
Abstract
Purpose of Review
The term “metabolic flexibility” denotes the dynamic responses of the cellular oxidative machinery in order to adapt to changes in energy substrate availability. A progressive loss of this adaptive capacity has been implicated in the development of obesity-related comorbidities. Mitochondria are dynamic intracellular organelles which play a fundamental role in energy metabolism, and the mitochondrial adaptation to environmental challenges may be viewed as the functional component of metabolic flexibility. Herein, we attempt to comprehensively review the available evidence regarding the role of mitochondrial adaptation and metabolic flexibility in the pathogenesis of obesity and related morbidities, namely insulin resistance states and non-alcoholic fatty liver disease (NAFLD).
Recent Findings
Overall, there is a concrete body of evidence to support the presence of impaired mitochondrial adaptation as a principal component of systemic metabolic inflexibility in conditions related to obesity. There are still many unresolved questions regarding the relationship between the gradual loss of mitochondrial adaptability and the progression of obesity-related complications, such as causality issues, the timely appearance and reversibility of the described disturbances, and the generalizability of the findings to the mitochondrial content of every affected tissue or organ.
Summary
The evidence regarding the causality between the observed associations remains inconclusive, although most of the available data points towards a bidirectional, potentially mutually amplifying relationship. The spectrum of NAFLD is of particular interest, since functional and pathological changes in the course of its development closely mirror the progression of dysmetabolism, if not constituting a dynamic component of the latter.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the last few decades, the obesity epidemic rages unopposed across the globe and among all age groups [1, 2]. Despite an increasing understanding of the pathogenesis and advances in the pharmacotherapy of obesity [3], the prevalence of obesity-related and potentially preventable conditions including insulin resistance, type 2 diabetes mellitus (T2DM), and non-alcoholic fatty liver disease (NAFLD) continues to rise [4, 5].
From a bioenergetic standpoint, obesity can be considered as the consequence of a chronically positive energy equilibrium resulting from an increased energetic influx [6]. In turn, the dynamic responses of the cellular oxidative machinery in order to adapt to changes in energy substrate availability have been collectively termed “metabolic flexibility,” and a progressive loss of this adaptive capacity has been implicated in the development of obesity-related comorbidities [7, 8].
Mitochondria are dynamic intracellular organelles which play a fundamental role in energy metabolism. Most notably, they are responsible for the production of the vast majority of adenosine triphosphate (ATP) through the process of cellular respiration, namely the oxidative conversion of nutritional substrate chemical energy into ATP [9]. Oxidative phosphorylation (OXPHOS) by the enzyme ATP synthase of the electron transport chain (ETC), which takes place across the inner mitochondrial membrane [10], represents the ultimate step in this process, which originates in the production of acetyl coenzyme A from several nutritional substrates. Beta-oxidation of free fatty acids (FFAs) and the Krebs cycle, a crucial intermediate metabolic step through which the high-energy electron donors to the ETC are generated, are also carried out in the mitochondrial matrix [11]. In the setting of prolonged fasting and energy depletion, available acetyl-CoA enters the ketogenic pathway in liver mitochondria, so that the resulting ketone bodies can be used as energy substrates by energy-demanding tissues such as the heart muscle and the brain [12]. Furthermore, mitochondria are involved in a plethora of vital functions related to energy metabolism and they orchestrate the process of cellular respiration, namely the production of reactive oxygen species (ROS) in the ETC and their subsequent detoxification, thus regulating the intracellular redox status, intracellular signaling [13], and apoptosis [14]. Of note, mitochondria constitute the integrators of energy handling at the cellular level, both under static conditions and in dynamic response to fluctuations in energy demands and substrate availability. Τhe mitochondrial adaptation to energetically ever-changing environments may be viewed as the functional component of metabolic flexibility.
In the present review, we attempt to comprehensively review the available evidence regarding the role of mitochondrial adaptation and metabolic flexibility in the pathogenesis of obesity and other obesity-related complications, namely insulin resistance and NAFLD. To that end, important conceptual and methodological aspects that delineate the notions of mitochondrial adaptation and metabolic flexibility are also briefly presented.
Defining Metabolic Flexibility
Metabolic flexibility corresponds to the ability of tissues to adapt to fluctuations in energy demands by rapidly and efficiently switching between oxidations of different energy substrates depending on their availability. Although the notion of metabolic flexibility applies to a broad spectrum of physiological conditions, it could be simplified by denoting the switch between glucose and FFA oxidation [15]; examples include the transition from fasting to the postprandial state and vice versa, the postprandial secretion or exogenous administration of insulin, and the consumption of meals of different consistency levels with respect to carbohydrate and fat content. The oxidation of different macronutrients results in altered molar CO2 production per oxygen molecule consumed and can be assessed through indirect calorimetry as the respiratory quotient (RQ). Monitoring of RQ in response to physiological or experimental nutritional challenges may thus serve as an indirect, non-invasive measure of whole-body metabolic flexibility [16]. Teleologically, a switch from glucose to fat oxidation may spare glucose reserves during fasting periods, and an increased insulin-stimulated postprandial glucose utilization may serve to achieve euglycemic homeostasis [17]. Conversely, the inability to adapt to substrate availability (metabolic inflexibility) may lead to an aberrant mobilization and utilization of fat and glucose, leading to increased FFA concentrations and hyperglycemia, as is the case in insulin resistance and overt T2DM, respectively.
Features of systemic metabolic inflexibility within the obesity–insulin resistance–T2DM continuum are strikingly distinguishable in the study of isolated tissues that are crucial for dynamic responses to metabolic challenges, namely the liver [18, 19] and the skeletal muscle [20]. Furthermore, features of whole-body metabolic flexibility assessed by means of an euglycemic–hyperinsulinemic clamp may be reflected on that of isolated cells, outside their physiological tissue architecture and independently from their systemic neurohormonal environment [21]. As far as energy metabolism is concerned, mitochondria are, by far, the most prominent representatives among cell organelles at the subcellular level. Most interestingly, features of impaired metabolic flexibility (i.e., impaired inhibition of pyruvate flux into the Krebs cycle in the presence of FFA and vice versa) are evident in isolated mitochondria of rodents after 12 weeks of obesogenic feeding [22]. On the whole, it could be argued that features of metabolic flexibility are preserved across ascending orders of biological system organization in a fractal-like fashion; in order to view the transition from obesity to insulin resistance and overt T2DM as a manifestation of progressive development of metabolic inflexibility, a detailed study of mitochondrial function and their adaptation to energetically changing environmental conditions is of crucial importance.
Defining Mitochondrial Adaptation
Under everyday conditions, the delivery of energy substrates varies greatly in quantity and composition, both in the short (e.g., during the meal intake–fasting cycles) and in the long term (hypocaloric or hypercaloric energy intake, varying dietary habits). Accordingly, a substantial versatility of the mitochondrial energy production machinery is essential in order to adapt to the quantitatively and qualitatively wavering energy availability and the real-time cellular energy demands and thus sustain cellular homeostasis and tissue viability. Taken together, these adaptive mechanisms comprise the concept of mitochondrial adaptation, while a mitochondrial “maladaptation” would denote a failure to optimally adjust to the changing equilibrium between energy demands and availability. In the following section, a brief overview of the major adaptive functions of mitochondria will be presented.
Quantitative and Qualitative Aspects of Mitochondrial Adaptation
Mitochondrial Content
Mitochondrial content, namely the absolute number of mitochondria, does not remain stable during the cell life, but it is rather the net resultant of the mitochondrial turnover on the one hand and mitochondrial dynamics on the other.
Although there is no gold standard method for the quantification of mitochondrial content, both direct and indirect methods can be used for this purpose. The mitochondrial absolute and relative to total cellular area can be directly measured in histological sections by using fluorescent plain microscopy or transmission electron microscopy (TEM) [23]. Besides directly quantifying mitochondrial content, these techniques additionally allow for the assessment of mitochondrial morphology [23]. Further techniques employ measurements of mitochondria-derived or mitochondria-associated components to provide an approximation of mitochondrial content; these include the quantification of mitochondrial DNA (mtDNA) through real-time polymerase chain reaction as well as the mtDNA-to-nuclear DNA ratio [24, 25]. The mtDNA content may accurately reflect mitochondrial mass and respiratory activity [24], although this finding is not unequivocal among studies [26]. Citrate synthase activity (CSA), a key enzyme of the Krebs cycle, has also been used as an index of mitochondrial density in skeletal muscle [26, 27], adipose [28, 29], and liver [30, 31] tissues. Furthermore, tissue cardiolipin content, a key phospholipid of the inner mitochondrial membrane, has been shown to correlate with adipose tissue and liver mitochondrial mass in experimental studies [32]. The quantification of protein elements crucial for mitochondrial function and/or structure (such as peroxisome proliferator–activated receptor gamma coactivator 1-alpha (PGC-1a), nuclear respiratory factor 1/2 (NRF-1/2), ATP synthase subunit c, cytochrome c oxidase subunit II) through Western Blot analysis may provide indirect evidence regarding cellular mitochondrial mass [23].
Mitochondrial Turnover (Biogenesis and Mitophagy)
Mitochondrial turnover denotes the equilibrium between the biogenesis of new and the autophagy of existing mitochondria. The synthesis of new mitochondria is a natural response to environmental bioenergetic stress, such as physical exercise, caloric restriction, temperature changes, cell division, and differentiation [33]. Mitochondrial biogenesis increases the cellular oxidative and redox capacity for a given amount of energy production. The coupling of mitochondrial biogenesis with current energetic demands is achieved through the induction or post-translational modification of critical transcriptional regulators of mitochondrial genome replication such as PGC-1a [34], NRF-1/2 [34], or mitochondrial transcription factor A (TFAM) [35], which are triggered by fluctuations of intracellular energy intermediates [36]. For example, during energy deprivation (e.g., fasting), the depletion of intracellular ATP, and the rise in AMP/ATP ratio, in conjunction with the changes of circulating hormones (rise in glucagon, noradrenalin, drop of insulin), activate AMP kinase (AMPK), which, in turn, phosphorylates and activates PGC-1a [37]. Under the same conditions, a reduced substrate flux into the Krebs cycle results in a rise of the NAD+/NADH ratio and a subsequent activation of sirtuin 1 (SIRT1), which deacetylates PGC-1, thereby increasing its transcriptional activity [38, 39]. Similarly, the energy-demanding and energy-depleted environment achieved in skeletal muscle during high-intensity exercise is met with increased mitochondrial biogenesis through the PGC-1a signaling pathway [40, 41]. In the other direction, in the chronic setting of excess energy intake and increased energy availability observed in obesity, the inactivation of AMPK and SIRT1 by the decline in AMP/ATP and NAD+/NADH ratios, respectively, leads to decreased PGC-1a transcriptional activity and reduced downstream signaling [37].
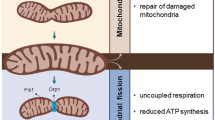
The opposing function of mitophagy, namely the selective lysosomal clearance of damaged and dysfunctional mitochondria, serves as a quality control mechanism to preserve a structurally and functionally intact mitochondrial inventory [42]. Despite the counter-regulatory role of antioxidant systems, mitochondria are prone to oxidative damage induced by accumulating ROS produced during electron flux across the ETC, which may collectively impair their ability to efficiently oxidize FFA and give rise to toxic intracellular fatty acid accumulation [43]. Considering that the oxidatively damaged mitochondria may be chief sources of further intracellular free radical production [44], an intact mitophagy function is essential to prevent the vicious cycle of further mitochondrial oxidative damage.
Mitochondrial Dynamics
Mitochondria are structurally dynamic, in the sense that they are able to change their shape, size, and numbers in the short term through the processes of fusion and fission. In the fusion process, mitochondria merge their outer and inner membranes and they exchange their contents [45, 46]. Mitochondrial fusion is chiefly mediated by mitofusins 1 and 2 (Mfn1 and Mfn2, respectively), G proteins of the outer mitochondrial membrane, and dynamin-like protein of the inner mitochondrial membrane (OPA1) [47]. In fission, mitochondria undergo fragmentation in a process that requires the recruitment of cytosolic dynamin-related protein 1 through binding to fission protein 1 (Fis1) of the outer mitochondrial membrane [47]. By fission, the contents of the initial mitochondrion are distributed into two new functionally independent organelles, in a sequence reminiscent of prokaryote cell division. Mitochondrial fission is tightly coupled with the cell cycle, as it reaches its peak during the pre-mitotic phase [45].
The dynamic processes of fusion and fission ensure that mitochondria react to environmental challenges as a functional whole, rather than as isolated units. Fused mitochondria display higher content and activity of dimerized ATP synthase, being hence more bioenergetically efficient [48]. The exchange of mitochondrial content through fusion allows the repair of damaged components, including mtDNA [46], or inversely, allows the selective segregation of irreparable components through fusion and subsequent fission [49], thus rendering the process of mitophagy more targeted towards damaged structures and, hence, more efficient. The state of obesity and excess energy availability has been shown to promote mitochondrial fission [37, 50]. In support of this statement, subjects with obesity display reduced levels of Mfn2 in skeletal muscle compared to their lean counterparts, while the shift of the fusion–fission balance towards fission results in substantial decreases of glucose oxidation and oxygen consumption [51]. In the same direction, obesity and insulin resistance have been associated with increased mitochondrial fission in skeletal muscle [52].
Functional Aspects of Mitochondrial Adaptation–Mitochondrial Plasticity
The term “plasticity” describes mitochondrial adaptation from a functional perspective. Among different aspects of mitochondrial adaptation, the one which is conceptually most directly related to metabolic flexibility is that of mitochondrial plasticity, since it highlights the sum of acute and chronic changes of mitochondrial content and function in response to metabolic challenges and energetically changing environmental conditions. These adaptive changes of mitochondrial content and function (turnover and dynamics) are principally shaped through changes of electron flux across the mitochondrial respiratory chain.
Under resting conditions, resting mitochondrial activity determines the cellular ATP content dependent on energy substrate and oxygen availability [53]. The amount of ATP produced for a given substrate entering the tricarboxylic acid (TCA) cycle reflects mitochondrial bioenergetic efficiency, which is strongly related to the coupling of OXPHOS with ATP synthesis. This electrochemical coupling is, in turn, dependent upon the presence of uncoupling proteins (UCPs) in the inner mitochondrial membrane, which allow for transmembrane proton transport by bypassing ATP synthase [54]. The bioenergetically futile proton cycling across the inner mitochondrial membrane is of particular importance for certain tissues (e.g., brown adipose tissue), and although inefficient from a thermodynamic point of view, the induction of uncoupled respiration in white adipose or skeletal muscle tissue has been shown to augment energy expenditure, protect against diet-induced obesity, and promote improved insulin sensitivity and redox status in transgenic mice [55,56,57].
In the setting of acutely increased energetic demands causing a depletion in intracellular energy intermediates, an increased proton flux through ATP synthase is expected to occur in an effort to replete the acutely diminished ATP stores; the resultant rise of AMP/ATP and NAH+/NADH ratios leads to increased activity of β-oxidation [58, 59], Krebs cycle [60], and OXPHOS activity. Likewise, in the postprandial state (or during a hyperinsulinemic clamp), the insulin-stimulated glucose uptake by skeletal muscle leads to a temporary intracellular ATP depletion due to the action of hexokinase, UTP-glucose-1-phosphate uridylyltransferase, and protein synthesis. The subsequently increased ATP production by mitochondria is known as insulin-stimulated ATP synthesis [61, 62].
Methods for Assessment of Mitochondrial Plasticity
In overview, the major methods for the assessment of mitochondrial plasticity either provide estimates of resting ATP content or ATP synthetic rates following metabolic challenges in whole tissues (in vivo methods) or provide estimates of OXPHOS activity by measuring oxygen consumption rates in cells or isolated mitochondria under tightly controlled conditions of oxygen and energy substrate availability (ex vivo methods).
In Vivo Methods
These include the quantification of intracellular phosphorus metabolites (mainly ATP or phosphocreatine (PCr)) by means of phosphorus magnetic resonance spectroscopy (31P-MRS) [63] under resting conditions or in response to metabolic challenges. By these techniques, mitochondrial activity can be evaluated in the setting of intact whole-body physiology that includes physiological substrate availability and inter-tissue interactions. Their non-invasive nature allows for serial applications in order to evaluate the effectiveness of potential lifestyle or pharmaceutical strategies in states characterized by loss of mitochondrial plasticity. On the other hand, these methods may be confounded by the variable availability of energy substrates and oxygen in tissues, and additionally, they provide no direct data on mitochondrial content or the functional integrity of isolated mitochondria or specific components of the ETC.
-
(i)
Resting OXPHOS activity (liver, skeletal muscle): This can be quantified by 31P-MRS as ATP content or else unidirectional ATP synthase flux under resting conditions [53, 64]. It is dependent upon energy substrate availability and oxygen delivery.
-
(ii)
Hepatic submaximal ADP-stimulated OXPHOS activity measured by 31P-MRS after intravenous [65, 66] or oral [67] fructose loading, which causes an intracellular ATP depletion through fructokinase enzymatic activity. The fractional ATP recovery (ratio between final and nadir ATP content) is the measured outcome variable.
-
(iii)
Skeletal muscle submaximal ADP-stimulated OXPHOS activity measured by 31P-MRS as the rate of PCr recovery following exercise or the rate of ATP synthesis during an euglycemic–hyperinsulinemic clamp [61, 68].
Ex Vivo Methods
These refer to the estimation of basal and maximal ADP-stimulated OXPHOS activity and are mainly represented by high-resolution respirometry. The oxidative capacity in whole or permeabilized cells or isolated mitochondria can be quantified under tightly controlled conditions that include supraphysiological, saturating energy substrate fluxes in the Krebs cycle [69] and β-oxidation [70]. By this technique, specific functional aspects of the mitochondrial respiratory chain can be quantified without the confounding effects of differential oxygen or energy substrate delivery that may obscure certain in vivo techniques. On the other hand, this technique depends upon invasive procedures (biopsies) to obtain considerable amounts of tissue to allow for thorough mitochondrial analyses.
In total, in vivo and ex vivo techniques are highly complementary, incorporating different approaches for mitochondrial functional assessment. Additional important information can be obtained through laboratory (in vitro) measurements of specific ETC enzyme activities, mitochondrial respiratory by-products (ROS, lipid peroxidation products), and antioxidant defense mechanisms (catalase activity, reduced/oxidized glutathione ratio) [57, 71, 72].
Metabolic Flexibility and Mitochondrial Adaptation in Obesity
The key studies investigating mitochondrial plasticity in the skeletal muscle and liver of humans with obesity are presented in Table 1.
Skeletal Muscle
It has been hypothesized that the high lipid accumulation observed in the skeletal muscle of humans with obesity may lead to impaired aspects of mitochondrial function through the generation of high levels of ROS; an inverse relationship is also possible, implicating altered mitochondrial function as the first step towards reduced FFA oxidation and increased ectopic lipid storage in skeletal muscle, predisposing humans with obesity to lipotoxicity-related insulin resistance [85]. Several studies have investigated the features of metabolic flexibility and mitochondrial plasticity in individuals with obesity with the use of in vivo non-invasive techniques.
Individuals with obesity have been reported to display higher fasting muscle RQ and a blunted increase in RQ after insulin stimulation compared to lean controls, in parallel with reduced skeletal muscle fat oxidation despite unaffected FFA uptake. Of note, ΔRQ correlated positively with insulin sensitivity and was increased after weight loss [74]. According to more recent studies, both fasting and post-exercise lipid oxidation has been found to be impaired in extremely obese (mean BMI 40.8 kg/m2) as well as obese subjects following weight loss of at least 45 kg (mean BMI 33.7 kg/m2), compared with never-obese lean controls [75]. In contrast with these data, similar OXPHOS activity assessed by 31P-MRS was observed between formerly overweight or obese normal-weight individuals (mean BMI 24.1 kg/m2 after a mean loss of 11.1 kg of body weight) and lean sedentary controls in another study [76]. In this study, diet-induced weight loss did not affect OXPHOS; however, a higher mitochondrial oxidative capacity was noted among lean, trained participants compared with formerly obese subjects [76]. In line with the latter study, markers of oxidative metabolism did not differ between formerly overweight (initial mean BMI 28.6, range 27–30 kg/m2 reduced to a mean of 23.8 kg/m2) and never-overweight females [77]. The above observations may indicate that the degree of physical activity may at least partly drive features of muscle mitochondrial oxidative function irrespective of the degree of adiposity.
A significant body of research has focused on pediatric obesity, highlighting the important role that excess adiposity in young age may be implicated in the future development of T2DM and related comorbidities [86]. In one study, adolescents with obesity and overweight exhibited higher insulin resistance and longer PCr recovery after exercise compared with lean controls, matched for age and physical activity [78]. In this study, an impaired mitochondrial adaptation to exercise correlated significantly with higher BMI, insulin resistance, and serum triglycerides, but not significantly with intramyocellular lipids (IMCLs); in multivariate regression analysis, only BMI remained as a significant independent predictor of PCr recovery [78]. Data of other research groups have been conflicting. Fleischman et al. [79] examined 37 normal-weight and 37 overweight children without a family history of T2DM; muscle mitochondrial plasticity expressed as PCr recovery after exercise did not differ between groups. Nonetheless, insulin-resistant overweight participants had longer PCr recovery time than their overweight counterparts with normal insulin sensitivity, highlighting the importance of insulin resistance as an important intermediate link between adiposity and defective muscle mitochondrial adaptation [79]. Insulin-resistant females with obesity were also shown to present lower rates of post-exercise PCr recovery than insulin-sensitive girls with obesity in another study, although there were no differences in the degree of adiposity, visceral fat, and IMCL [81]. Contrary to these data, a recent study in 17 lean and 21 adolescents with obesity failed to show any evidence of impaired skeletal muscle mitochondrial adaptation to exercise assessed by 31P-MRS in those with obesity, despite their having more profound insulin resistance and higher IMCL content [80]. Although in this study post-exercise OXPHOS capacity did not differ between lean subjects and subjects with obesity without diabetes, a significant impairment of mitochondrial plasticity was observed in adolescents with T2DM in comparison to lean and obese controls in another study from the same study group; furthermore, in the sum of the studied cohort, whole-body insulin resistance was associated with decreased mitochondrial OXPHOS capacity and lack of FFA suppression during an euglycemic–hyperinsulinemic clamp independent of hemoglobin A1c (HbA1c) [82]. Certain differences in the design of these studies, regarding participant composition by age and gender, selection of control groups, varying severities of obesity in the studied populations, and method of insulin resistance (IR) assessment (HOMA [78, 79, 81] or euglycemic–hyperinsulinemic clamp [80, 82]), may have at least in part contributed to these seemingly inconsistent results. In view of the evidence summarized above, alterations of skeletal muscle mitochondrial adaptation may be observed in obesity as long as it is complicated by the development of insulin resistance, and are not necessarily related to adiposity per se or increased IMCL.
Liver
About one-third of individuals with obesity suffer from NAFLD, and half of patients with NAFLD have obesity [87]. Obesity and NAFLD are therefore tightly inter-related. In this section, we briefly review the available data of human studies regarding hepatic mitochondrial adaptation in individuals with obesity, in whom hepatic steatosis was either absent or not taken into consideration. Data from human research on the association of NAFLD with mitochondrial maladaptation will be discussed separately.
Nair et al. [66] examined hepatic mitochondrial plasticity in 5 lean, 7 overweight, and 7 participants with obesity, applying 31P-MRS before and after an intravenous fructose challenge; post-infusion ATP recovery rate did not differ between groups; basal ATP was, however, lower in individuals with obesity and overweight than in lean controls and correlated inversely with BMI [66]. A different approach was applied by Iozzo et al. [84] with the use of 11C-palmitate positron emission tomography (PET) imaging. Enhanced FFA oxidation by the liver of individuals with obesity was reported, whereas the rates of FFA uptake and esterification between lean subjects and participants with obesity were similar [84].
Metabolic Flexibility and Mitochondrial Adaptation in Insulin Resistance
The hypothesis of impaired mitochondrial function in patients with T2DM, as well as insulin resistance states without overt T2DM, has been formed more than two decades ago. In the following sections, we present data on mitochondrial plasticity in individuals with IR (T2DM and beyond) assessed by in vitro techniques. Table 2 summarizes the studies investigating mitochondrial plasticity in the skeletal muscle and liver of humans with IR.
Skeletal Muscle
A considerable number of relevant studies have focused on quantitative and functional alterations of skeletal muscle mitochondria and the accompanying impairment of metabolic flexibility, highlighting the important role of skeletal muscle in the pathogenesis of systemic IR [110].
Basal ATP content in skeletal muscle cells assessed by 31P-MRS in patients with T2DM has been found to be lower than that of controls with normal glucose tolerance; an inverse correlation between PCr content and glycemic measures such as fasting plasma glucose (FPG) and glycated HbA1c has been also reported [89]. The evidence suggesting muscle mitochondrial maladaptation underlying impaired metabolic flexibility is mainly provided by studies applying metabolic challenges. A study by Schrauwen-Hinderling et al. [111] revealed a 45% longer half-time of PCr recovery measured by 31P-MRS following exercise in overweight participants with T2DM compared with BMI-matched healthy controls. In this study, PCr recovery half-time correlated positively with FPG and HbA1c, suggesting that the magnitude of muscle mitochondrial adaptation defects in T2DM relates to the degree of metabolic impairment. Of note, the IMCL content measured by 1H-MRS did not differ between groups in this study, implying that mitochondrial dysfunction may be a more critical step for the pathogenesis of T2DM than IMCL accumulation [111].
In the same direction, patients with T2DM displayed lower increases in RQ after insulin infusion and longer PCr recovery half-time than healthy controls in a study using both 31P-MRS and indirect calorimetry to assess mitochondrial plasticity [91]. A similarly defective muscle PCr recovery rate accompanied by impaired metabolic flexibility as assessed by blunted changes of RQ following insulin stimulation was ascertained in individuals with T2DM by Meex et al. [88]. Interestingly, these differences were attenuated after a 12-week course of combined aerobic/resistance training and were paralleled by improvements in IR, suggesting a reversible component of mitochondrial plasticity impairment in T2DM, at least in skeletal muscle cells [88].
In agreement with these findings, fasting ATP synthesis in skeletal muscle was 27% lower in patients with T2DM compared with young healthy controls, and fasting ATP in muscle correlated with fasting FFA and waist-to-hip ratio. Insulin-stimulated flux through ATP synthase was impaired in patients with T2DM and correlated positively with peripheral insulin sensitivity, independent of glucose transport or phosphorylation and IMCL accumulation [90].
Of note, Chomentowski et al. [92] observed that metabolic flexibility, as assessed by ΔRQ during an euglycemic–hyperinsulinemic clamp, was significantly lower not only in patients with T2DM but also in non-diabetic insulin-resistant participants; this was paralleled by a lower inter-myofibrillar mitochondrial content among participants with T2DM and IR compared with healthy controls. Mitochondrial content showed a significantly positive correlation with ΔRQ (R = 0.38, p = 0.02) [92]. In contrast with these data, Phielix et al. [93] ascertained a solely functional, rather than quantitative, component regarding the loss of muscle mitochondrial adaptation in T2DM; in their study, they enrolled 10 patients with T2DM with a mean BMI within the overweight range (29.1 kg/m2), 12 first-degree relatives of patients with T2DM, and 16 healthy controls [93]. The in vivo assessment of mitochondrial adaptation using 31P-MRS revealed a prolonged half-time of PCr recovery in subjects with T2DM versus controls and a non-significant trend in the first-degree relatives (p = 0.08), while there was no difference between groups in mitochondrial density assessed by ex vivo techniques [93]. Taking all the above data into account, it is likely that decreases in mitochondrial numbers contribute to the functional impairment of the mitochondrial machinery in IR states. However, differences in the applied methodologies for the quantitative mitochondrial assessment may be at least partly responsible for the discordant reported findings.
In contrast to the abovementioned studies, there have been reports that do not support the relationship between compromised muscle mitochondrial plasticity and T2DM. No significant differences were observed in PCr recovery kinetics in patients with T2DM and IR without T2DM compared to healthy controls in a study using 31P-MRS pre- and post-exercise [94]. Likewise, patients with well-controlled T2DM had similar basal ATP content and post-exercise PCr recovery time with age-, gender-, and weight-matched non-diabetic individuals. No change was further observed in muscle mitochondrial adaptation after 8 weeks of training, apart from an enhanced lipid oxidation in the group with T2DM [95]. The adequate glycemic control of the enrolled patients with T2DM, the lack of discontinuation of medical therapy contrary to other studies, and the relatively low intensity of the training intervention (increased daily walking) should be taken into account for the interpretation of these results. Of note, Larsen et al. [96] reported that the impaired mitochondrial function in subjects with T2DM versus non-diabetic subjects was limited to leg muscles, whereas no difference was found in arm muscle measurements. In addition, Bajpeyi et al. [97] reported a significant correlation between PCr recovery rates and insulin sensitivity within their total study sample including subjects with varying degrees of IR; the relationship was, however, non-significant in the subgroup of participants with T2DM. In this study, the measures of mitochondrial oxidative capacity in patients with T2DM overlapped, to a greater or lesser extent, with those of young healthy sedentary participants and active controls [97].
Of particular interest are the results of studies regarding metabolic flexibility and mitochondrial plasticity in subjects with IR and without overt hyperglycemia, in order to circumvent the potentially confounding effects of a glucotoxic environment. Apostolopoulou et al. [98] observed that ΔRQ assessed by indirect calorimetry among 136 near-normoglycemic individuals with recently diagnosed T2DM had a positive correlation with whole-body insulin sensitivity. This correlation was abolished after adjustment for circulating FFA, implying that increased circulating FFA concentration and the resulting lipotoxic milieu by itself may contribute to metabolic inflexibility [98]. The findings of three other studies conducted in non-T2DM populations with IR similarly point towards a direct relationship of defective skeletal muscle mitochondrial plasticity with IR independent of hyperglycemia; in the first one, lean elderly individuals (mean age 70 years) were found to have ~40% lower muscle mitochondrial oxidative activity compared with their younger controls (mean age 27 years) in conjunction with IR and increased IMCL content, hence highlighting a concomitant development of age-related mitochondrial functional changes and of age-associated IR [64]. In the second study, 14 lean but insulin-resistant offspring of patients with T2DM and 12 controls were investigated; basal mitochondrial OXPHOS activity in skeletal muscle assessed by 31MRS was 30% lower in the insulin-resistant offspring versus insulin-sensitive controls, and this finding was accompanied by a 80% increase in IMCL [100]. The aforementioned study measured OXPHOS capacity in a steady state, while in a third study conducted by the same research group, insulin-resistant offspring of patients with T2DM were subjected to exogenous insulin stimulation. In the latter study, the insulin-resistant offspring were found to have significantly lower rates of insulin-stimulated glucose uptake and a twofold increase in IMCL compared with the control population. The insulin-stimulated changes in ATP synthesis correlated with intramyocellular Pi concentrations, implying that changes in myocellular phosphate transport may constitute an additional critical step for the emergence of the observed phenomena [68]. Similarly, and in further support of a role of mitochondrial content in the setting of IR, Ukropcova et al. [101] reported lower skeletal muscle mitochondrial content (estimated by mtDNA quantification) among sedentary male T2DM offspring compared with matched controls with a negative family history for T2DM. This was interestingly accompanied by a higher sleep RQ following 3 days of high-fat diet and a trend towards lower insulin-stimulated changes in RQ [101].
In contrast with the above data suggesting impaired muscle mitochondrial plasticity in non-diabetic IR states, no significant differences were observed in PCr recovery time post-exercise between healthy males born with low birth weight (LBW) and healthy controls, despite the ascertainment of hepatic insulin resistance and a variety of other pre-diabetic abnormalities such as elevated FPG and insulin levels in LBW subjects [102]. In addition, Mensink et al. [103] reported similar values of post-exercise RQ, total carbohydrate, and fat oxidation in subjects with T2DM and impaired glucose tolerance (IGT), although a reduced skeletal muscle FFA uptake and oxidation was evident in both IGT and participants with T2DM compared to controls.
As clearly suggested by the data summarized above, IR is not limited to patients with T2DM; accumulating evidence suggests that IR may also be present in T1DM [112] and related to impaired skeletal muscle mitochondrial adaptation in response to exercise [104, 105]. Another group of investigators reported that fasting muscle ATP content was similar in patients with T1DM and healthy insulin-sensitive controls, but the insulin-stimulated flux through ATP synthase was lower in T1DM, possibly due to an impaired myocellular glucose transport and phosphorylation in these individuals [106].
Although definite conclusions regarding the causal relationship between impaired muscle mitochondrial adaptation and IR cannot be drawn based on the presented evidence, special reference should be made to the results of a unique study that examined 7 patients with severe congenital IR due to mutations in the insulin receptor. In this study, patients with congenital IR had prolonged PCr recovery rate following exercise compared to healthy controls [107]. Based on this finding, a speculation towards an impaired mitochondrial adaptability driven by IR itself could be made. Nevertheless, although interesting, the above observations were made in cases representing physiological extremes and cannot be extrapolated to the total spectrum of IR states.
Liver
A relatively limited amount of data extracted by in vivo non-invasive techniques is available about the plasticity of liver mitochondria in IR states, compared with the numerous studies of skeletal muscle mitochondrial function. Hepatic ATP turnover was found to be 42% lower in patients with T2DM than in age- and BMI-matched controls [53]; in this study, hepatic ATP production showed a positive correlation with hepatic and whole-body insulin sensitivity and a negative correlation with hepatocellular lipid content, waist circumference, BMI, HbA1c, and FPG. Of note, the hepatocellular Pi content explained 56% of the variation in hepatic ATP production [53]. In an additional study by Szendroedi et al. [108], patients with well-controlled T2DM were compared with two groups of controls: a group of participants matched for BMI and age and a second group of young, lean subjects. Patients with T2DM were found to have lower hepatic ATP content and Pi compared with both control groups. Importantly, independent of the hepatic lipid content, both hepatic ATP and Pi content showed a negative correlation with hepatic, but not with whole-body insulin sensitivity, indicating that the development of hepatic insulin resistance may actually precede hepatic steatosis [108]. The complexity of the phenomena underlying the development of metabolic inflexibility and metabolic dysregulation is further highlighted in a study by Fritsch et al. [83], in which hepatic ATP and Pi content in the fasted and postprandial state were compared across three groups of lean and individuals with obesity with and without T2DM. In this study, despite similarly increased postprandial FFA and triglyceride concentrations between lean and participants with obesity, a significant postprandial increase of hepatic ATP was evident solely among individuals with obesity without T2DM, but not in those with T2DM [83]. Even though a respirometric assessment of hepatic mitochondrial oxidative function was not provided, the results of this study point towards an upregulated hepatic mitochondrial oxidative metabolism in obesity, which is, however, not evident or lost with the development of overt T2DM. Of interest, in further support of the presence of adaptational mechanisms prone to metabolic fatigue, daily dietary fructose consumption may also tend to impair the plasticity of hepatic mitochondria in subjects with T2DM; a lower baseline hepatic ATP and a lower ATP recovery rate following an intravenous fructose challenge has been reported in patients with T2DM on a high-fructose diet compared with those consuming lower amounts of fructose [109].
Metabolic Flexibility and Mitochondrial Adaptation in NAFLD
NAFLD and Insulin Resistance
The spectrum of NAFLD ranges in severity from simple steatosis to advanced fibrosis, cirrhosis, and end-stage liver disease, which can, in some cases, lead to the development of hepatocellular carcinoma. A crucial transitional step towards advanced NAFLD is that of non-alcoholic steatohepatitis (NASH), which denotes the presence of hepatocellular necro-inflammation with or without liver enzyme elevations [113, 114]. The manifestations and severity of NAFLD share a close pathogenetic relationship with obesity, IR, and overt T2DM; NAFLD is highly prevalent among individuals with obesity, and its presence correlates strongly with the degree of obesity [115]. The estimates regarding NAFLD prevalence in patients with T2DM vary considerably depending on detection methodology and geographical area; it is noteworthy, however, that the prevalence of NASH reaches an alarming 40% within this population [116]. Furthermore, the presence of NAFLD in patients with T2DM is associated with the presence and severity of coronary artery disease [117,118,119] independent of traditional risk factors, including glycemic control and features of the metabolic syndrome [118, 119], although this does not necessarily translate into increased mortality or incidence of cardiovascular complications [117, 119].
The major studies that have investigated the features of metabolic flexibility and mitochondrial adaptation in NAFLD are presented in Table 3.
Metabolic Flexibility in NAFLD
The accumulated fat in human hepatocytes originates mainly from adipose tissue–derived FFA (59%), followed by de novo intrahepatic lipogenesis (26%) and dietary fat intake (15%) [124]. Consequently, an obvious link between the pathogenesis of obesity, IR, and NAFLD would include the increased availability of circulating FFA derived from the insulin-resistant adipose tissue [125]. Furthermore, in the setting of increased circulating FFA levels irrespective of source (“lipotoxic” environment), a defective switch towards fat catabolism for energy production or ketogenesis (a feature of metabolic inflexibility) would favor its aberrant storage in hepatocytes. Studies in humans have demonstrated typical features of defective metabolic flexibility among individuals with NAFLD. The interpretation of these results warrants caution however, since the confounding effects of obesity and IR or the severity of NAFLD itself have not always been considered. Samantha et al. [120] demonstrated a delayed switch to fat oxidation as assessed by a drop in RQ following a high-fat meal among individuals with NAFLD in comparison to healthy controls. Conversely, an impaired transition to carbohydrate oxidation suggested by a less profound increase in RQ during an euglycemic–hyperinsulinemic clamp has been ascertained among adolescents with NAFLD in comparison to age- and BMI-matched controls without hepatic steatosis [19]. Similarly, a significantly lower increase of RQ during an euglycemic–hyperinsulinemic clamp has been observed among adults with obesity and NAFLD or NASH in comparison to lean or counterparts with obesity and without NAFLD [30]. On the other hand, the physiologically expected decrease in RQ during fasting is preserved in NAFLD irrespective of the degree of hepatic steatosis [121]. Taken together, these data imply that whole-body metabolic inflexibility in NAFLD mainly manifests as an impaired ability to manage additional substrate flux in the setting of an energy storage overload. In this context, whole-body fat utilization indicated by a lower RQ under basal conditions appears diminished in NAFLD [19, 122, 126], despite a higher resting energy expenditure which is observed among obese individuals with NAFLD compared with their age-matched, gender-matched, BMI-matched, and fat mass–matched counterparts without hepatic steatosis [126]. Taken together, these observations may denote a qualitatively imbalanced metabolic state, with a shift away from fat oxidation in NAFLD despite an overall increase of oxidative activity. Furthermore, Croci et al. [122] demonstrated that fasting RQ correlates significantly with the histological severity of NAFLD, independent of BMI, visceral, and total adiposity. Of note, in this study, maximal fat oxidation during physical exercise showed no correlation with NAFLD histological features, implying a differential course of metabolic inflexibility development in liver and skeletal muscle [122].
Mitochondrial Adaptation in NAFLD
From a teleological point of view, the adaptation of hepatocyte mitochondrial function in obesity and IR would ideally serve to eventually maximize fat catabolism to acetyl-CoA through beta-oxidation and its further diversion through the TCA cycle towards ETC for ATP production. An increased liver fat oxidation in NAFLD is not an unequivocal finding among studies; it has been demonstrated in individuals with obesity using 11C-palmitate PET [84] and in subjects with NASH using 13C-octanoate breath tests [127]. However, Kotronen et al. [128] reported no differences in hepatic fat oxidation between NAFLD and controls, albeit using the more indirect and downstream metabolite 3-hydroxybutyrate acid as a marker of hepatic lipid oxidation.
Likewise, the induction of the TCA cycle in NAFLD has been debated among studies. In line with preclinical data supporting an upregulated TCA flux in mice with diet-induced hepatic steatosis and IR [129, 130], Sunny et al. [131] demonstrated an almost twofold increased TCA flux in humans with hepatic steatosis, indicating an upregulation of components of mitochondrial respiration. In the same study, TCA flux strongly positively correlated with hepatic fat content [131]. In contrast with these data, Petersen et al. [132] reported similar hepatic mitochondrial oxidation rates between individuals with and without NAFLD. However, it is important to consider that these two studies employed different methods for assessing mitochondrial oxidation (ex vivo 13C-NMR spectra analysis after stable isotope administration vs in vivo dynamic 13C-MRS, respectively) [133]. Furthermore, it should be noted that these studies were conducted in vastly different populations: participants in the study by Sunny et al. [131] had obesity and demonstrated features of the metabolic syndrome, while those in the study by Petersen et al. [132] were lean or only minimally overweight. Data on the histological severity of NAFLD were available in none of these studies. In total, it is likely that the presence of obesity and a dysmetabolic milieu plays a decisive role for the induction of the TCA cycle in the setting of hepatic steatosis. This is further supported by the ascertainment of increased TCA cycle activity in individuals with morbid obesity and NAFLD, as shown in a recent study using a genome-scale metabolic modeling analysis [134].
The eventual upregulation of hepatic mitochondrial oxidative function in NAFLD does not necessarily imply an efficient coupling to increased energetic demands. On the contrary, features of progressive impairment of mitochondrial adaptation are encountered in NAFLD. In a comparative study conducted in a relatively small sample of patients with NASH (n = 8) and age- and gender-matched controls, the submaximal ADP-stimulated hepatic OXPHOS activity assessed by ATP repletion after an intravenous fructose challenge was shown to be impaired in NASH [123]. Fletcher et al. [121] further demonstrated that individuals with NAFLD exhibited an upregulated TCA cycle flux in conjunction with impaired ketogenesis and increased gluconeogenetic hepatic flux following 24-h fasting compared to controls, to a magnitude mirroring the degree of hepatic steatosis.
In a study incorporating multiple measures of mitochondrial content and function, conducted in three groups of individuals with obesity (with and without hepatic steatosis, and NASH) and lean healthy controls, participants with NAFLD exhibited features of whole-body metabolic inflexibility as shown by a blunted rise in RQ during an euglycemic–hyperinsulinemic clamp, combined with gradual worsening of hepatic and peripheral IR, which were most evident in the NASH group [30]. The applied ex vivo high-resolution respirometry in whole liver tissue and isolated mitochondria from liver biopsy specimens revealed elevated β-oxidation–related and TCA-related respiration rates in the groups with obesity with and without hepatic steatosis compared to controls, possibly denoting a counter-regulatory increase of hepatic mitochondrial oxidative capacity in response to chronically increased FFA availability in the setting of obesity. Despite these findings of quantitatively upregulated mitochondrial respiration in obesity, measures of mitochondrial bioenergetic efficacy (respiratory control ratio) showed a significant impairment and markers of uncoupling respiration (leak control ratio) showed a progressive increase across obese groups without NAFLD and with NAFLD and NASH. Specifically among individuals with NASH, maximal respiration rates in isolated mitochondria were 31–40% lower compared with those among individuals with obesity with and without steatosis. Importantly, this compromise of mitochondrial respiratory function was observed in conjunction with hepatic IR, increased hepatocellular oxidative damage, augmented hepatic inflammation, and decreased ETC complex expression, despite an increased mitochondrial mass [30]. These findings are in line with previous observations of significant impairments of ETC complex activity in NASH, irrespective of unchanged mitochondrial content as assessed by CSA [135]. Similar trends towards a defective hepatic mitochondrial oxidative metabolism in advanced NAFLD, suggested by reduced oxygen consumption rates in the higher extremes of liver fat content, were also shown in the previously mentioned study by Fletcher et al. [121].
The unaltered or even increased mitochondrial mass observed in NASH does not necessarily guarantee a healthy mitochondrial turnover; in fact, the aforementioned study in patients with very severe obesity and NAFLD provided clear evidence of impaired mitochondrial biogenesis in NASH (as shown by reduced expression of TFAM, PGC-1a, NRF-1), indicating that mitochondrial mass in NASH is preserved through a defective mitophagy of dysfunctional and oxidatively damaged mitochondria [30]. Accordingly, hepatic mitochondria from patients with NASH exhibit structural abnormalities of functionally important structures such as loss of inner membrane folds, which are not observed in normal controls or individuals with simple steatosis [136], again implying defective mitochondrial quality control mechanisms in advanced NAFLD.
Another important component of mitochondrial adaptation in NAFLD is the diversion of a proportion of excess energy intermediates in futile cycles of uncoupled respiration. In preclinical studies, an upregulation of mitochondrial uncoupled respiration through uncoupling protein 2 (UCP-2) has been demonstrated in hepatocytes of rodent models of obesity [137], while the in vitro exposure of rat hepatocytes to lipid emulsions leads to increased UCP-2 expression, likely through a mechanism mediated by excessive ROS production [138]. In human NAFLD, this phenomenon peaks in the setting of established NASH [30] and is also mediated by an increased expression of UCP-2, which coincides with evidence of severe oxidative stress and damage [139]. This fuels the hypothesis that increased uncoupled respiration in advanced NAFLD serves as a “relief valve” against the energetic overstrain of ETC and the subsequent redox disequilibrium. However, this occurs by definition at the expense of reduced bioenergetic efficiency, which may partly account for the reduced hepatic energy-generating capacity in NASH [123], rendering hepatocytes more vulnerable to acute energy-demanding challenges, such as ischemic injury [139].
Summary and Critical Appraisal of the Evidence
Overall, there is a concrete body of evidence to support the presence of impaired mitochondrial adaptation as a principal component of systemic metabolic inflexibility in conditions related to the spectrum of “diabesity.” The question that still remains under debate is the direction of causality between the observed quantitative and qualitative changes delineating the concept of mitochondrial maladaptation and IR. This question harbors important implications for a potential future application of novel obesity and T2DM therapeutics, aiming to restore the abolished features of mitochondrial plasticity.
Essentially, the design of the majority of available studies does not allow for safe conclusions to be drawn regarding the timely appearance of the respective abnormalities, since measures of IR and mitochondrial function have been examined cross-sectionally, even in studies with a prospective design [88]; in the latter case, mutual reversibility is not helpful in determining causality. Nevertheless, a confident statement can be made on the parallel escalation of mitochondrial flexibility loss and severity of IR based on the findings of several studies [30, 90, 97, 121]. Perhaps the most prominent evidence of the scalability of these phenomena is provided by studies conducted in individuals with NAFLD, a condition which, by itself, represents a continuum with relatively well-defined and variably measurable stages of severity [30, 121].
The existing evidence suggests a bidirectional, mutually amplifying relationship between IR and altered mitochondrial function [140]; data from animal studies support a crucial role of components of insulin signaling in the maintenance of mitochondrial functional integrity [141,142,143,144]. Among humans, evidence in favor of this notion is provided by observations of mitochondrial functional impairment in skeletal muscle among individuals with congenital insulin resistance due to insulin receptor defects [107], although it is questionable whether these findings are generalizable to other insulin resistance states. Conversely, toxic lipid metabolite accumulation (ceramides, diacylglycerols) and excessive ROS production by a dysfunctional and energetically overloaded ETC in the setting of mitochondrial functional impairment [145, 146] may induce or exacerbate IR. It is also likely that ROS production under these circumstances may further aggravate mitochondrial structural and functional damage [147]. On the other hand, the production of ROS from ETC and other sources may also exhibit a positive effect on insulin signaling and enhance insulin sensitivity [148]. The threshold between a “healthy” and a “deleterious” load of ROS for insulin signaling is difficult to determine.
With issues of causality being further obscured by reactive adaptive mechanisms at a subcellular and inter-tissue interaction level, an integrated presentation of IR and overt T2DM as manifestations of altered mitochondrial plasticity is impossible based on actual data. A linear and simplified approach would include the presence of obesity as a necessary step in a cascade of events eventually leading to metabolic decompensation and overt T2DM, driven by the resulting lipotoxic environment (Fig. 1). As adipocytes reach a crucial threshold of triglyceride storage, a failure of insulin to suppress lipolysis leads to an increased release of FFA into the circulation [149]. Attempts to attribute a central role regarding these phenomena to functional changes of adipocyte mitochondria have yielded mixed results [150, 151]. An initially increased skeletal muscle FFA uptake and utilization are eventually fatigued, with a parallel development of IR at the myocyte level [149]. At this stage, features of metabolic inflexibility and impaired mitochondrial plasticity in skeletal muscle may already be evident and the burden of managing the energetic excess in the form of FFA and glycolytic intermediates (glycerol) is undertaken solely by the liver. As a last resort, under the influence of increased high-energy substrate influx, the hepatic mitochondrial oxidative and ATP-producing capacity is transiently upregulated, albeit at the expense of a decreased bioenergetic efficiency of the ETC and an increased burden of ROS, which may overwhelm cellular antioxidant defense mechanisms and ultimately lead to oxidative stress and hepatocyte inflammation [152]. Furthermore, the increased ATP production may facilitate the unwanted effect of its utilization in aberrantly activated energy-demanding procedures such as de novo lipogenesis [153] or gluconeogenesis [121, 131], despite increased insulin levels. These features contribute to worsening hepatocellular fat accumulation and pave the way for the transition from simple steatosis to NASH. The increasing burden of ROS production mobilizes a counter-regulatory increase of uncoupled respiration at the cost of decreased capability of hepatocytes to respond to acute fluctuations of energy demands [139]. The FFA overload may eventually overcome the upregulated hepatic mitochondrial oxidative capacity, facilitated by the fatigue of the oxidative machinery in the later stages of NAFLD [30]. Consequently, the available FFA may be redirected to triglyceride biosynthesis [154] and furthermore to alternative metabolic pathways, being used as substrates for potentially toxic metabolites such as ceramides and diacylglycerols [155], which may further functionally compromise the ETC [156] and promote hepatic IR [157,158,159,160].

An overview of the major aspects of mitochondrial function and lipid metabolism in distinct stages across the pathophysiological spectrum of insulin resistance (progression from obesity and insulin resistance to clinically overt hyperglycemia). An inter-tissue crosstalk among different target tissues for insulin (adipose tissue, skeletal muscle, liver, and pancreatic β-cells) should be also considered. As explicitly stated in the manuscript, the relationship between mitochondrial function alterations and insulin resistance is mutual and bidirectional, and its causality remains poorly understood based on the available evidence. Abbreviations: ATP adenosine triphosphate; DAG diacylglycerols; FFA free fatty acids; IMCL intramyocellular lipids; NAFL non-alcoholic fatty liver; NASH non-alcoholic steatohepatitis; Pi inorganic phosphate, ROS reactive oxygen species; T2DM type 2 diabetes mellitus; TCA tricarboxylic acid cycle; TG triglycerides
It may be further hypothesized that the turning points for the metabolic decompensation that mark the transition from a systemic dysmetabolic state to overt T2DM are the progression of NAFLD and the saturation of its inherent mitochondrial adaptation beyond a critical threshold. Evidence in favor of this hypothesis includes the increasing whole-body IR [30] and hepatic glucose production [121]. Most interestingly, T2DM may be characterized by marked structural and functional mitochondrial changes at the level of pancreatic beta-cell [161, 162], suggesting a potential role of mitochondrial maladaptation of the beta-cell in the development of late stages of T2DM.
Clearly, such a linear course of parallel development is not to be assumed, especially in the setting of whole-body metabolism where the presumable bidirectional relationship of progressively worsening dysmetabolism and mitochondrial impairment and superimposed inter-tissue crosstalk would enable the emergence of vicious circles at various levels. A putative common link behind most theoretical and experimental approaches appears to be the presence of increased FFA availability in the sense of the establishment of a lipotoxic milieu (Fig. 1). This notion could be exploited for the design of future studies with a mechanistic component to investigate the cause–consequence relationship between altered mitochondrial function and IR; a theoretical example could implicate the acute establishment of a lipotoxic environment among individuals without apparent insulin resistance, through the intravenous infusion of lipid emulsions or orally supplemented lipid preparations, aiming to approximate the FFA concentrations in plasma commonly observed in obesity and insulin resistance states. The serial assessment of components of mitochondrial functional status (using in vivo, ex vivo, and in vitro techniques as described above) and insulin resistance at cellular level in repeated biopsy specimens (serine/threonine phosphorylation of insulin receptor substrate proteins [163], PKC isoform activity [164], etc.) complemented by whole-body insulin resistance measured through an euglycemic–hyperinsulinemic clamp could serve to identify the sequential appearance of respective disorders. Furthermore, under such controlled experimental conditions, the potential contribution of factors that mediate the crosstalk between the key implicated tissues (adipokines, myokines among other candidate hormonal signals) could also be investigated.
Conclusion
There are still many unresolved questions regarding the relationship between the gradual loss of mitochondrial adaptability and the progression of IR towards overt T2DM. Important issues pertain to the causal nature of the observed correlations, the direction of the causality, the timely appearance and reversibility of the described disturbances, and the generalizability of the findings to the mitochondrial content of every affected crucial tissue or organ. The spectrum of NAFLD is of particular interest, since functional and pathological changes in the course of its development closely mirror the progression of dysmetabolism, if not constituting a dynamic component of the latter.
The aforementioned aspects present a challenging field for future mechanistic studies to focus on, by utilizing combinations of available methods of mitochondrial function assessment. Following a detailed and thorough characterization of the role of mitochondria in the pathogenesis and progression of IR, the effect of existing and emerging therapies on the implicated mitochondrial mechanisms would be next to evaluate. Nonetheless, a future with pharmacological interventions tailored to modulate functional and/or structural mitochondrial targets is still a long way ahead.
References
Malik VS, Willet WC, Hu FB. Nearly a decade on - trends, risk factors and policy implications in global obesity. Nat Rev Endocrinol. 2020;16:615–6. https://doi.org/10.1038/s41574-020-00411-y.
Guarisco G, Leonetti F. Covid-19 and diabesity: when a pandemia cross another pandemia. Eating and Weight Disorders: EWD. 2020:1–4. https://doi.org/10.1007/s40519-020-00958-9.
Tsilingiris D, Liatis S, Dalamaga M, Kokkinos A. The fight against obesity escalates: new drugs on the horizon and metabolic implications. Curr Obes Rep. 2020;9:136–49. https://doi.org/10.1007/s13679-020-00378-x.
Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, Shaw JE, Bright D, Williams R, IDF Diabetes Atlas Committee Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract 2019;157:107843. https://doi.org/10.1016/j.diabres.2019.107843
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (Baltimore, Md). 2016;64:73–84. https://doi.org/10.1002/hep.28431.
Hill JO, Wyatt HR, Peters JC. Energy balance and obesity. Circulation. 2012;126:126–32. https://doi.org/10.1161/circulationaha.111.087213.
Corpeleijn E, Saris WH, Blaak EE. Metabolic flexibility in the development of insulin resistance and type 2 diabetes: effects of lifestyle. Obesity Reviews: an official journal of the International Association for the Study of Obesity. 2009;10:178–93. https://doi.org/10.1111/j.1467-789X.2008.00544.x.
Gastaldelli A. Insulin resistance and reduced metabolic flexibility: cause or consequence of NAFLD? Clinical Science (London, England: 1979). 2017;131:2701–4. https://doi.org/10.1042/cs20170987.
Wikström M, Sharma V, Kaila VR, Hosler JP, Hummer G. New perspectives on proton pumping in cellular respiration. Chem Rev. 2015;115:2196–221. https://doi.org/10.1021/cr500448t.
Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci. 2016;41:261–73. https://doi.org/10.1016/j.tibs.2016.01.001.
Osellame LD, Blacker TS, Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab. 2012;26:711–23. https://doi.org/10.1016/j.beem.2012.05.003.
Newman JC, Verdin E. β-Hydroxybutyrate: a signaling metabolite. Annu Rev Nutr. 2017;37:51–76. https://doi.org/10.1146/annurev-nutr-071816-064916.
Tait SW, Green DR. Mitochondria and cell signalling. J Cell Sci. 2012;125:807–15. https://doi.org/10.1242/jcs.099234.
Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet. 2009;43:95–118. https://doi.org/10.1146/annurev-genet-102108-134850.
Smith RL, Soeters MR, Wüst RCI, Houtkooper RH. Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr Rev. 2018;39:489–517. https://doi.org/10.1210/er.2017-00211.
Galgani JE, Moro C, Ravussin E. Metabolic flexibility and insulin resistance. Am J Phys Endocrinol Metab. 2008;295:E1009–17. https://doi.org/10.1152/ajpendo.90558.2008.
Muoio DM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159:1253–62. https://doi.org/10.1016/j.cell.2014.11.034.
Burke SJ, Batdorf HM, Huang TY, Jackson JW, Jones KA, Martin TM, et al. One week of continuous corticosterone exposure impairs hepatic metabolic flexibility, promotes islet β-cell proliferation, and reduces physical activity in male C57BL/6 J mice. J Steroid Biochem Mol Biol. 2019;195:105468. https://doi.org/10.1016/j.jsbmb.2019.105468.
Lee S, Rivera-Vega M, Alsayed HM, Boesch C, Libman I. Metabolic inflexibility and insulin resistance in obese adolescents with non-alcoholic fatty liver disease. Pediatr Diabetes. 2015;16:211–8. https://doi.org/10.1111/pedi.12141.
Thyfault JP, Rector RS, Noland RC. Metabolic inflexibility in skeletal muscle: a prelude to the cardiometabolic syndrome? Journal of the Cardiometabolic Syndrome. 2006;1:184–9. https://doi.org/10.1111/j.1559-4564.2006.05629.x.
Ukropcova B, McNeil M, Sereda O, de Jonge L, Xie H, Bray GA, et al. Dynamic changes in fat oxidation in human primary myocytes mirror metabolic characteristics of the donor. J Clin Invest. 2005;115:1934–41. https://doi.org/10.1172/jci24332.
Jørgensen W, Rud KA, Mortensen OH, Frandsen L, Grunnet N, Quistorff B. Your mitochondria are what you eat: a high-fat or a high-sucrose diet eliminates metabolic flexibility in isolated mitochondria from rat skeletal muscle. Physiological Reports. 2017;5. https://doi.org/10.14814/phy2.13207.
Medeiros DM. Assessing mitochondria biogenesis. Methods (San Diego, Calif). 2008;46:288–94. https://doi.org/10.1016/j.ymeth.2008.09.026.
D'Erchia AM, Atlante A, Gadaleta G, Pavesi G, Chiara M, De Virgilio C, et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion. 2015;20:13–21. https://doi.org/10.1016/j.mito.2014.10.005.
Chiappini F, Barrier A, Saffroy R, Domart MC, Dagues N, Azoulay D, et al. Exploration of global gene expression in human liver steatosis by high-density oligonucleotide microarray. Laboratory Investigation; a journal of technical methods and pathology. 2006;86:154–65. https://doi.org/10.1038/labinvest.3700374.
Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590:3349–60. https://doi.org/10.1113/jphysiol.2012.230185.
Vigelsø A, Andersen NB, Dela F. The relationship between skeletal muscle mitochondrial citrate synthase activity and whole body oxygen uptake adaptations in response to exercise training. International Journal of Physiology, Pathophysiology and Pharmacology. 2014;6:84–101.
Christe M, Hirzel E, Lindinger A, Kern B, von Flüe M, Peterli R, et al. Obesity affects mitochondrial citrate synthase in human omental adipose tissue. ISRN Obesity. 2013;2013:826027–8. https://doi.org/10.1155/2013/826027.
Laye MJ, Rector RS, Warner SO, Naples SP, Perretta AL, Uptergrove GM, et al. Changes in visceral adipose tissue mitochondrial content with type 2 diabetes and daily voluntary wheel running in OLETF rats. J Physiol. 2009;587:3729–39. https://doi.org/10.1113/jphysiol.2009.172601.
Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–46. https://doi.org/10.1016/j.cmet.2015.04.004.
Lee K, Haddad A, Osme A. Hepatic mitochondrial defects in a nonalcoholic fatty liver disease mouse model are associated with increased degradation of oxidative phosphorylation subunits. 2018;17:2371–86. https://doi.org/10.1074/mcp.RA118.000961.
Koekemoer TC, Oelofsen W. Properties of porcine white adipose tissue and liver mitochondrial subpopulations. Int J Biochem Cell Biol. 2001;33:889–901. https://doi.org/10.1016/s1357-2725(01)00064-4.
Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. https://doi.org/10.1042/bse0470069.
Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459–66. https://doi.org/10.1016/j.tem.2012.06.006.
Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7:39–44. https://doi.org/10.1016/j.mito.2006.11.017.
Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–70. https://doi.org/10.1016/j.cmet.2005.05.004.
Gao AW, Cantó C, Houtkooper RH. Mitochondrial response to nutrient availability and its role in metabolic disease. EMBO Mol Med. 2014;6:580–9. https://doi.org/10.1002/emmm.201303782.
Tang BL. Sirt1 and the mitochondria. Molecules and Cells. 2016;39:87–95. https://doi.org/10.14348/molcells.2016.2318.
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8. https://doi.org/10.1038/nature03354.
Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. The American Journal of Clinical Nutrition. 2011;93:884 s-90. https://doi.org/10.3945/ajcn.110.001917.
Groennebaek T, Vissing K. Impact of resistance training on skeletal muscle mitochondrial biogenesis, content, and function. Front Physiol. 2017;8:713. https://doi.org/10.3389/fphys.2017.00713.
Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20:31–42. https://doi.org/10.1038/cdd.2012.81.
Su Z, Nie Y, Huang X, Zhu Y, Feng B, Tang L, et al. Mitophagy in hepatic insulin resistance: therapeutic potential and concerns. Front Pharmacol. 2019;10:1193. https://doi.org/10.3389/fphar.2019.01193.
Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–50. https://doi.org/10.1152/physrev.00026.2013.
Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15:634–46. https://doi.org/10.1038/nrm3877.
Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–9. https://doi.org/10.1016/j.cell.2010.02.026.
Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. https://doi.org/10.1146/annurev.cellbio.22.010305.104638.
Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–98. https://doi.org/10.1038/ncb2220.
Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46. https://doi.org/10.1038/sj.emboj.7601963.
Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si Y, Las G, et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 2014;33:418–36. https://doi.org/10.1002/embj.201385014.
Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–7. https://doi.org/10.1074/jbc.M212754200.
Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–50. https://doi.org/10.2337/diabetes.51.10.2944.
Schmid AI, Szendroedi J, Chmelik M, Krssák M, Moser E, Roden M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care. 2011;34:448–53. https://doi.org/10.2337/dc10-1076.
Busiello RA, Savarese S, Lombardi A. Mitochondrial uncoupling proteins and energy metabolism. Front Physiol. 2015;6:36. https://doi.org/10.3389/fphys.2015.00036.
Klaus S, Keipert S, Rossmeisl M, Kopecky J. Augmenting energy expenditure by mitochondrial uncoupling: a role of AMP-activated protein kinase. Genes Nutr. 2012;7:369–86. https://doi.org/10.1007/s12263-011-0260-8.
Keipert S, Voigt A, Klaus S. Dietary effects on body composition, glucose metabolism, and longevity are modulated by skeletal muscle mitochondrial uncoupling in mice. Aging Cell. 2011;10:122–36. https://doi.org/10.1111/j.1474-9726.2010.00648.x.
Adjeitey CN, Mailloux RJ, Dekemp RA, Harper ME. Mitochondrial uncoupling in skeletal muscle by UCP1 augments energy expenditure and glutathione content while mitigating ROS production. Am J Phys Endocrinol Metab. 2013;305:E405–15. https://doi.org/10.1152/ajpendo.00057.2013.
Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–73. https://doi.org/10.1111/j.1432-1033.1997.00259.x.
Ye X, Li M, Hou T, Gao T, Zhu WG, Yang Y. Sirtuins in glucose and lipid metabolism. Oncotarget. 2017;8:1845–59. https://doi.org/10.18632/oncotarget.12157.
Martinez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102. https://doi.org/10.1038/s41467-019-13668-3.
Brehm A, Krssak M, Schmid AI, Nowotny P, Waldhäusl W, Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes. 2006;55:136–40.
Chen RH, Maher VM, Brouwer J, van de Putte P, McCormick JJ. Preferential repair and strand-specific repair of benzo[a]pyrene diol epoxide adducts in the HPRT gene of diploid human fibroblasts. Proc Natl Acad Sci U S A. 1992;89:5413–7. https://doi.org/10.1073/pnas.89.12.5413.
Valkovič L, Chmelík M, Krššák M. In-vivo 31P-MRS of skeletal muscle and liver: a way for non-invasive assessment of their metabolism. Anal Biochem. 2017;529:193–215. https://doi.org/10.1016/j.ab.2017.01.018.
Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science (New York, NY). 2003;300:1140–2. https://doi.org/10.1126/science.1082889.
Oberhaensli RD, Galloway GJ, Taylor DJ, Bore PJ, Radda GK. Assessment of human liver metabolism by phosphorus-31 magnetic resonance spectroscopy. Br J Radiol. 1986;59:695–9. https://doi.org/10.1259/0007-1285-59-703-695.
Nair S, V PC, Arnold C, Diehl AM. Hepatic ATP reserve and efficiency of replenishing: comparison between obese and nonobese normal individuals. Am J Gastroenterol. 2003;98:466–70. https://doi.org/10.1111/j.1572-0241.2003.07221.x.
Bawden SJ, Stephenson MC, Ciampi E, Hunter K, Marciani L, Macdonald IA, et al. Investigating the effects of an oral fructose challenge on hepatic ATP reserves in healthy volunteers: A (31)P MRS study. Clinical Nutrition (Edinburgh, Scotland). 2016;35:645–9. https://doi.org/10.1016/j.clnu.2015.04.001.
Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. https://doi.org/10.1371/journal.pmed.0020233.
Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol. 2009;41:1837–45. https://doi.org/10.1016/j.biocel.2009.03.013.
Ojuka E, Andrew B, Bezuidenhout N, George S, Maarman G, Madlala HP, et al. Measurement of β-oxidation capacity of biological samples by respirometry: a review of principles and substrates. Am J Phys Endocrinol Metab. 2016;310:E715–23. https://doi.org/10.1152/ajpendo.00475.2015.
El-Hafidi M, Franco M, Ramírez AR, Sosa JS, Flores JAP, Acosta OL, et al. Glycine Increases insulin sensitivity and glutathione biosynthesis and protects against oxidative stress in a model of sucrose-induced insulin resistance. Oxidative Med Cell Longev. 2018;2018:2101562–12. https://doi.org/10.1155/2018/2101562.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. https://doi.org/10.1042/bj20081386.
Mihalik SJ, Michaliszyn SF, de las Heras J, Bacha F, Lee S, Chace DH, et al. Metabolomic profiling of fatty acid and amino acid metabolism in youth with obesity and type 2 diabetes: evidence for enhanced mitochondrial oxidation. Diabetes Care. 2012;35:605–11. https://doi.org/10.2337/dc11-1577.
Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol. 1999;277:E1130–41. https://doi.org/10.1152/ajpendo.1999.277.6.E1130.
Thyfault JP, Kraus RM, Hickner RC, Howell AW, Wolfe RR, Dohm GL. Impaired plasma fatty acid oxidation in extremely obese women. Am J Phys Endocrinol Metab. 2004;287:E1076–81. https://doi.org/10.1152/ajpendo.00177.2004.
Larson-Meyer DE, Newcomer BR, Hunter GR, McLean JE, Hetherington HP, Weinsier RL. Effect of weight reduction, obesity predisposition, and aerobic fitness on skeletal muscle mitochondrial function. Am J Phys Endocrinol Metab. 2000;278:E153–61. https://doi.org/10.1152/ajpendo.2000.278.1.E153.
Newcomer BR, Larson-Meyer DE, Hunter GR, Weinsier RL. Skeletal muscle metabolism in overweight and post-overweight women: an isometric exercise study using (31)P magnetic resonance spectroscopy. International Journal of Obesity and Related Metabolic Disorders: journal of the International Association for the Study of Obesity. 2001;25:1309–15. https://doi.org/10.1038/sj.ijo.0801673.
Wells GD, Banks L, Caterini JE, Thompson S, Noseworthy MD, Rayner T, et al. The association among skeletal muscle phosphocreatine recovery, adiposity, and insulin resistance in children. Pediatr Obes. 2017;12:163–70. https://doi.org/10.1111/ijpo.12123.
Fleischman A, Kron M, Systrom DM, Hrovat M, Grinspoon SK. Mitochondrial function and insulin resistance in overweight and normal-weight children. J Clin Endocrinol Metab. 2009;94:4923–30. https://doi.org/10.1210/jc.2009-1590.
Cree-Green M, Wiromrat P, Stuppy JJ, Thurston J, Bergman BC, Baumgartner AD, et al. Muscle insulin resistance in youth with obesity and normoglycemia is associated with altered fat metabolism. Obesity (Silver Spring, Md). 2019;27:2046–54. https://doi.org/10.1002/oby.22658.
Slattery MJ, Bredella MA, Thakur H, Torriani M, Misra M. Insulin resistance and impaired mitochondrial function in obese adolescent girls. Metab Syndr Relat Disord. 2014;12:56–61. https://doi.org/10.1089/met.2013.0100.
Cree-Green M, Gupta A, Coe GV, Baumgartner AD, Pyle L, Reusch JE, et al. Insulin resistance in type 2 diabetes youth relates to serum free fatty acids and muscle mitochondrial dysfunction. J Diabetes Complicat. 2017;31:141–8. https://doi.org/10.1016/j.jdiacomp.2016.10.014.
Fritsch M, Koliaki C, Livingstone R, Phielix E, Bierwagen A, Meisinger M, et al. Time course of postprandial hepatic phosphorus metabolites in lean, obese, and type 2 diabetes patients. Am J Clin Nutr. 2015;102:1051–8. https://doi.org/10.3945/ajcn.115.107599.
Iozzo P, Bucci M, Roivainen A, Någren K, Järvisalo MJ, Kiss J, et al. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology. 2010;139:846-56, 56.e1-6. https://doi.org/10.1053/j.gastro.2010.05.039.
Wells GD, Noseworthy MD, Hamilton J, Tarnopolski M, Tein I. Skeletal muscle metabolic dysfunction in obesity and metabolic syndrome. The Canadian Journal of Neurological Sciences Le journal canadien des sciences neurologiques. 2008;35:31–40. https://doi.org/10.1017/s0317167100007538.
D'Adamo E, Caprio S. Type 2 diabetes in youth: epidemiology and pathophysiology. Diabetes Care. 2011;34(Suppl 2):S161–5. https://doi.org/10.2337/dc11-s212.
Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol. 2017;23:8263–76. https://doi.org/10.3748/wjg.v23.i47.8263.
Meex RC, Schrauwen-Hinderling VB, Moonen-Kornips E, Schaart G, Mensink M, Phielix E, et al. Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes. 2010;59:572–9. https://doi.org/10.2337/db09-1322.
Ripley EM, Clarke GD, Hamidi V, Martinez RA, Settles FD, Solis C, et al. Reduced skeletal muscle phosphocreatine concentration in type 2 diabetic patients: a quantitative image-based phosphorus-31 MR spectroscopy study. Am J Phys Endocrinol Metab. 2018;315:E229–e39. https://doi.org/10.1152/ajpendo.00426.2017.
Szendroedi J, Schmid AI, Chmelik M, Toth C, Brehm A, Krssak M, et al. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med. 2007;4:e154. https://doi.org/10.1371/journal.pmed.0040154.
van de Weijer T, Sparks LM, Phielix E, Meex RC, van Herpen NA, Hesselink MK, et al. Relationships between mitochondrial function and metabolic flexibility in type 2 diabetes mellitus. PLoS One. 2013;8:e51648. https://doi.org/10.1371/journal.pone.0051648.
Chomentowski P, Coen PM, Radiková Z, Goodpaster BH, Toledo FG. Skeletal muscle mitochondria in insulin resistance: differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J Clin Endocrinol Metab. 2011;96:494–503. https://doi.org/10.1210/jc.2010-0822.
Phielix E, Schrauwen-Hinderling VB, Mensink M, Lenaers E, Meex R, Hoeks J, et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes. 2008;57:2943–9. https://doi.org/10.2337/db08-0391.
De Feyter HM, van den Broek NM, Praet SF, Nicolay K, van Loon LJ, Prompers JJ. Early or advanced stage type 2 diabetes is not accompanied by in vivo skeletal muscle mitochondrial dysfunction. Eur J Endocrinol. 2008;158:643–53. https://doi.org/10.1530/eje-07-0756.
Trenell MI, Hollingsworth KG, Lim EL, Taylor R. Increased daily walking improves lipid oxidation without changes in mitochondrial function in type 2 diabetes. Diabetes Care. 2008;31:1644–9. https://doi.org/10.2337/dc08-0303.
Larsen S, Ara I, Rabøl R, Andersen JL, Boushel R, Dela F, et al. Are substrate use during exercise and mitochondrial respiratory capacity decreased in arm and leg muscle in type 2 diabetes? Diabetologia. 2009;52:1400–8. https://doi.org/10.1007/s00125-009-1353-4.
Bajpeyi S, Pasarica M, Moro C, Conley K, Jubrias S, Sereda O, et al. Skeletal muscle mitochondrial capacity and insulin resistance in type 2 diabetes. J Clin Endocrinol Metab. 2011;96:1160–8. https://doi.org/10.1210/jc.2010-1621.
Apostolopoulou M, Strassburger K, Herder C, Knebel B, Kotzka J, Szendroedi J, et al. Metabolic flexibility and oxidative capacity independently associate with insulin sensitivity in individuals with newly diagnosed type 2 diabetes. Diabetologia. 2016;59:2203–7. https://doi.org/10.1007/s00125-016-4038-9.
Corpeleijn E, Mensink M, Kooi ME, Roekaerts PM, Saris WH, Blaak EE. Impaired skeletal muscle substrate oxidation in glucose-intolerant men improves after weight loss. Obesity (Silver Spring, Md). 2008;16:1025–32. https://doi.org/10.1038/oby.2008.24.
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–71. https://doi.org/10.1056/NEJMoa031314.
Ukropcova B, Sereda O, de Jonge L, Bogacka I, Nguyen T, Xie H, et al. Family history of diabetes links impaired substrate switching and reduced mitochondrial content in skeletal muscle. Diabetes. 2007;56:720–7. https://doi.org/10.2337/db06-0521.
Brøns C, Jensen CB, Storgaard H, Alibegovic A, Jacobsen S, Nilsson E, et al. Mitochondrial function in skeletal muscle is normal and unrelated to insulin action in young men born with low birth weight. J Clin Endocrinol Metab. 2008;93:3885–92. https://doi.org/10.1210/jc.2008-0630.
Mensink M, Blaak EE, van Baak MA, Wagenmakers AJ, Saris WH. Plasma free Fatty Acid uptake and oxidation are already diminished in subjects at high risk for developing type 2 diabetes. Diabetes. 2001;50:2548–54. https://doi.org/10.2337/diabetes.50.11.2548.
Cree-Green M, Newcomer BR, Brown MS, Baumgartner AD, Bergman B, Drew B, et al. Delayed skeletal muscle mitochondrial ADP recovery in youth with type 1 diabetes relates to muscle insulin resistance. Diabetes. 2015;64:383–92. https://doi.org/10.2337/db14-0765.
Crowther GJ, Milstein JM, Jubrias SA, Kushmerick MJ, Gronka RK, Conley KE. Altered energetic properties in skeletal muscle of men with well-controlled insulin-dependent (type 1) diabetes. Am J Phys Endocrinol Metab. 2003;284:E655–62. https://doi.org/10.1152/ajpendo.00343.2002.
Kacerovsky M, Brehm A, Chmelik M, Schmid AI, Szendroedi J, Kacerovsky-Bielesz G, et al. Impaired insulin stimulation of muscular ATP production in patients with type 1 diabetes. J Intern Med. 2011;269:189–99. https://doi.org/10.1111/j.1365-2796.2010.02298.x.
Sleigh A, Raymond-Barker P, Thackray K, Porter D, Hatunic M, Vottero A, et al. Mitochondrial dysfunction in patients with primary congenital insulin resistance. J Clin Invest. 2011;121:2457–61. https://doi.org/10.1172/jci46405.
Szendroedi J, Chmelik M, Schmid AI, Nowotny P, Brehm A, Krssak M, et al. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology (Baltimore, Md). 2009;50:1079–86. https://doi.org/10.1002/hep.23093.
Abdelmalek MF, Lazo M, Horska A, Bonekamp S, Lipkin EW, Balasubramanyam A, et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology (Baltimore, Md). 2012;56:952–60. https://doi.org/10.1002/hep.25741.
Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98:2133–223. https://doi.org/10.1152/physrev.00063.2017.
Schrauwen-Hinderling VB, Kooi ME, Hesselink MK, Jeneson JA, Backes WH, van Echteld CJ, et al. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia. 2007;50:113–20. https://doi.org/10.1007/s00125-006-0475-1.
Yki-Järvinen H, Koivisto VA. Natural course of insulin resistance in type I diabetes. N Engl J Med. 1986;315:224–30. https://doi.org/10.1056/nejm198607243150404.
Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis. 2004;24:3–20. https://doi.org/10.1055/s-2004-823098.
Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol. 2013;28(Suppl 1):68–76. https://doi.org/10.1111/jgh.12212.
Divella R, Mazzocca A, Daniele A, Sabbà C, Paradiso A. Obesity, nonalcoholic fatty liver disease and adipocytokines network in promotion of cancer. Int J Biol Sci. 2019;15:610–6. https://doi.org/10.7150/ijbs.29599.
Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J Hepatol. 2019;71:793–801. https://doi.org/10.1016/j.jhep.2019.06.021.
Wong VW, Wong GL, Yeung JC, Fung CY, Chan JK, Chang ZH, et al. Long-term clinical outcomes after fatty liver screening in patients undergoing coronary angiogram: a prospective cohort study. Hepatology (Baltimore, Md). 2016;63:754–63. https://doi.org/10.1002/hep.28253.
Targher G, Bertolini L, Padovani R, Rodella S, Tessari R, Zenari L, et al. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care. 2007;30:1212–8. https://doi.org/10.2337/dc06-2247.
Wong VW, Wong GL, Yip GW, Lo AO, Limquiaco J, Chu WC, et al. Coronary artery disease and cardiovascular outcomes in patients with non-alcoholic fatty liver disease. Gut. 2011;60:1721–7. https://doi.org/10.1136/gut.2011.242016.
Samantha M, Elisa M, Db D, Francesco P, Cristina R, Yvelise F. Impaired fat oxidation and reduced resting energy expenditure after a fat load in individuals with liver steatosis. J Nutr Food Sci. 2018;8:1–7. https://doi.org/10.4172/2155-9600.1000665.
Fletcher JA, Deja S, Satapati S, Fu X, Burgess SC, Browning JD. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight. 2019;5. https://doi.org/10.1172/jci.insight.127737.
Croci I, Byrne NM, Choquette S, Hills AP, Chachay VS, Clouston AD, et al. Whole-body substrate metabolism is associated with disease severity in patients with non-alcoholic fatty liver disease. Gut. 2013;62:1625–33. https://doi.org/10.1136/gutjnl-2012-302789.
Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. 1999;282:1659–64. https://doi.org/10.1001/jama.282.17.1659.
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–51. https://doi.org/10.1172/jci23621.
Sears B, Perry M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015;14:121. https://doi.org/10.1186/s12944-015-0123-1.
Reddavide R, Cisternino AM, Inguaggiato R, Rotolo O, Zinzi I, Veronese N, et al. Non-alcoholic fatty liver disease is associated with higher metabolic expenditure in overweight and obese subjects: a case-control study. Nutrients. 2019;11. https://doi.org/10.3390/nu11081830.
Miele L, Grieco A, Armuzzi A, Candelli M, Forgione A, Gasbarrini A, et al. Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am J Gastroenterol. 2003;98:2335–6. https://doi.org/10.1111/j.1572-0241.2003.07725.x.
Kotronen A, Seppälä-Lindroos A, Vehkavaara S, Bergholm R, Frayn KN, Fielding BA, et al. Liver fat and lipid oxidation in humans. Liver International: official journal of the International Association for the Study of the Liver. 2009;29:1439–46. https://doi.org/10.1111/j.1478-3231.2009.02076.x.
Patterson RE, Kalavalapalli S, Williams CM, Nautiyal M, Mathew JT, Martinez J, et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am J Phys Endocrinol Metab. 2016;310:E484–94. https://doi.org/10.1152/ajpendo.00492.2015.
Satapati S, Sunny NE, Kucejova B, Fu X, He TT, Mendez-Lucas A, et al. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res. 2012;53:1080–92. https://doi.org/10.1194/jlr.M023382.
Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–10. https://doi.org/10.1016/j.cmet.2011.11.004.
Petersen KF, Befroy DE, Dufour S, Rothman DL, Shulman GI. Assessment of hepatic mitochondrial oxidation and pyruvate cycling in NAFLD by (13)C magnetic resonance spectroscopy. Cell Metab. 2016;24:167–71. https://doi.org/10.1016/j.cmet.2016.06.005.
Befroy DE, Perry RJ, Jain N, Dufour S, Cline GW, Trimmer JK, et al. Direct assessment of hepatic mitochondrial oxidative and anaplerotic fluxes in humans using dynamic 13C magnetic resonance spectroscopy. Nat Med. 2014;20:98–102. https://doi.org/10.1038/nm.3415.
Hyötyläinen T, Jerby L, Petäjä EM, Mattila I, Jäntti S, Auvinen P, et al. Genome-scale study reveals reduced metabolic adaptability in patients with non-alcoholic fatty liver disease. Nat Commun. 2016;7:8994. https://doi.org/10.1038/ncomms9994.
Pérez-Carreras M, Del Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology (Baltimore, Md). 2003;38:999–1007. https://doi.org/10.1053/jhep.2003.50398.
Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–92. https://doi.org/10.1053/gast.2001.23256.
Chavin KD, Yang S, Lin HZ, Chatham J, Chacko VP, Hoek JB, et al. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J Biol Chem. 1999;274:5692–700. https://doi.org/10.1074/jbc.274.9.5692.
Cortez-Pinto H, Zhi Lin H, Qi Yang S, Odwin Da Costa S, Diehl AM. Lipids up-regulate uncoupling protein 2 expression in rat hepatocytes. Gastroenterology. 1999;116:1184–93. https://doi.org/10.1016/s0016-5085(99)70022-3.
Serviddio G, Bellanti F, Tamborra R, Rollo T, Capitanio N, Romano AD, et al. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut. 2008;57:957–65. https://doi.org/10.1136/gut.2007.147496.
Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab. 2010;21:589–98. https://doi.org/10.1016/j.tem.2010.06.005.
Cheng Z, Guo S, Copps K, Dong X, Kollipara R, Rodgers JT, et al. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009;15:1307–11. https://doi.org/10.1038/nm.2049.
Long YC, Cheng Z, Copps KD, White MF. Insulin receptor substrates Irs1 and Irs2 coordinate skeletal muscle growth and metabolism via the Akt and AMPK pathways. Mol Cell Biol. 2011;31:430–41. https://doi.org/10.1128/mcb.00983-10.
Liu S, Okada T, Assmann A, Soto J, Liew CW, Bugger H, et al. Insulin signaling regulates mitochondrial function in pancreatic beta-cells. PLoS One. 2009;4:e7983. https://doi.org/10.1371/journal.pone.0007983.
Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97.
Sokolowska E, Blachnio-Zabielska A. The role of ceramides in insulin resistance. Front Endocrinol. 2019;10:577. https://doi.org/10.3389/fendo.2019.00577.
Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. https://doi.org/10.1210/er.2001-0039.
Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. https://doi.org/10.1172/jci32601.
Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–72. https://doi.org/10.1016/j.cmet.2009.08.009.
Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–77. https://doi.org/10.1038/nrm2391.
Petrick HL, Foley KP, Zlitni S, Brunetta HS, Paglialunga S, Miotto PM, et al. Adipose tissue inflammation is directly linked to obesity-induced insulin resistance, while gut dysbiosis and mitochondrial dysfunction are not required. Function. 2020;1. https://doi.org/10.1093/function/zqaa013.
Woo CY, Jang JE, Lee SE, Koh EH, Lee KU. Mitochondrial dysfunction in adipocytes as a primary cause of adipose tissue inflammation, vol. 43; 2019. p. 247–56. https://doi.org/10.4093/dmj.2018.0221.
Satapati S, Kucejova B, Duarte JA, Fletcher JA, Reynolds L, Sunny NE, et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest. 2015;125:4447–62. https://doi.org/10.1172/jci82204.
Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–35. https://doi.org/10.1053/j.gastro.2013.11.049.
Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Comprehensive Physiology. 2017;8:1–8. https://doi.org/10.1002/cphy.c170012.
Petersen MC, Shulman GI. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol Sci. 2017;38:649–65. https://doi.org/10.1016/j.tips.2017.04.004.
Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. 1997;272:24154–8. https://doi.org/10.1074/jbc.272.39.24154.
Pagadala M, Kasumov T, McCullough AJ, Zein NN, Kirwan JP. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol Metab. 2012;23:365–71. https://doi.org/10.1016/j.tem.2012.04.005.
Reali F, Morine MJ, Kahramanoğulları O, Raichur S, Schneider HC, Crowther D, et al. Mechanistic interplay between ceramide and insulin resistance. Sci Rep. 2017;7:41231. https://doi.org/10.1038/srep41231.
Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, et al. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 2017;19:1997–2004. https://doi.org/10.1016/j.celrep.2017.05.035.
Chen Z, Yu R, Xiong Y, Du F, Zhu S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017;16:203. https://doi.org/10.1186/s12944-017-0572-9.
Anello M, Lupi R, Spampinato D, Piro S, Masini M, Boggi U, et al. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia. 2005;48:282–9. https://doi.org/10.1007/s00125-004-1627-9.
Haythorne E, Rohm M. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. 2019;10:2474. https://doi.org/10.1038/s41467-019-10189-x.
Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012;55:2565–82. https://doi.org/10.1007/s00125-012-2644-8.
Keildson S, Fadista J, Ladenvall C, Hedman AK, Elgzyri T, Small KS, et al. Expression of phosphofructokinase in skeletal muscle is influenced by genetic variation and associated with insulin sensitivity. Diabetes. 2014;63:1154–65. https://doi.org/10.2337/db13-1301.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of Interest
All authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Metabolism
Rights and permissions
About this article

Cite this article
Tsilingiris, D., Tzeravini, E., Koliaki, C. et al. The Role of Mitochondrial Adaptation and Metabolic Flexibility in the Pathophysiology of Obesity and Insulin Resistance: an Updated Overview. Curr Obes Rep 10, 191–213 (2021). https://doi.org/10.1007/s13679-021-00434-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-021-00434-0