal muscle, increased lipid accumulation triggers the production of long-chain CoA and elevates DAG levels. This surge in DAG is linked to diminished fat oxidation due to mitochondrial dysfunction, hindering glucose uptake by muscle cells. Beyond its role in facilitating triglyceride synthesis for cellular lipid storage, increased DAG levels activate PKCθ. This, in turn, enhances IRS-1 phosphorylation, disrupting insulin signaling. Consequently, the impairment of the PI3K/AKT pathway, crucial for glucose transport and glycogen synthesis, results in diminished glucose transport and glycogen synthesis in muscle cells [48,49,50,51].
As obesity-induced IR worsens, it contributes to inflammation, impairs glucose transport in adipose tissue, and triggers an increase in the volume of β-cell mass. This is a compensatory mechanism to meet the increased demand for insulin secretion caused by the reduced effectiveness of insulin due to IR. However, the increased insulin secretion eventually leads to hyperinsulinemia, exacerbating the accumulation of lipids in both the liver and throughout the body (systemic lipid accumulation). Furthermore, during IR, increased glycolysis due to high insulin levels leads to more pyruvate and NADH production [52]. This excess pyruvate can turn into lactate, especially when glycolysis is exaggerated during hyperinsulinemia. This lactate is then released into the bloodstream and utilized as a substrate for hepatic lipogenesis, contributing to further lipid buildup. Lastly, as a consequence, the β-cells undergo collapse, resulting in a deficiency of insulin secretion, ultimately leading to hyperglycemia or high blood sugar levels [48].
Genetic Overlap Between Obesity and T2D
Until 2007, genetic mapping of complex diseases such as T2D and obesity relied primarily on genetic linkage analyses or candidate gene association studies, limited by design flaws and small sample sizes [15]. However, technological advancements in array-based genotyping facilitated Genome-Wide Association Studies (GWASs), leading to rapid progress in identifying common variants associated with these diseases [53], including obesity [54, 55] and T2D [56, 57] (Supp Tables 1.1 and 1.2). The first GWAS for obesity traits, published in 2007, found that common variations in the FTO gene are linked to BMI [58]. To date, GWAS has uncovered 78 obesity-related variants and 53 genes (Supp Table 1.1). By early 2014, 90 genetic loci were firmly established as T2D risk loci, with the TCF7L2 locus showing the most significant association [59]. GWAS has uncovered T2D related 780 variants and 434 genes [60, 61, 62•] (Supp Table 1.2). Amongst these, 12 genes (Fig. 3a) and 3 variants (Fig. 3b) are overlapping between T2D and Obesity. Amongst three variants, rs1421085 of the FTO gene are also associated with body mass index (BMI) [63], and blood pressure (BP) [64]. Interestingly, rs1421085 is present in highly conserved regions, thereby suggesting its potential functionality [65].

Obesity and T2D associated with gene and its variants associated with identified by GWAS: a 12 overlapping genes, b 3 variants, c Of the three variants, rs1421085 in the FTO gene is linked with obesity related traits, like body mass index and blood pressure
Recent study [66] has also demonstrated that genetic variations in the FTO gene are linked to obesity and dietary habits. Using 71 adults in Jakarta, Indonesia, the authors examined the association between rs1421085 and BMI, macronutrients, and fatty acid intake using various genetic models. The results revealed a significant increase in BMI only among individuals with the CC genotypes. Moreover, those with the minor C allele exhibited higher fat intake across different genetic models and increased consumption of monounsaturated and saturated fatty acids for TC/CC genotypes compared to TT genotype carriers. This underscores the significant role of the FTO gene in food preference and its impact on body weight. The rs1421085 C allele of the FTO gene has also been associated with an increased risk of obesity and IR [67, 68]. Specifically, the rs1421085 variant was positively associated with insulin sensitivity measures, such as fasting insulin levels and HOMA-IR (a marker of IR) [67, 68]. The influence of the rs1421085 variant on insulin sensitivity appears to be mediated through its effects on adiposity, as the associations were attenuated after adjusting for BMI. This suggests the variant may impact IR partly by influencing body weight and fat accumulation [68]. Studies have also linked the rs1421085 C allele to an increased risk of polycystic ovary syndrome (PCOS), which is often accompanied by IR [69, 70]. This provides further evidence for the potential role of rs1421085 in Obesity, IR, and T2D.
Recent studies have also shed light on population-specific T2D loci, for instance, KCNQ1 and C2CD4A in Japanese individuals [71,72,73]. Similarly, other studies have identified obesity-specific risk variants in Asian or African populations, such as the PCSK1 [74, 75]. The protein encoded by PCSK1 is crucial for cleaving peptide hormone precursors in regulating food intake, glucose, and energy homeostasis, such as proopiomelanocortin, proinsulin, proglucagon, and proghrelin [76]. SNP rs155971, located in intron 6 of PCSK1, was associated with obesity in the Chinese population [77]. Another SNP, rs261967, was linked to BMI in East Asians, while rs2570467 showed a weak association with waist circumference in individuals of African ancestry [74, 78]. While these SNPs didn't cause changes in the protein sequence, mutations within introns could potentially impact gene expression and splicing [79]. Thus, comparing genetic associations across diverse ethnic backgrounds may provide valuable insights into both shared and distinct genetic susceptibilities [80]. However, most of the GWAS studies were carried out in the European population [81]. Also, GWAS can explain only a small proportion of the heritability of many complex traits, leaving a substantial portion unaccounted for. This "missing heritability" could be due to rare variants, gene-gene interactions, gene-environment interactions, and epigenetic factors not captured by GWAS. Recently developed evolutionary medicine offers a powerful framework for understanding and addressing complex health challenges by integrating evolutionary principles with biomedical knowledge and practice [82,83,84,85, 86•]. It has significant potential to transform how we approach prevention, diagnosis, and treatment across a wide range of medical domains. Evolutionary medicine may also offer a valuable framework for designing and interpreting rare variant association studies, leveraging insights into the genetic architecture of complex diseases across different populations and the functional consequences of rare variants to improve the detection and clinical application of these important genetic factors [17, 87].
Evolutionary Hypothesis Proposing the Link Between Obesity and Type 2 Diabetes
In 1962, Neel proposed the concept of the thrifty genotype to explain the increasing occurrence of diabetes in developed countries [23]. Since then, this concept has since been recognized as a potential explanation for the worldwide surge in obesity and T2D [23, 24]. Subsequently, several evolutionary hypotheses, like the Pima Paradox and Drifty hypothesis (Table 1), have been proposed, focusing on the origin and causes of obesity and T2D.
Thrifty Genotypes
In 1962, the American population geneticist, JV Neel, introduced a novel theory to explain the increasing prevalence of diabetes in the Western world [23]. He hypothesized that individuals predisposed to diabetes possessed a "thrifty genotype" that would have provided a survival advantage under historical "feast-and-famine" conditions. Specifically, Neel proposed that this thrifty genotype allowed for more efficient fat storage during food abundance, providing an energy reserve that could be utilized during periods of food scarcity [23, 98]. As in the twentieth century, people around the world transitioned towards the Westernized lifestyle, such as less physically active lifestyles, consuming high-fat & low-fiber diets, and intake of excess calories. This might induce the thrifty genes to accumulate more fat. However, since there is no food shortage or famine situation, this led to obesity and obesity-associated disorders, including T2D (Fig. 4). The hypothesis of Neel was subsequently used to clarify the epidemics of obesity, diabetes, and associated conditions that occurred in several population groups as they produced the rapid shift from traditional (agriculturalist or hunter-gatherer) to a Westernized lifestyle throughout the twentieth century [99]. For instance, the ancestors of modern Polynesians undertook extensive open ocean voyages, facing challenges such as cold stress and periods of starvation during their migration across the Pacific. These harsh conditions could have led to the proliferation of thrifty alleles among Polynesians, potentially contributing to the elevated rates of T2D and obesity observed in Polynesian populations today [100,101,102,103]. Earlier one study [89] in the Samoan population, indigenous Polynesian people of the Samoan Islands, identified a genetic variant, rs373863828 (p.Arg457Gln) in the CREBRF gene, which is common among Samoans and associated with increased fat storage and decreased energy use. This variant may have been advantageous in the past for Samoans facing periods of food scarcity, aligning with the "thrifty gene" hypothesis. However, in the contemporary obesogenic environment characterized by readily available high-calorie foods and reduced physical activity, this genetic predisposition contributes to the high prevalence of obesity observed in Samoans today. Recently, the Gly482Ser variant in the PARGC1A (Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha) gene was also examined as a potential "thrifty gene" in Pacific populations [88].

Thrifty gene hypothesis: The hunter-gatherer’s food metabolism in feast and famine environment helps them to survive. Modern individuals following a western lifestyle with less physical activity are highly prone to obesity and T2D
In addition to the thrifty genotype hypothesis focused on efficient fat storage, other mechanisms by which IR could have aided survival under historical "feast-and-famine" conditions have also been suggested [26, 104]. While Neel's thrifty gene hypothesis proposes that cycles of feast and famine led to the selection of a "quick insulin trigger" mechanism characterized by postprandial hyperinsulinemia. This mechanism was advantageous as it facilitated the storage of fat during periods of food abundance and made it available during times of food scarcity. In contrast, Reaven [105] hypothesized that muscle IR, rather than a "quick insulin trigger," was the relevant physiological abnormality that would have conferred a survival advantage. The rationale behind Reaven's hypothesis is that muscle IR would have helped conserve glucose by minimizing gluconeogenesis (the process of producing glucose from non-carbohydrate precursors) and redirected glucose utilization to prioritize the insulin-independent tissues like the brain, fetus, and mammary gland, facilitating their survival during food scarcity. Different tissues have varying degrees of dependence on insulin for glucose uptake. By modulating insulin levels and sensitivity, the body can fine-tune the allocation of energy resources to various organs. High IR or low insulin levels would divert more glucose to the insulin-independent brain, providing a survival advantage under starvation conditions. Beyond just efficient fat storage, this differential energy allocation mechanism has been proposed as another potential way IR could have been advantageous in historical "feast-and-famine" environments.
Sometimes, the thrifty genotype was also used an umbrella term for a set of hypotheses about genes that may have been selectively favored because of their effects in certain environments [106]. The best support for this concept comes from the work of Kagawa et al. [107], who linked single nucleotide polymorphisms to a broad range of local ecological, climatic, and agricultural stress worldwide. While this hypothesis may seem intuitive, it's crucial to note that despite the widespread adoption of Western lifestyles across the globe, only a minority of the population, approximately 20%–30%, actually experience obesity [108]. One of the major criticisms of the thrifty gene hypothesis is the lack of evidence supporting the notion that our ancestors faced frequent, high-mortality famines required to drive the selection of thrifty genes [109]. Consequently, discussions surrounding thrifty genes remain debatable.
Pima Paradox
Pima Paradox refers to the stark contrast in obesity and diabetes rates between the Pima Indians living traditional lifestyles in Mexico versus those living a more Westernized lifestyle in the U.S [90,91,92]. The author notes in a 1998 report in the New Yorker magazine (http://www.mendosa.com/ir_history.htm) stated that Pima Indians, who live in Arizona, are more prone to obesity in the world, and half of them have diabetes above the age of 35 [110]. Pima Indians of Arizona have limited European admixture [110], and their diabetes tends to be predominantly T2D (with little proof of the autoimmunity typical of T1D [111]). The absence of T1D and low admixture in this population may also mean that there is little genetic and environmental heterogeneity in the etiology of T2D in Pima Indians, thereby allowing T2D-associated susceptibility loci to be more easily identified. Diabetes is also familial in Pima Indians of Arizona, and the degree of familiarity is higher at younger onset ages relative to older onset ages [112]. Pima Indians diagnosed with T2D exhibit typical clinical characteristics shared by most populations affected by this disease [113], including obesity, IR, impaired insulin secretion, and elevated rates of endogenous glucose production [112]. Interestingly, Pima Indians in the Mexican state of Sierra Madre do not struggle with obesity or its related diseases, like T2D (https://www.surgicalgroupofjc.com/morbid_obesity_causes.shtml) [114, 115]. Thus, understanding of what makes these two Pima populations, i.e., Pima Indians of Arizona (experience obesity) and Mexico (experience no obesity), distinct from each other may not only unmask the molecular mechanism associated with the IR in Pima Indians but also in almost all human populations across the globe [114].
According to the prevailing view [114], this contrast may arise from the differing lifestyles embraced by the two Pima groups: while the Mexican Pima uphold a traditional lifestyle characterized by substantial physical activity, the Arizona Pima have adopted a more sedentary, Westernized way of living with decreased levels of physical activity. Moderate to heavy physical activity levels were observed roughly 2.5 times higher among Mexican men and seven times higher among Mexican women compared to their Arizona Pima counterparts. The heightened levels of physical activity observed among the Mexican Pima population seem to exert a significant protective influence against IR and the onset of T2D, regardless of their genetic predisposition.
Drifty Hypothesis
In 2008, Speakman critiqued the "thrifty gene" hypothesis as an attractive yet flawed concept. He introduced an alternative theory known as the "drifty gene" hypothesis. According to Speakman's proposal, most mutations found in genes related to obesity are likely neutral and have undergone genetic drift over evolutionary time, rather than being positively selected for as suggested by the thrifty gene theory. Speakman argued that if the thrifty genes were genuinely advantageous, they would have become more prevalent in the population over extended evolutionary periods, even if they provided only minimal benefits. Instead, he posited that alleles associated with obesity may have spread through genetic drift mechanisms rather than being primarily driven by positive selection for a "thrifty" phenotype [25].
Genetic drift [116] occurs when mutant alleles are not under active selection but are selectively neutral. For instance, our body's regulation of obesity typically involves upper and lower intervention limits [117,118,119,120]. The risk of starvation determines the lower intervention limit, while the upper intervention limit is influenced by the risk of predation. Speakman proposed that approximately 2 million years ago, as humans adapted to social life, developed arms, and controlled fire, our vulnerability to predation decreased, leading to an increase in body mass. Significant evidence also suggests that many wild animals, including mammals and birds, regulate body mass by balancing the risks of starvation (lower intervention point) and predation (upper intervention point) [121, 122]. Thus, separating these intervention points may be a key mechanism contributing to human predisposition to obesity. One of the scenarios explaining the prevalence of the obesity crisis, among many, is the predation release hypothesis, which focuses on the drifting of unselected genes.
Another possible explanation for the "drifty genotype" is that historically, humans have not been exposed to such high levels of dietary fat [25]. Consequently, mutations impairing the function of genes controlling fat oxidation could have occurred randomly and drifted in frequency. As a result, individuals would have varied fat oxidation capacities, which would not be problematic as long as fat intake remained moderate. However, individuals with these mutations would be at risk of obesity if dietary fat levels were too high. This hypothesis aligns with the observation that differences in baseline fat oxidation rates strongly predict susceptibility to obesity in both humans and animals [123,124,125,126,127,128,129,130].
In both scenarios, the underlying argument does not rely on the belief that obesity was traditionally advantageous. Instead, it suggests the opposite: that there was no positive selection for genes predisposing to obesity. Random mutations occur in these genes, and the frequencies of mutant alleles drift randomly. These genes may be considered "drifty genes" because their presence in the gene pool predisposes individuals to conditions with detrimental consequences in contemporary culture. Conversely, it is speculated that obesity-related genes were not subject to positive selection. In this case, mutations arise randomly in genes, and the frequency of resulting mutant alleles fluctuates randomly. Some individuals inherit a predisposition to diseases like obesity due to the impact of these genes - genes that could be termed "drifty" [25].
However, researchers have argued that none of the aforementioned models adequately capture the complexity associated with IR in industrialized countries [131, 132]. Furthermore, given the vastly different evolutionary selection processes encountered by the ancestors of various ethnic groups, a single evolutionary principle such as genetic drift or selection for thrifty genes cannot fully explain the genetic predisposition to obesity [131, 132].
A Behavioral Switch Hypothesis
If neither genetic drift nor natural selection for thrifty genes can account for the prevalence of obesity in developed nations, what else may be the reason? Earlier, Watve and Yajnik proposed that IR is a "socio-ecological adaptation that causes two phenotypic transitions, (i) a transition in reproductive strategy from 'r' (a large number of offspring with little investment in each) to 'K' (smaller number of offspring with more investment in each) and (ii) a transition from 'stronger to smarter' or 'soldier to diplomat,' i.e., from relatively more muscle dependent to brain dependent lifestyle" (Fig. 5) [26]. In the first scenario, authors hypothesized that the ideal environmental factors for the first two transitions are largely overlapping and interdependent, a common switch, e.g., the insulin signaling pathway, may have evolved for the two transitions [26]. For instance, the placenta is an organ that does not need insulin. Maternal IR during pregnancy may lead to increased nutrient absorption by the placenta. This results in a larger infant size at birth, independent of the mother's weight gain. This is proposed as a potential adaptive mechanism facilitated by IR during pregnancy [23, 133]. Additionally, it has been discovered that overexpression of the Klotho gene, recognized for extending lifespan, operates by enhancing IR [93]. Conversely, studies have also shown that IR may inhibit ovulation [134,135,136,137] and cause polycystic ovary syndrome (PCOS) [138, 139]. Thus, IR may act as a double-edged sword, decreasing fertility on one hand while boosting fetal growth on the other. While this dual effect is widely recognized, several crucial linkages in the process remain unknown.

A behavioral switch hypothesis: General relationships amongst environmental factors and the two transitions via ‘r’ (large number of offspring with little investment in each) to ‘K’ (smaller number of offspring with more investment in each). The excess food consumption adapted by the evolutionary social behaviour of soldier/diplomat showing IR as well as obesity that causes these two phenotypic transitions. This image is redraw based on information present in [26] under the creative common license CC BY 2.0
In the case of transitioning from a "stronger to smarter" lifestyle, IR redirects energy resources from muscle to brain tissues. This shift has significant implications, potentially enhancing cognitive function, memory, and learning due to increased glucose supply to the brain and the effects of hyperinsulinemia associated with IR. In younger individuals, the cognitive benefits of IR may have been evolutionarily selected for. Additionally, developmental factors, like intrauterine malnutrition, can influence the balance between physical and brain development, with IR possibly helping to maintain a preference for brain growth. Furthermore, the transition from a "soldier" to a "diplomat" lifestyle could be an adaptive response to varying nutritional conditions, with muscle strength prioritized during times of food scarcity and brain development favored during times of abundance or malnutrition [26].
Thus, the behavioral switch hypothesis attributes the surge in metabolic diseases to intense environmental factors such as population density, urbanization, social rivalry, easy access to high-calorie foods, and sedentary lifestyles, unprecedented in human history. Like the "thrifty" hypothesis, it suggests that physiological adaptations that were once beneficial are now detrimental in modern settings. Consequently, it advocates for a novel clinical and epidemiological approach, emphasizing social reforms to address root causes. It anticipates higher rates of obesity and diabetes in densely populated regions with increased socioeconomic competition. Mitigating urban overcrowding, reducing wealth disparities, and fostering social equality are proposed as strategies to curb the escalating insulin-related issues driving these metabolic disorders [140].
Carnivore Connection
In 1994, Miller and Colagiuri postulated that the amount and quality of carbohydrates in one's diet had a significant influence on the development of T2D [141]. Prior to the Pleistocene era, our prehuman ancestors in Africa lived in a warm, moist environment where carbohydrate-rich fruits and berries were an important energy source. However, the onset of the first severe Ice Ages around 2.5 million years ago dramatically altered the landscape, as the African forests gave way to drier, open woodlands and savannas. This environmental shift forced hominids to become increasingly carnivorous, supplementing their vegetarian diet with scavenged or hunted meat. The appearance of the first stone tools around 2 million years ago, and the active hunting and use of fire by Homo erectus 1.5 million years ago, further indicate this transition to a diet low in carbohydrates and high in protein. Even during warmer interglacial periods, many regions remained cold, and humans maintained a hunting and fishing existence with diets almost devoid of readily absorbable carbohydrates. While wild plant foods provided some carbohydrates, much of it was unavailable and elicited a low glycemic response due to high fiber content. This prolonged exposure to a low-carbohydrate, high-protein diet may have facilitated the development of IR as an adaptive mechanism, as the body sought to maintain glucose homeostasis for critical tissues like the brain and reproductive organs. The advent of agriculture 12,000 years ago, with its increased cereal consumption and high intakes of available starch, marked a significant dietary shift for many human populations, though some groups like the Australian Aborigines never adopted agriculture, maintaining their traditional low-carbohydrate, high-protein diets [94].
Considering this, the "Carnivore Connection Hypothesis" by Brand-Miller and Colagiuri suggests that during the Ice Ages, when human diets were low in carbohydrates and high in protein, there was selective pressure for IR genes. As agriculture increased carbohydrate intake, particularly in populations like Europeans, who have had high-carbohydrate diets for millennia, this selection pressure for IR genes may have been relaxed [94]. Thus, the prevalence of genes producing IR may be lower in the European population and any other group exposed to high-carbohydrate intake for a sufficiently long period of time. The prolonged consumption of high carbohydrates throughout millennia might have also led to increased positive selection for genes associated with elevated β-cell mass. In populations such as Swedish and Finnish, variants in 11 genes (TCF7L2, PPARG, FTO, KCNJ11, NOTCH2, WFS1, CDKAL1, IGF2BP2, SLC30A8, JAZF1, and HHEX) have shown significant associations with the risk of T2D independent of clinical risk factors [142]. Additionally, variants in eight of these genes have been linked to impaired β-cell function.
The Carnivore Connection hypothesis was also examined in sample populations from the Asian steppes [141]. Comparisons were made between traditional herders (pastoralists with a high-protein diet) and farmers (agriculturalists with a high-carbohydrate diet) regarding ten candidate genes associated with IR, anthropometry, and physiological measures, including HOMA IR. While none of the genes tested exhibited causal mutations with higher frequency in herders, neutrality tests indicated that some genes (SLC30A8, LEPR, and KCNQ1) might have been involved in past adaptations to diet. In line with the Carnivore Connection hypothesis, the prevalence of IR was significantly higher in herders compared to farmers, despite no major differences in their current diet. Environmental pressures such as geographic isolation and/or starvation may have further contributed to increases in the prevalence of IR gene(s) in specific population groups. Genetic bottlenecks and reduced genetic diversity have been observed in populations like the Nauruans and Pima Indians, who have also experienced food shortages and starvation in recent history [43]. Similar to low carbohydrate intake, starvation necessitates increased gluconeogenesis and peripheral IR [44], potentially selecting for individuals with profound IR that subsequent generations could inherit. Women with polycystic ovarian syndrome, known for exceptional IR, might also represent a group that was highly fertile in a low-carbohydrate environment [45]. Specific mechanisms for coping with low carbohydrate intake, rather than total dietary energy, may likely conferred the greatest reproductive and survival advantages. Thus, efforts to identify the gene(s) associated with IR and T2D have focused on genome-wide scans and other research avenues. However, given the complexity of human metabolism, multiple gene systems are likely involved, influencing insulin sensitivity and β-cell function [51]. For instance, PC-1 interferes with insulin receptor tyrosine kinase activity, thereby inhibiting cellular signaling [52], while reduced expression of the "susceptibility" gene CAPN10 has been linked to decreased glucose uptake in skeletal muscle [54]. The ACAD10 gene, associated with fatty-acid-induced IR, may catalyze mitochondrial fatty-acid oxidation [55].
The Adjustable Threshold Hypothesis
At the onset of 1871, Henry P. Bowditch observed that muscles respond to electrical stimuli in an all-or-nothing manner, where the stimulus either triggers the largest possible muscular contraction or no contraction at all [97]. Subsequently, similar dynamics were noted in other processes, such as nerve impulse propagation (action potential) along nerve trunks and the bursting electrical activity within pancreatic beta cells [143], leading to significant discoveries. Therefore, it is plausible that the dynamical approach would offer valuable insights into IR and T2D. Insights from such dynamical viewpoints prompt questions such as "How does a typical cell, the fundamental unit of life, respond to insulin?" Is there a gradual increase in response intensity, or does it surge abruptly, akin to muscle fiber contraction, when subjected to electrical current? Previous findings have indicated a gradual progression in our body's response to insulin [144], suggesting that cells may exhibit a similar graded response to insulin (the graded response view). In this context, the adjustable threshold hypothesis, proposed by Wang, suggests that in response to insulin, most cells either fully express or completely suppress their sensitivity to the hormone. At two threshold insulin concentrations, IRon and IRoff, hysteresis is observed: a delayed switch-on to safeguard brain glucose levels and a delayed switch-off to prevent hyperglycemia. Hence, despite being a "pathologic" condition contributing to T2D, IR serves a beneficial purpose.
Interestingly, under certain metabolic conditions, even extreme IR (to the point of causing diabetes) may be essential. For example, many women without a history of diabetes experience elevated blood glucose levels during pregnancy. Gestational diabetes, a condition with many unresolved questions [145], may be understood through the adjustable threshold theory, which posits that insulin is designed to protect brain glucose levels. During pregnancy, there are effectively two brains present—the mother's and the fetus's—and in cases of multiple pregnancies, additional brains are prepared [146]. As the fetal brain develops, the mother's muscles require a higher IRon content to prevent damage. The placenta may assist in this by secreting placental growth factor, which inhibits IRS-PI3K interaction, thereby increasing the threshold IRon. Since brain mass steadily increases throughout pregnancy, it follows that IRon levels will also progressively rise, resulting in a constant right-shift in the body's insulin response, similar to the shift induced by obesity. However, the right-shift often does not elevate plasma glucose levels since the growing fetal brain utilizes any additional glucose for its needs. Nonetheless, there exists a potential mismatch between fetal development and the right-shift, leading to the development of gestational diabetes if fetal growth lags behind. In most cases, gestational diabetes may be reversed after pregnancy, with placental growth factor levels returning to normal.
It could be argued that increasing dietary intake during each meal would sufficiently meet the pregnant woman's brain demands, with the expectation that the additional glucose would solely benefit the baby. However, there are three reasons why this strategy is unlikely to succeed. Firstly, starvation has plagued humanity throughout history; even in modern affluent civilizations, hunger persists. Secondly, even with an abundant food supply, most of the additional glucose would likely be absorbed by the mother's cells due to the higher overall insulin levels. Finally, a higher-carbohydrate diet may exacerbate reactive hypoglycemia, posing a risk of fetal death from severe hypoglycemia. Therefore, it is crucial to further develop and evaluate the adjustable threshold hypothesis, as emphasized by Wang. Subsequently, Okoduwa also observed a similar pattern of IR in a rat model [147]. They hypothesized that IR often accompanies a high-fat diet (HFD), leading to hyperinsulinemia. Comparing the pathophysiology between the water or fortified diet feed (FDF) group and the normal-diet-feed (NDF) group, they found that insulin levels in the blood were higher in an insulin-resistant state compared to healthy control conditions. This observation aligns with Wang's Hypothesis, suggesting that IR prevents the brain from depleting its glucose supply by limiting glucose absorption by muscles [97]. In another study, Akhtar et al. [148] used a combination of cellular studies and mathematical models to investigate the bistability (IRon and IRoff) hypothesis. Based on their findings, they proposed that bistability enables the body to simultaneously avoid hypoglycemia and hyperglycemia. Although the model described by Akhtar et al. was significant [148], they highlighted the need for a more generalized mathematical model of glucose-insulin homeostasis, as the one they used was too narrowly focused. Several physiological processes influence insulin dynamics, including insulin breakdown and pancreatic insulin production in response to glucose stimulation. The latter is particularly intriguing in light of the discovery of a new mechanism called "dynamic compensation" (where regulated glucose dynamics reciprocally control pancreatic cell mass) to account for the accuracy and stability of glucose dynamics despite significant changes in physiological parameters [149]. Unfortunately, including these complex systems in their study would have introduced too many unknowns, thus they were omitted from the modeling. This limitation could be addressed by creating a more comprehensive mathematical model for glucose-insulin homeostasis in the future, building upon the existing model (the bistable insulin response) and incorporating the aforementioned physiological processes [148].
Thrifty Phenotype
Hales and Barker also disputed the idea of a "thrifty genotype" proposed by Neel [23, 96], and instead proposed the "thrifty phenotype" hypothesis. The thrifty phenotype hypothesis emphasizes the role of early-life environmental factors and developmental plasticity in shaping metabolic adaptations, in contrast to the thrifty genotype hypothesis, which focused more on genetic predisposition. The thrifty phenotype theory suggests that all individuals have the potential for a range of metabolic outcomes shaped by the interaction between genes and the environment during development [54] (Fig. 6). In support of this, recently Li [150] reported that early embryogenesis is the essential stage for the re-establishment of genome-wide epigenetic patterns. A single mismatch during embryogenesis may result in developmental malformations, embryonic lethality and increased risk for certain diseases. On the contrary, a healthy rewriting of the early-life epigenome may result in desirable effects such as illness prevention. Thus, early epigenetic reprogramming mechanisms can be influenced by maternal nutritional status, resulting in adjustments in gene expression in adipogenesis and metabolisms contributing to altered phenotypes like different vulnerabilities in adult life to overweight and obesity-related metabolic disorders [150]. A recent animal study discovered that consuming probiotics during pregnancy and lactation reduced the adverse nutritional programming of offspring obesity induced by a high-fat diet (HFD) in maternal obesity. This finding implies that modifying maternal gut microbiota through additional microbiota manipulations could potentially serve as an effective strategy to enhance metabolic outcomes for both mothers and offspring [151]. Preliminary data from a human investigation also suggests that prenatal probiotic supplementation might influence the DNA methylation status of specific promoters associated with genes related to obesity and weight gain in both mothers and offspring [152]. These findings lend support to the hypothesis that there is an interconnection between gut microbiota composition, epigenetic regulation, and the occurrence of obesity in humans [150].

Thrifty phenotype: The thrifty phenotype hypothesis highlights the influence of early-life environments on metabolic adaptations, suggesting that genetic and environmental interactions during development shape individual metabolic outcomes
Earlier, Venniyoor [95•] proposes that the gene PTEN (phosphatase and tensin homolog) may be a likely candidate for a "thrifty gene" as it has functions spanning metabolism, cancer, and reproduction - all of which are deranged in obesity and IR. The activity of PTEN can be calibrated in utero by the availability of nutrients, through the methylation arm of the epigenetic pathway. Deficiency of specific nutrients like protein and choline has been shown to upregulate DNA methyltransferases (DNMT), which can then suppress PTEN expression by methylating its promoter region. This suggests that the "dose" of PTEN, which negatively regulates the insulin-PI3K/AKT/mTOR pathway, is tuned proportional to the availability of specific nutrients during early development. This "fixes" the metabolic capacity of the organism to a specific postnatal level of nutrition. However, when this "fixed" metabolic capacity is faced with a discordant, nutrient-rich environment later in life, it can lead to the development of obesity-related diseases. Overall, the thrifty phenotype hypothesis provides a framework for understanding how developmental plasticity in response to early-life conditions can shape an individual's long-term metabolic health, with important implications for disease prevention and management.
Therapeutic and Preventive Aspects Based on EM and Obesity/T2D
By considering the evolutionary origins of human biology and behavior, EM may provide novel insights that can significantly transform healthcare approaches in tackling the obesity-related disorders pandemic [153]. Also, this can guide the development of targeted interventions to address the root causes of obesity and T2D, along with treating the symptoms.
Population-Specific Drug Development
A key insight from EM is that susceptibility to conditions like obesity and T2D can vary significantly across different populations, often due to their unique evolutionary histories. Certain ethnic and racial groups appear more prone to these metabolic disorders, likely originating from a complex interplay between genetic adaptations and modern lifestyle factors [132]. Thus, by considering an individual's genetic makeup and evolutionary background [154], healthcare providers can tailor treatment plans to address specific needs and optimize therapeutic outcomes for conditions like obesity and T2D. Earlier pharmacogenomic studies have identified genetic variants that can influence an individual's response to anti-obesity and anti-diabetic medications [155,156,157,158,159]. The FTO gene is one such example, where certain variants, particularly the rs9939609 SNP, may increase obesity risk and alter the response to weight loss interventions [58, 160, 161]. Individuals carrying the alleles A and AA of the rs9939609 polymorphism tend to consume approximately 1231 kilojoules more calories than those with the TT allele [162]. Another study found a correlation between the rs9939609 genotype and physical activity, showing that passive carriers of the A allele had a BMI 1.95 kg/m2 higher than TT carriers. This suggests that low physical activity may worsen rs9939609's impact on fat accumulation in the body [161]. However, adolescents with high physical activity levels might mitigate the negative impacts of the rs9939609 polymorphism [163]. Likewise, earlier, several population-specific T2D variants were identified. Ng and his research team identified 13 SNPs across seven genes-TCF7L2, CDKN2A/CDKN2B, SLC30A8, CDKAL1, HHEX, IGF2BP2, and FTO-as significant risk factors for T2D among individuals of Asian descent from Korea and Hong Kong [164]. Studies on SLC30A8 have demonstrated that the rs13266634 polymorphism reduces the activity of the ZnT8 zinc transporter. This reduction in activity results in abnormal synthesis, maturation, and secretion of insulin, leading to decreased efficiency in glucose metabolism and ultimately contributing to the development of diabetes [165]. Another GWAS has identified the rs5219 polymorphism in the KCNJ11 gene as a significant risk factor for T2D in Caucasians and Syrian populations but not in Mongolian, Indian, or Ashkenazi Jewish populations. KCNJ11, located at 11p15.1, encodes the inward-rectifier potassium ion channel (Kir6.2), a component of the KATP channel. This channel regulates insulin production and secretion by modulating glucose metabolism. The rs5219 polymorphism disrupts KATP channel function, impairing insulin production and secretion, thereby contributing to T2D development in humans [166]. Recently, one study aggregated genetic data from over 2.5 million individuals across diverse ancestries, identifying 1,289 independent signals associated with T2D. Eight distinct clusters associated with unique cardiometabolic trait associations and cell-specific chromatin enrichments were identified. Cluster specific polygenic scores in ~279,000 individuals revealed obesity-related vascular outcomes across different ancestry group. This in turn suggests potential optimizations for global diabetes care access by leveraging genetic insights and understanding the diverse etiological factors contributing to T2D [167]. While this information provides a proximate view of the population-specific risk variant, understanding how nature has shaped the function of these variants throughout human evolution and if these variants were advantageous or deleterious in our ancestors may help us to understand what happened during the course of time that certain contemporary human population became prone to obesity and T2D while others not.
Preventive Aspects
Considering the long human lifespan and inherent genetic complexity, elucidating the evolutionary origins and mechanisms of complex traits like obesity and T2D is extremely challenging. Model organisms like C. elegans, Drosophila, and zebrafish have significantly shorter lifespans and generation times compared to humans [168,169,170,171,172]. This facilitates researchers to rapidly investigate the impact of genetic and environmental factors on phenotypes such as obesity across numerous generations, offering vital insights into the underlying evolutionary dynamics. Thus, studying model organisms may facilitate a more comprehensive investigation of the evolutionary origins and mechanisms underlying obesity. For example, earlier study has reported that Plutella xylostella caterpillars raised on carbohydrate-rich diets, like a chemically defined artificial diet or high-starch Arabidopsis mutant, learned to consume excess carbohydrates without storing them as fat over generations, indicating a fitness cost to excess fat storage. Conversely, those raised in carbohydrate-deficient environments tended to store carbohydrates as fat. Additionally, caterpillars from low-starch Arabidopsis mutants preferred laying eggs on this plant, contrasting with no such preference in those from high-starch mutants [173]. In a rat model, the adaptive hypothesis that an obese-prone genotype offers a fitness advantage during food scarcity and locomotion challenges was tested. Obese-prone rats (cp/cp) showed dramatically enhanced survival and endurance compared to lean-prone counterparts (+/?) when exposed to restricted meals and voluntary wheel running. Biochemical measures revealed better energy maintenance in obese-prone rats, suggesting a lower stress response. These findings support the idea that an obese-prone genotype is advantageous in times of food scarcity but detrimental in food-abundant environments [174]. Earlier, we also observed that T2D genes in Drosophila are ancient and evolving under purifying selection. However, some sites in membrane proteins encoded by specific T2D genes, such as ZnT35C, continue to evolve under positive selection in specific circumstances. This may reflect adaptive responses to external factors like disease and ecological changes and internal mechanisms like compensatory mutations and co-evolution [169]. Thus, EM approached may also be utilized to develop preventive strategies to address human diseases [175, 176], explicitly focusing on understanding the evolutionary origins of human metabolism and how modern environments may mismatch these adaptations. One key preventive strategy is promoting nutrition and physical activity consistent with our evolutionary past, such as emphasizing whole foods, diverse diets, and regular physical activity [177]. Previous evidence suggests that consuming low-energy-dense (LED) foods, such as fish, lean meat, fruits, and vegetables, may decrease hunger sensations and energy intake, potentially aiding in weight loss [178]. Sophie and her colleagues conducted a randomized crossover study comparing low-energy-dense ready meals (LEDRMs) with higher-energy-dense ready meals (HEDRM) on satiety and food intake [179]. While low-energy-dense ready meals (LEDRMs) did not result in reduced energy intake, they did enhance feelings of fullness and have the potential to enhance the nutritional quality of meals by reducing the consumption of saturated fat. Similarly, diets containing whole grains have been shown to prevent obesity by regulating metabolic functions and reducing pro-inflammatory states. Wei-Yi and colleagues conducted another intervention study to assess the impact of consuming Dehulled Adlay on lipid metabolism and inflammation in overweight and obese individuals. After a 6-week intervention, it was found that consuming 60 g of dehulled adlay powder daily significantly improved body fat mass and blood levels of total cholesterol, triglycerides, and inflammatory markers [180].
Early life interventions targeting maternal nutrition, breastfeeding, and childhood diet and activity levels can also influence lifelong metabolic health [181, 182]. The offspring of women exposed to famine during early gestation, despite being born with normal birth weight, exhibited an elevated risk of obesity and a threefold increase in the likelihood of coronary heart disease as adults. Conversely, the offspring of women exposed to famine in mid-and late gestation were born smaller than unexposed babies and faced an increased risk of impaired glucose tolerance in adulthood [183]. In a cross-sectional study by Martha and colleagues [184], the abundance of Bacteroidetes and Actinobacteria in human milk was found to be directly and significantly associated with the adiposity of women before pregnancy and during lactation. This suggests that intestinal microbiota may play a crucial role in mediating the impact of the nutritional status of mothers on their offspring. Environmental modifications, like creating walkable communities and improving access to fresh foods, can also reduce exposure to obesogenic factors such as processed foods and sedentary behaviors. At the population level, policies and interventions such as food labeling regulations, and urban planning for physical activity can address the obesity epidemic from a broader societal perspective [185].
Limitation and Future Perspective of the Evolutionary Hypothesis
The above-outlined evolutionary medical perspective not only helps us to understand the ultimate reason why obesity and T2D exists in humans but also its pathogenesis. There are, however, certain limitations to this method. To begin with, the theory of evolutionary medicine relies on several fundamental concepts that are difficult to demonstrate experimentally. As an example, to date, no specific thrifty genes have been detected, which in turn limits the involvement of the thrifty gene in the diagnosis and treatment of T2D. Also, due to their long lifespans, mammals make it almost hard to scientifically establish or witness the impacts of natural selection as well as the trade-offs between early and later life [186]. We may overcome this limitation by studying human diseases in model organisms (e.g., Drosophila) and creating a contrasting artificial environment. For instance, to understand the thrifty genotype, we may expose Drosophila under feast & famine condition and what happens if any of the feast or famine condition is disrupted. Since Drosophila has a very short life cycle, we have more power to capture the situation, which may also help us understand how the change in the human environment influences human diseases, including T2D [169]. Furthermore, given that research in EM encompasses human health, pharmacokinetics, various temporal scales, cultural and population diversity, and environmental science, interdisciplinary collaboration is crucial for comprehensively understanding human diseases' mechanisms, such as T2D. Additionally, it is imperative to broaden our approach beyond population genomic studies, which typically focus solely on humans, to include other methodologies such as phylogenetic, co-evolutionary, and sexual selection approaches. This holistic approach will deepen our understanding of the pathophysiology of various human diseases, including T2D, cancer, and immune system disorders [187,188,189]. However, most of the other EM approaches are still in infancy. Thus, more research and funding may be required to grow this field [187]. Additionally, most of the EM hypotheses are based on the comparison between two human groups, e.g., modern people vs. ancestors or carnivorous vs. non-carnivorous. However, EM studies of different ethnicities are very important to unmask the disease pathogenesis more precisely. For instance, earlier studies have reported that the genetic landscape and signatures of selection in the genome are important for mapping susceptibility variations and understanding the development of diseases/traits [190]. They reported that the variants in genes like TCF7L2 show evidence of selection and that may have helped Africans adapt to dietary stresses by influencing glucose and lipid metabolism. Similarly, the low visceral adipose tissue (VAT) and high intermuscular adipose tissue (IAT) in people of African descent, as well as the high VAT-adiponectin connection, may reflect adaptive responses to the dietary and infectious illness stresses of the African environment. Thus, comparing different ethnicities using EM approaches may help us unmask human diseases more precisely. Additionally, it's crucial to study how IR affects different organs. Recent research, like tweaking genes in specific tissues, is helping us learn more. Interestingly, while IR in some organs like the liver and brain is bad, it seems to be good for health and lifespan in fat cells. We need to understand if the reasons behind IR affect organs differently. Answering these questions could change how we view basic biology and healthcare. Finally, while some universities have achieved success in including EM in medical school curricula, now, EM is mostly taught to evolutionary biologists. Only a small percentage of physicians learn about evolution in their premedical or medical training and even fewer get to really use it in their practice. If EM continues as it is, it will become focused only on research, and doctors will only be interested in the outcomes (or proximal causes). Thus, the involvement of experts from various fields will help us to understand the underlying mechanism associated with obesity and T2D using the EM approach more precisely.
Conclusion
Obesity is a major cause of the development of IR, thereby increasing the risk of chronic diseases such as type 2 diabetes. However, complete mechanism how obesity causes T2D remains elusive. Considering this, recently developed evolutionary medicine examines the genetic, behavioral, environmental, and microbial factors to understand how previous evolutionary processes have shaped the present-day human genome and made them vulnerable or protective against diseases. To date, several hypotheses have been proposed to understand the link between Obesity and T2D. However, these hypotheses give a possible reason why one group of individuals is prone to T2D. There are several points of uncertainty and contradiction in each hypothesis. Most of the hypotheses are developed on the theories and have not been proved experimentally. Furthermost of the research focused on ethnicity and cell/tissue-specific is still required, along with participation from a wide range of researchers, from medical practitioners to basic biology scientists. Since obesity-associated IR is a significant cause of morbidity and mortality worldwide currently, and evolutionary awareness has the ability to revolutionize the line of disease prevention and care, the information present in this article will be a valuable illustration of evolutionary biology's approach to medicine. In the near future, the information presented in the article will be useful in understanding disease and its management.
Data Availability
No datasets were generated or analysed during the current study.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Choi WG, Choi W, Oh TJ, Cha H-N, Hwang I, Lee YK, et al. Inhibiting serotonin signaling through HTR2B in visceral adipose tissue improves obesity-related insulin resistance. J Clin Invest [Internet]. 2021 [cited 2023 May 3]. Available from https://www.jci.org/articles/view/145331.
Friedrich MJ. Global obesity epidemic worsening. JAMA. 2017;318:603.
Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res. 2020;126:1549–64.
McAllister EJ, Dhurandhar NV, Keith SW, Aronne LJ, Barger J, Baskin M, et al. Ten putative contributors to the obesity epidemic. Crit Rev Food Sci Nutr. 2009;49:868–913.
Koliaki C, Dalamaga M, Liatis S. Update on the obesity epidemic: after the sudden rise, is the upward trajectory beginning to flatten? Curr Obes Rep. 2023;12:514–27.
Blüher M, Stumvoll M. Diabetes and obesity. In: Bonora E, DeFronzo RA, editors. Diabetes complications, comorbidities and related disorders [Internet]. Cham: Springer; 2020. p. 1–49. https://doi.org/10.1007/978-3-030-36694-0_1.
Smith GI, Mittendorfer B, Klein S. Metabolically healthy obesity: facts and fantasies. J Clin Invest. 2019;129:3978–89.
Wondmkun YT. Obesity, insulin resistance, and type 2 diabetes: associations and therapeutic implications. Diabetes Metab Syndr Obes Targets Ther. 2020;13:3611–6.
Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106:473–81.
Oliveri A, Rebernick RJ, Kuppa A, Pant A, Chen Y, Du X, et al. Comprehensive genetic study of the insulin resistance marker TG:HDL-C in the UK Biobank. Nat Genet. 2024;56:212–21.
Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med. 2005;56:45–62.
Ormazabal V, Nair S, Elfeky O, Aguayo C, Salomon C, Zuñiga FA. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol. 2018;17:122.
Wang CCL, Goalstone ML, Draznin B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes. 2004;53:2735–40.
Wu Y, Chen W, Zhao Y, Gu M, Gao Y, Ke Y, et al. Visit to visit transition in TXNIP gene methylation and the risk of type 2 diabetes mellitus: a nested case-control study. J Hum Genet. 2024;1–9.
Grarup N, Sandholt CH, Hansen T, Pedersen O. Genetic susceptibility to type 2 diabetes and obesity: from genome-wide association studies to rare variants and beyond. Diabetologia. 2014;57:1528–41.
Norris JM, Rich SS. Genetics of Glucose Homeostasis Implications for Insulin Resistance and Metabolic Syndrome. Arterioscler Thromb Vasc Biol. 2012;32:2091–6.
• Benton ML, Abraham A, LaBella AL, Abbot P, Rokas A, Capra JA. The influence of evolutionary history on human health and disease. Nat Rev Genet. 2021;22:269–83. Discuss how evolutionary medicine is making strides in fields of human diseases by applying evolutionary principles.
Moltzau Anderson J, Horn F. (Re-) Defining evolutionary medicine. Ecol Evol. 2020;10:10930–6.
Nesse RM, Schulkin J. An evolutionary medicine perspective on pain and its disorders. Philos Trans R Soc B Biol Sci. 2019;374:20190288.
Huppert FA, Baylis N, Keverne B, Nesse RM. Natural selection and the elusiveness of happiness. Philos Trans R Soc Lond B Biol Sci. 2004;359:1333–47.
Litman GW, Cooper MD. Why study the evolution of immunity? Nat Immunol. 2007;8:547–8.
Cooper MD, Herrin BR. How did our complex immune system evolve? Nat Rev Immunol. 2010;10:2–3.
Neel JV. Diabetes mellitus: A “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet. 1962;14:353–62.
O’Dea K. Overview of the thrifty genotype hypothesis. Asia Pac J Clin Nutr. 1995;4:339–40.
Speakman JR. Thrifty genes for obesity, an attractive but flawed idea, and an alternative perspective: the ‘drifty gene’ hypothesis. Int J Obes. 2008;32:1611–7.
Watve MG, Yajnik CS. Evolutionary origins of insulin resistance: a behavioral switch hypothesis. BMC Evol Biol. 2007;7:61.
Abarca-Gómez L, Abdeen ZA, Hamid ZA, Abu-Rmeileh NM, Acosta-Cazares B, Acuin C, et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet. 2017;390:2627–42.
World Health Organization. Prevalence of obesity among adults, BMI >= 30 (age-standardized estimate) (%) [cited 2022 Aug 30]. 2022. No Title.
Zhou B, Lu Y, Hajifathalian K, Bentham J, Cesare MD, Danaei G, et al. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4·4 million participants. Lancet. 2016;387:1513–30.
World Health Organization. Indicator: raised fasting blood glucose (>= 7.0 mmol/L) (age-standardized estimate) - sex: both sexes - age group: 18+ years. 2022. No Title.
Djalalinia S, Qorbani M, Peykari N, Kelishadi R. Health impacts of obesity. Pak J Med Sci. 2015;31:239–42.
Nuttall FQ. Body mass index: obesity, bmi, and health: a critical review. Nutr Today. 2015;50:117–28.
South A. Rnaturalearth: world map data from natural earth. R package version 0.1.0. 2017.
Wickham H. ggplot2. Wiley Interdiscip Rev Comput Stat. 2011;3:180–5.
Luo L, Liu M. Adipose tissue in control of metabolism. J Endocrinol. 2016;231:R77–99.
Skorobogatko Y, Dragan M, Cordon C, Reilly SM, Hung C-W, Xia W, et al. RalA controls glucose homeostasis by regulating glucose uptake in brown fat. Proc Natl Acad Sci. 2018;115:7819–24.
Zhu Y, Li N, Huang M, Bartels M, Dogné S, Zhao S, et al. Adipose tissue hyaluronan production improves systemic glucose homeostasis and primes adipocytes for CL 316,243-stimulated lipolysis. Nat Commun. 2021;12:4829.
Chadt A, Al-Hasani H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflugers Arch. 2020;472:1273–98.
Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444:847–53.
Grant RW, Stephens JM. Fat in flames: influence of cytokines and pattern recognition receptors on adipocyte lipolysis. Am J Physiol Endocrinol Metab. 2015;309:E205–13.
Girgis CM, Cheng K, Scott CH, Gunton JE. Novel links between HIFs, type 2 diabetes, and metabolic syndrome. Trends Endocrinol Metab. 2012;23:372–80.
He Q, Gao Z, Yin J, Zhang J, Yun Z, Ye J. Regulation of HIF-1α activity in adipose tissue by obesity-associated factors: adipogenesis, insulin, and hypoxia. Am J Physiol Endocrinol Metab. 2011;300:E877–85.
van Herpen NA, Schrauwen-Hinderling VB. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav. 2008;94:231–41.
Xu L, Li Y, Dai Y, Peng J. Natural products for the treatment of type 2 diabetes mellitus: Pharmacology and mechanisms. Pharmacol Res. 2018;130:451–65.
Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes. 2002;51:2005–11.
Tumova J, Andel M, Trnka J. Excess of free fatty acids as a cause of metabolic dysfunction in skeletal muscle. Physiol Res. 2016;65:193–207.
Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014;371:1131–41.
Ruze R, Liu T, Zou X, Song J, Chen Y, Xu R, et al. Obesity and type 2 diabetes mellitus: connections in epidemiology, pathogenesis, and treatments. Front Endocrinol [Internet]. 2023 [cited 2023 Jul 15]. https://doi.org/10.3389/fendo.2023.1161521.
Turcotte LP, Fisher JS. Skeletal muscle insulin resistance: roles of fatty acid metabolism and exercise. Phys Ther. 2008;88:1279–96.
Morales PE, Bucarey JL, Espinosa A. Muscle lipid metabolism: role of lipid droplets and perilipins. J Diabetes Res. 2017;2017:1789395.
Park SS, Seo Y-K. Excess accumulation of lipid impairs insulin sensitivity in skeletal muscle. Int J Mol Sci. 2020;21:1949.
Joseph A, Parvathy S, Varma KK. Hyperinsulinemia induced altered insulin signaling pathway in muscle of high fat- and carbohydrate-fed rats: effect of exercise. J Diabetes Res. 2021;2021:5123241.
Basile KJ, Johnson ME, Xia Q, Grant SFA. Genetic susceptibility to type 2 diabetes and obesity: follow-up of findings from genome-wide association studies. Int J Endocrinol. 2014;2014:769671.
Berndt SI, Gustafsson S, Mägi R, Ganna A, Wheeler E, Feitosa MF, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet. 2013;45:501–12.
Meyre D, Delplanque J, Chèvre J-C, Lecoeur C, Lobbens S, Gallina S, et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009;41:157–9.
Bonàs-Guarch S, Guindo-Martínez M, Miguel-Escalada I, Grarup N, Sebastian D, Rodriguez-Fos E, et al. Re-analysis of public genetic data reveals a rare X-chromosomal variant associated with type 2 diabetes. Nat Commun. 2018;9:321.
Domínguez-Cruz MG, de Lourdes Muñoz M, Totomoch-Serra A, García-Escalante MG, Burgueño J, Valadez-González N, et al. Pilot genome-wide association study identifying novel risk loci for type 2 diabetes in a Maya population. Gene. 2018;677:324–31.
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94.
Grant SFA, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–3.
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206.
Xue A, Wu Y, Zhu Z, Zhang F, Kemper KE, Zheng Z, et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun. 2018;9:2941.
• Suzuki K, Hatzikotoulas K, Southam L, Taylor HJ, Yin X, Lorenz KM, et al. Multi-ancestry genome-wide study in >2.5 million individuals reveals heterogeneity in mechanistic pathways of type 2 diabetes and complications. MedRxiv Prepr Serv Health Sci. 2023. https://doi.org/10.1101/2023.03.31.23287839. T2D’s heterogeneity requires understanding its genetic drivers via GWAS and cluster-specific polygenic scores forimproved prevention and management.
Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–8.
Takeuchi F, Akiyama M, Matoba N, Katsuya T, Nakatochi M, Tabara Y, et al. Interethnic analyses of blood pressure loci in populations of East Asian and European descent. Nat Commun. 2018;9:5052.
Dina C, Meyre D, Gallina S, Durand E, Körner A, Jacobson P, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–6.
Al-Jawadi AA, Priliani L, Oktavianthi S, Febinia CA, Daya M, Artika IM, et al. Association of FTO rs1421085 single nucleotide polymorphism with fat and fatty acid intake in Indonesian adults. BMC Res Notes. 2021;14:411.
Inandiklioğlu N, Yaşar A. Association between rs1421085 and rs9939609 polymorphisms of fat mass and obesity-associated gene with high-density lipoprotein cholesterol and triglyceride in obese Turkish children and adolescents. J Pediatr Genet. 2021;10:9–15.
Vámos A, Arianti R, Vinnai BÁ, Alrifai R, Shaw A, Póliska S, et al. Human abdominal subcutaneous-derived active beige adipocytes carrying FTO rs1421085 obesity-risk alleles exert lower thermogenic capacity. Front Cell Dev Biol [Internet]. 2023 [cited 2024 Apr 3]. https://doi.org/10.3389/fcell.2023.1155673.
Chen Y, Fang S. Potential genetic polymorphisms predicting polycystic ovary syndrome. Endocr Connect. 2018;7:R187–95.
Xue H, Zhao H, Zhao Y, Liu X, Chen Z, Ma J. Association of common variants of FTO in women with polycystic ovary syndrome. Int J Clin Exp Pathol. 2015;8:13505–9.
Yamauchi T, Hara K, Maeda S, Yasuda K, Takahashi A, Horikoshi M, et al. A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nat Genet. 2010;42:864–8.
Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008;40:1098–102.
Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet. 2008;40:1092–7.
Wen W, Cho YS, Zheng W, Dorajoo R, Kato N, Qi L, et al. Meta-analysis identifies common variants associated with body mass index in East Asians. Nat Genet. 2012;44:307–11.
Monda KL, Chen GK, Taylor KC, Palmer C, Edwards TL, Lange LA, et al. A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat Genet. 2013;45:690–6.
Ramos-Molina B, Martin MG, Lindberg I. PCSK1 variants and human obesity. Prog Mol Biol Transl Sci. 2016;140:47–74.
Chang YC, Chiu YF, Shih KC, Lin MW, Sheu WHH, Donlon T, et al. Common PCSK1 haplotypes are associated with obesity in the Chinese population. Obes Silver Spring Md. 2010;18:1404–9.
Liu C-T, Monda KL, Taylor KC, Lange L, Demerath EW, Palmas W, et al. Genome-wide association of body fat distribution in African ancestry populations suggests new loci. PLoS Genet. 2013;9:e1003681.
Stijnen P, Ramos-Molina B, O’Rahilly S, Creemers JWM. PCSK1 mutations and human endocrinopathies: from obesity to gastrointestinal disorders. Endocr Rev. 2016;37:347–71.
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20:467–84.
Fitipaldi H, Franks PW. Ethnic, gender and other sociodemographic biases in genome-wide association studies for the most burdensome non-communicable diseases: 2005–2022. Hum Mol Genet. 2023;32:520–32.
Hood E, Jenkins KP. Evolutionary medicine: a powerful tool for improving human health. Evol Educ Outreach. 2008;1:114–20.
Ellison PT. Evolutionary tradeoffs. Evol Med. Public Health. 2014;2014:93–93.
Alcock J, Schwartz MD. A clinical perspective in evolutionary medicine: what we wish we had learned in medical school. Evol Educ Outreach. 2011;4:574–9.
Perry GH. Evolutionary medicine. eLife. 2021;10:e69398.
• Lenover MB, Shenk MK. Evolutionary medicine approaches to chronic disease: The case of irritable bowel syndrome. Evol Anthropol Issues News Rev. 2024;33:e22010. Highlights how the evolutionary medicine perspective can provide valuable insights into the understanding andmanagement of chronic diseases.
Gorlov IP, Gorlova OY, Frazier ML, Spitz MR, Amos CI. Evolutionary evidence of the effect of rare variants on disease etiology. Clin Genet. 2011;79:199–206.
Myles S, Lea RA, Ohashi J, Chambers GK, Weiss JG, Hardouin E, et al. Testing the thrifty gene hypothesis: the Gly482Ser variant in PPARGC1A is associated with BMI in Tongans. BMC Med Genet. 2011;12:10.
Minster RL, Hawley NL, Su C-T, Sun G, Kershaw EE, Cheng H, et al. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat Genet. 2016;48:1049–54.
Bogardus C, Lillioja S, Ravussin E. The pathogenesis of obesity in man: results of studies on Pima Indians. Int J Obes. 1990;14(Suppl 1):5–13; discussion 13–15.
Pettitt DJ, Nelson RG, Saad MF, Bennett PH, Knowler WC. Diabetes and obesity in the offspring of Pima Indian women with diabetes during pregnancy. Diabetes Care. 1993;16:310–4.
Schulz LO, Bennett PH, Ravussin E, Kidd JR, Kidd KK, Esparza J, et al. Effects of traditional and western environments on prevalence of type 2 diabetes in Pima Indians in Mexico and the US. Diabetes Care. 2006;29:1866–71.
Unger RH. Klotho-induced insulin resistance: a blessing in disguise? Nat Med. 2006;12:56–7.
Brand-Miller JC, Griffin HJ, Colagiuri S. The carnivore connection hypothesis: revisited. J Obes. 2011;2012:e258624.
• Venniyoor A. PTEN: a thrifty gene that causes disease in times of plenty? Front Nutr. 2020 [cited 2022 Dec 21]. https://doi.org/10.3389/fnut.2020.00081. PTEN, termed a “thrifty gene,” is suppressed during fetal nutrient deficiency, favoring fat storage, but detrimental inobesogenic environments.
Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5–20.
Wang G. Raison d’être of insulin resistance: the adjustable threshold hypothesis. J R Soc Interface. 2014;11:20140892.
Lindsay RS, Bennett PH. Type 2 diabetes, the thrifty phenotype – an overview. Br Med Bull. 2001;60:21–32.
Knowler WC, Pettitt DJ, Saad MF, Bennett PH. Diabetes mellitus in the Pima Indians: incidence, risk factors and pathogenesis. Diabetes Metab Rev. 1990;6:1–27.
Zimmet PZ. Kelly West lecture 1991 challenges in diabetes epidemiology—from west to the rest. Diabetes Care. 1992;15:232–52.
Houghton P. The adaptive significance of Polynesian body form. Ann Hum Biol. 1990;17:19–32.
Baker P. Migrations, genetics, and the degenerative diseases of South Pacific islanders. In: Migration and mobility. Routledge; 2022. p. 209–39.
Bindon JR, Baker PT. Bergmann’s rule and the thrifty genotype. Am J Phys Anthropol. 1997;104:201–10.
Swinburn BA. The thrifty genotype hypothesis: how does it look after 30 years? Diabet Med J Br Diabet Assoc. 1996;13:695–9.
Reaven GM. Hypothesis: muscle insulin resistance is the (“not-so”) thrifty genotype. Diabetologia. 1998;41:482–4.
Wells JCK. The thrifty phenotype as an adaptive maternal effect. Biol Rev. 2007;82:143–72.
Kagawa Y, Yanagisawa Y, Hasegawa K, Suzuki H, Yasuda K, Kudo H, et al. Single nucleotide polymorphisms of thrifty genes for energy metabolism: evolutionary origins and prospects for intervention to prevent obesity-related diseases. Biochem Biophys Res Commun. 2002;295:207–22.
Goh KP. Chapter 15 - gene–environment interaction in the pathogenesis of type 2 diabetes. In: Bagchi D, Nair S, editors. Nutritional and therapeutic interventions for diabetes and metabolic syndrome. 2nd ed. Academic Press; 2018 [cited 2022 Dec 8]. p. 193–205 Available from https://www.sciencedirect.com/science/article/pii/B9780128120194000155.
Stipp D. Linking nutrition, maturation and aging: from thrifty genes to the spendthrift phenotype. Aging. 2011;3:85–93.
Williams RC, Knowler WC, Pettitt DJ, Long JC, Rokala DA, Polesky HF, et al. The magnitude and origin of European-American admixture in the Gila River Indian Community of Arizona: a union of genetics and demography. Am J Hum Genet. 1992;51:101–10.
Dabelea D, Hanson RL, Bennett PH, Roumain J, Knowler WC, Pettitt DJ. Increasing prevalence of Type II diabetes in American Indian children. Diabetologia. 1998;41:904–10.
Hanson RL, Elston RC, Pettitt DJ, Bennett PH, Knowler WC. Segregation analysis of non-insulin-dependent diabetes mellitus in Pima Indians: evidence for a major-gene effect. Am J Hum Genet. 1995;57:160–70.
Bogardus C, Lillioja S, Howard BV, Reaven G, Mott D. Relationships between insulin secretion, insulin action, and fasting plasma glucose concentration in nondiabetic and noninsulin-dependent diabetic subjects. J Clin Invest. 1984;74:1238–46.
Schulz LO, Chaudhari LS. High-risk populations: The Pimas of Arizona and Mexico. Curr Obes Rep. 2015;4:92–8.
Esparza J, Fox C, Harper IT, Bennett PH, Schulz LO, Valencia ME, et al. Daily energy expenditure in Mexican and USA Pima Indians: low physical activity as a possible cause of obesity. Int J Obes. 2000;24:55–9.
Kimura M. The neutral theory of molecular evolution. Cambridge University Press; 1983.
Peacock WL, Speakman JR. Effect of high-fat diet on body mass and energy balance in the bank vole. Physiol Behav. 2001;74:65–70.
Król E, Redman P, Thomson PJ, Williams R, Mayer C, Mercer JG, et al. Effect of photoperiod on body mass, food intake and body composition in the field vole. Microtus Agrestis J Exp Biol. 2005;208:571–84.
El-Bakry HA, Plunkett SS, Bartness TJ. Photoperiod, but not a high-fat diet, alters body fat in Shaw’s jird. Physiol Behav. 1999;68:87–91.
Speakman JR. A nonadaptive scenario explaining the genetic predisposition to obesity: the “predation release” hypothesis. Cell Metab. 2007;6:5–12.
Cuthill IC, Maddocks SA, Weall CV, Jones EKM. Body mass regulation in response to changes in feeding predictability and overnight energy expenditure. Behav Ecol. 2000;11:189–95.
Gosler AG, Greenwood JJD, Perrins C. Predation risk and the cost of being fat. Nature. 1995;377:621–3.
Zurlo F, Lillioja S, Esposito-Del Puente A, Nyomba BL, Raz I, Saad MF, et al. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Physiol. 1990;259:E650–7.
Marra M, Scalfi L, Contaldo F, Pasanisi F. Fasting respiratory quotient as a predictor of long-term weight changes in non-obese women. Ann Nutr Metab. 2004;48:189–92.
Frisancho AR. Reduced rate of fat oxidation: a metabolic pathway to obesity in the developing nations. Am J Hum Biol Off J Hum Biol Counc. 2003;15:522–32.
Kunz I, Schorr U, Römmling K, Klaus S, Sharma AM. Habitual fat intake and basal fat oxidation in obese and non-obese Caucasians. Int J Obes. 2002;26:150–6.
Ravussin E, Gautier JF. Metabolic predictors of weight gain. Int J Obes. 1999;23:S37–41.
Tataranni PA. From physiology to neuroendocrinology: a reappraisal of risk factors of body weight gain in humans. Diabetes Metab. 1998;24:108–15.
Chang S, Graham B, Yakubu F, Lin D, Peters JC, Hill JO. Metabolic differences between obesity-prone and obesity-resistant rats. Am J Physiol. 1990;259:R1103–10.
Ji H, Friedman MI. Reduced capacity for fatty acid oxidation in rats with inherited susceptibility to diet-induced obesity. Metab Clin Exp. 2007;56:1124–30.
Waterson MJ, Horvath TL. Neuronal regulation of energy homeostasis: beyond the hypothalamus and feeding. Cell Metab. 2015;22:962–70.
Sellayah D, Cagampang FR, Cox RD. On the evolutionary origins of obesity: a new hypothesis. Endocrinology. 2014;155:1573–88.
Kehl RJ, Krew MA, Thomas A, Catalano PAA. Fetal growth and body composition in infants of women with diabetes mellitus during pregnancy. J Matern Fetal Med. 1996;5:273–80.
Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab. 1980;50:113–6.
Dunaif A, Book CB. Insulin resistance in the polycystic ovary syndrome. In: Draznin B, Rizza R, editors. Clinical research in diabetes and obesity [Internet]. Humana Press; 1997 [cited 2022 Dec 7]. p. 249–74. https://doi.org/10.1007/978-1-4757-3906-0_14.
Dale PO, Tanbo T, Haug E, Abyholm T. The impact of insulin resistance on the outcome of ovulation induction with low-dose follicle stimulating hormone in women with polycystic ovary syndrome. Hum Reprod. 1998;13:567–70.
Homburg R. The management of infertility associated with polycystic ovary syndrome. Reprod Biol Endocrinol. 2003;1:109.
Greisen S, Ledet T, Ovesen P. Effects of androstenedione, insulin and luteinizing hormone on steroidogenesis in human granulosa luteal cells. Hum Reprod. 2001;16:2061–5.
Toprak S, Yönem A, Çakır B, Güler S, Azal Ö, Özata M, et al. Insulin resistance in nonobese patients with polycystic ovary syndrome. Horm Res Paediatr. 2001;55:65–70.
Genné-Bacon EA. Thinking evolutionarily about obesity. Yale J Biol Med. 2014;87:99–112.
Brand Miller JC, Colagiuri S. The carnivore connection: dietary carbohydrate in the evolution of NIDDM. Diabetologia. 1994;37:1280–6.
Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–32.
Sherman A. Dynamical systems theory in physiology. J Gen Physiol. 2011;138:13–9.
Bonadonna RC, Groop L, Kraemer N, Ferrannini E, Del Prato S, DeFronzo RA. Obesity and insulin resistance in humans: a dose-response study. Metabolism. 1990;39:452–9.
Metzger BE, Coustan DR. Summary and recommendations of the Fourth International Workshop-Conference on Gestational Diabetes Mellitus. The organizing committee. Diabetes Care. 1998;21(Suppl 2):B161–7.
Dekaban AS. Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann Neurol. 1978;4:345–56.
Okoduwa SIR, Umar IA, James DB, Inuwa HM. Appropriate insulin level in selecting fortified diet-fed, streptozotocin-treated rat model of type 2 diabetes for anti-diabetic studies. PLoS ONE. 2017;12:e0170971.
Akhtar J, Han Y, Han S, Lin W, Cao C, Ge R, et al. Bistable insulin response: The win-win solution for glycemic control. iScience. 2022;25:105561.
Karin O, Swisa A, Glaser B, Dor Y, Alon U. Dynamical compensation in physiological circuits. Mol Syst Biol. 2016;12:886.
Li Y. Epigenetic mechanisms link maternal diets and gut microbiome to obesity in the offspring. Front Genet [Internet]. 2018 [cited 2021 Jan 26]. https://doi.org/10.3389/fgene.2018.00342/full.
Paul HA, Bomhof MR, Vogel HJ, Reimer RA. Diet-induced changes in maternal gut microbiota and metabolomic profiles influence programming of offspring obesity risk in rats. Sci Rep. 2016;6:20683.
Vähämiko S, Laiho A, Lund R, Isolauri E, Salminen S, Laitinen K. The impact of probiotic supplementation during pregnancy on DNA methylation of obesity-related genes in mothers and their children. Eur J Nutr. 2019;58:367–77.
Natterson-Horowitz B, Aktipis A, Fox M, Gluckman PD, Low FM, Mace R, et al. The future of evolutionary medicine: sparking innovation in biomedicine and public health. Front Sci [Internet]. 2023 [cited 2023 Jul 27]. https://doi.org/10.3389/fsci.2023.997136/full.
Ségurel L, Austerlitz F, Toupance B, Gautier M, Kelley JL, Pasquet P, et al. Positive selection of protective variants for type 2 diabetes from the Neolithic onward: a case study in Central Asia. Eur J Hum Genet. 2013;21:1146–51.
Sivadas A, Sahana S, Jolly B, Bhoyar RC, Jain A, Sharma D, et al. Landscape of pharmacogenetic variants associated with non-insulin antidiabetic drugs in the Indian population. BMJ Open Diabetes Res Care. 2024;12: e003769.
Hocking S, Sumithran P. Individualised prescription of medications for treatment of obesity in adults. Rev Endocr Metab Disord. 2023;24:951–60.
Venkatachalapathy P, Padhilahouse S, Sellappan M, Subramanian T, Kurian SJ, Miraj SS, et al. Pharmacogenomics and personalized medicine in type 2 diabetes mellitus: potential implications for clinical practice. Pharmacogenomics Pers Med. 2021;14:1441–55.
Ang MY, Takeuchi F, Kato N. Deciphering the genetic landscape of obesity: a data-driven approach to identifying plausible causal genes and therapeutic targets. J Hum Genet. 2023;68:823–33.
Goodarzi MO. Genetics of obesity: what genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endocrinol. 2018;6:223–36.
Reinehr T, Wolters B, Roth CL, Hinney A. FTO gene: association to weight regain after lifestyle intervention in overweight children. Horm Res Paediatr. 2014;81:391–6.
Kalantari N, Doaei S, Keshavarz-Mohammadi N, Gholamalizadeh M, Pazan N. Review of studies on the fat mass and obesity-associated (FTO) gene interactions with environmental factors affecting on obesity and its impact on lifestyle interventions. ARYA Atheroscler. 2016;12:281–90.
Freathy RM, Timpson NJ, Lawlor DA, Pouta A, Ben-Shlomo Y, Ruokonen A, et al. Common variation in the FTO gene alters diabetes-related metabolic traits to the extent expected given its effect on BMI. Diabetes. 2008;57:1419–26.
Ruiz JR, Labayen I, Ortega FB, Legry V, Moreno LA, Dallongeville J, et al. Attenuation of the effect of the FTO rs9939609 polymorphism on total and central body fat by physical activity in adolescents: the HELENA study. Arch Pediatr Adolesc Med. 2010;164:328–33.
Ng MCY, Park KS, Oh B, Tam CHT, Cho YM, Shin HD, et al. Implication of genetic variants near TCF7L2, SLC30A8, HHEX, CDKAL1, CDKN2A/B, IGF2BP2, and FTO in type 2 diabetes and obesity in 6,719 Asians. Diabetes. 2008;57:2226–33.
Gupta MK, Vadde R. Insights into the structure-function relationship of both wild and mutant zinc transporter ZnT8 in human: a computational structural biology approach. J Biomol Struct Dyn. 2020;38:137–51.
Gupta MK, Vadde R. A computational structural biology study to understand the impact of mutation on structure–function relationship of inward-rectifier potassium ion channel Kir6. 2 in human. J Biomol Struct Dyn. 2021;39:1447–60.
Suzuki K, Hatzikotoulas K, Southam L, Taylor HJ, Yin X, Lorenz KM, et al. Genetic drivers of heterogeneity in type 2 diabetes pathophysiology. Nature. 2024;627:347–57.
Bertile F, Matallana-Surget S, Tholey A, Cristobal S, Armengaud J. Diversifying the concept of model organisms in the age of -omics. Commun Biol. 2023;6:1–4.
Gupta MK, Vadde R. Divergent evolution and purifying selection of the Type 2 diabetes gene sequences in Drosophila: a phylogenomic study. Genetica. 2020;148:269–82.
Musselman LP, Kühnlein RP. Drosophila as a model to study obesity and metabolic disease. Suarez RK, Hoppeler HH, editors. J Exp Biol. 2018;221:jeb163881.
Smolińska K, Sobczyński J, Szopa A, Wnorowski A, Tomaszewska E, Muszyński S, et al. Innovative high fat diet establishes a novel zebrafish model for the study of visceral obesity. Sci Rep. 2024;14:3012.
Kopp R, Billecke N, Legradi J, den Broeder M, Parekh SH, Legler J. Bringing obesity to light: Rev-erbα, a central player in light-induced adipogenesis in the zebrafish? Int J Obes. 2016;40:824–32.
Warbrick-Smith J, Behmer ST, Lee KP, Raubenheimer D, Simpson SJ. Evolving resistance to obesity in an insect. Proc Natl Acad Sci. 2006;103:14045–9.
Pierce WD, Diane A, Heth CD, Russell JC, Proctor SD. Evolution and obesity: resistance of obese-prone rats to a challenge of food restriction and wheel running. Int J Obes. 2010;34:589–92.
Brüne M, Hochberg Z. Evolutionary medicine – the quest for a better understanding of health, disease and prevention. BMC Med. 2013;11:116.
Hochberg ME, Thomas F, Assenat E, Hibner U. Preventive evolutionary medicine of cancers. Evol Appl. 2013;6:134–43.
Basile AJ, Renner MW, Hidaka BH, Sweazea KL. An evolutionary mismatch narrative to improve lifestyle medicine: a patient education hypothesis. Evol Med Public Health. 2021;9:eoab010.
Jiao J. The role of nutrition in obesity. Nutrients. 2023;15:2556.
Hannon SC, Hillier SE, Thondre PS, Clegg ME. Lower energy-dense ready meal consumption affects self-reported appetite ratings with no effect on subsequent food intake in women. Nutrients. 2021;13:4505.
Cheng W-Y, Yeh W-J, Ko J, Huang Y-L, Yang H-Y. Consumption of dehulled adlay improved lipid metabolism and inflammation in overweight and obese individuals after a 6-week single-arm pilot study. Nutrients. 2022;14:2250.
Gupta MK, Peng H, Li Y, Xu C-J. The role of DNA methylation in personalized medicine for immune-related diseases. Pharmacol Ther. 2023;250:108508.
Alves JGB, Alves LV. Early-life nutrition and adult-life outcomes. J Pediatr (Rio J). 2024;100:S4–9.
Inadera H. Developmental origins of obesity and type 2 diabetes: molecular aspects and role of chemicals. Environ Health Prev Med. 2013;18:185–97.
Chavoya-Guardado MA, Vasquez-Garibay EM, Ruiz-Quezada SL, Ramírez-Cordero MI, Larrosa-Haro A, Castro-Albarran J. Firmicutes, bacteroidetes and actinobacteria in human milk and maternal adiposity. Nutrients. 2022;14:2887.
Westbury S, Oyebode O, van Rens T, Barber TM. Obesity stigma: causes, consequences, and potential solutions. Curr Obes Rep. 2023;12:10–23.
Aoshiba K, Tsuji T, Itoh M, Yamaguchi K, Nakamura H. An evolutionary medicine approach to understanding factors that contribute to chronic obstructive pulmonary disease. Respiration. 2015;89:243–52.
Nesse RM, Bergstrom CT, Ellison PT, Flier JS, Gluckman P, Govindaraju DR, et al. Making evolutionary biology a basic science for medicine. Proc Natl Acad Sci USA. 2010;107:1800–7.
• Grunspan DZ, Nesse RM, Barnes ME, Brownell SE. Core principles of evolutionary medicine: A Delphi study. Evol Med Public Health. 2018;2018:13–23. Provides a comprehensive framework of the core principles that define the evolutionary medicine approach and its applications.
Gupta MK, Vadde R. Next-generation development and application of codon model in evolution. Front Genet. 2023;14:1091575.
Meeks KAC, Bentley AR, Adeyemo AA, Rotimi CN. Evolutionary forces in diabetes and hypertension pathogenesis in Africans. Hum Mol Genet. 2021;30:R110–8.
Acknowledgements
Authors would like to thank Mr. Jan Benzenberg, Operations Manager at Deutsche Telekom, (Germany), and MakerSpace Bonn e.V (Germany), for providing a computation facility for the literature survey.
Funding
None.
Author information
Authors and Affiliations
Contributions
Conceptualization and design: MKG and RV. Data collection and analysis: MKG and GG. Writing – original draft: MKG. Writing – review and editing: All co-authors. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gupta, M.K., Gouda, G. & Vadde, R. Relation Between Obesity and Type 2 Diabetes: Evolutionary Insights, Perspectives and Controversies. Curr Obes Rep 13, 475–495 (2024). https://doi.org/10.1007/s13679-024-00572-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-024-00572-1