
Abstract
Epigallocatechin-3-gallate (EGCG), an antioxidant present in green tea, could play an important role in the prevention of cancer. However, its bioavailability is low due to its instability in gastrointestinal environment. β-Lactoglobulin (β-Lg), the major protein in whey, is known for its capacity to bind bioactive molecules and could protect them from oxidation. Interaction between β-Lg and EGCG was investigated at pH 7.0 and 4.0 using fluorescence and Fourier transform infrared spectroscopy, and its impact on EGCG antioxidant activity was determined using the ferric-reducing antioxidant power method. EGCG bound to native or heat-denatured β-Lg by hydrogen bonding and possibly hydrophobic interaction at pH 7.0 and 4.0. The affinity of EGCG for heat-denatured β-Lg at pH 7.0 was greater than for native β-Lg and greater than its affinity for the protein in either state at pH 4.0. Complexing with β-Lg decreased the antioxidant activity of EGCG under all conditions investigated. However, the protein provided limited protection of EGCG against degradation over time, the heat-denatured form being slightly more effective at pH 7.0. This study provides insight into the characteristics of β-Lg binding with a flavonoid and its impact on the antioxidant activity of flavonoids in food systems.
茶多酚和β-乳球蛋白的相互作用:分子表征和生物学意义
摘要 表没食子儿茶素没食子酸酯(EGCG)是绿茶中类黄酮类多酚物质。它具有抗氧化活性,可预防和延缓癌症的发生。然而,由于不稳定性,EGCG的生物利用度比较低。β-乳球蛋白(β-LG)是牛奶中的主要乳清蛋白,可结合生物活性分子,并降低它们的氧化降解。本研究采用荧光光谱法和傅里叶变换红外光谱法研究β-LG和EGCG的相互作用,并通过铁离子还原/抗氧化能力(FRAP)法表征了β-LG对EGCG抗氧化活性的影响。当溶液pH在7.0和4.0时,EGCG与天然和热变性的β-Lg反应生成复合物。氢键是两者结合的驱动力。此外,疏水作用也可能发生在茶多酚和蛋白质间。当pH为7.0时,EGCG与热变性蛋白质间的结合力高于天然态蛋白质,也高于它与两种蛋白质在pH4.0时的结合作用。虽然β- Lg-EGCG复合物的形成降低EGCG的抗氧化活性,但是β-LG在某种程度上可以延缓EGCG的降解。β-LG的延缓作用在热变性态和中性pH时是稍更有效的。这些研究结果为β-LG与类黄酮化合物的结合作用以及其对食品体系中类黄酮的抗氧化性的影响提供了理论基础和实验数据。
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
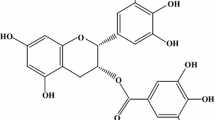
Flavonoids are biologically active polyphenolic compounds widely distributed in plants and occurring in fruits, stems, nuts, seeds, and roots. Plant-based beverages like wine and tea are also major sources of flavonoids. Green tea is arousing growing interest because of its antioxidant activity and hence potential health benefit of preventing cancer and cardiovascular disease. This activity is attributed mainly to catechins, which represent approximately 80% of the total flavonoids of green tea and include epicatechin, epigallocatechin, epicatechin-gallate and epigallocatechin-3-gallate (EGCG). Catechins have strong ROO˙ scavenging activity, being ten times more effective than l-ascorbate and β-carotene (Nakao et al. 1998). EGCG (Fig. 1) is one of the most abundant catechins in green tea and contributes most of its antioxidant activity (Fourneau et al. 1998). Both the catechin and gallate moieties contribute to antioxidant activity by virtue of phenolic hydroxyl groups (Harbowy and Balentine 1997).

Chemical structure of EGCG
The majority of polyphenols must be absorbed in the small intestine in order to exert a therapeutic antioxidant effect. Absorption of EGCG is low because of its instability in the gastrointestinal environment (Zhu et al. 1997). Other factors that affect the absorption and activity of polyphenols include molecular structure, extent of bioconversion in the intestine, and interactions with macromolecules like proteins causing a variation on their antioxidant capacity (Arts et al. 2002). Polyphenols interact with proteins primarily through hydrogen bonding (Rawel et al. 2001) and hydrophobic interactions (Baxter et al. 1997), and this not only may have negative effects on their antioxidant capacity (Rohn et al. 2004) but could also have synergistic effects (Almajano et al. 2007a).
Beta-lactoglobulin (β-Lg), the major whey protein in cow milk, is a small globular protein folded into a calyx (Fig. 2) formed by eight antiparallel β-strands and a three-turn α-helix located at the outer surface of the β-barrel (Kontopidis et al. 2002). It is widespread in our diet and has a significant nutritional role as well as valuable functional properties. Its capacity for binding molecules, such as fatty acids (Wu et al. 1999), retinol, and β-carotene (Dufour and Haertlé 1991), phospholipids (Lefèvre and Subirade 2001), vitamin D (Wang et al. 1997), polyphenolic compounds (Liang et al. 2008; Rawel et al. 2001; Almajano et al. 2007b), and folic acid (Liang and Subirade 2010), is well known. Three sites have been proposed for the binding of ligands to β-Lg: the internal cavity of the β-barrel, the surface hydrophobic pocket in a groove between the α-helix and the β-barrel, and the outer surface near Trp19-Arg124 (Papiz et al. 1986; Sawyer et al. 1998).

Representation of β-lactoglobulin secondary structure. The β strands and joining loops are labelled (Brownlow et al. 1997)
Binding of ligands to β-Lg has been reported to depend on the structure and oligomeric state of the protein (Wang et al. 1998; Riihimäki et al. 2008), which are related to environmental factors such as pH and temperature. At neutral pH and room temperature, β-Lg exists as a dimer, while at pH below 3 or above 8 it dissociates into monomers. Between pH 3.5 and 5.5, β-Lg tends to form octamers (Sawyer and Kontopidis 2000). As the temperature increases from 50 to 80 °C, β-Lg dimers dissociate to monomers, and the latter then undergo conformational modifications (Cayot and Lorient 1998), which may destroy the three specific ligand-binding sites of native β-Lg and expose more hydrophobic residues to the solvent phase. This protein may therefore be considered heat sensitive.
Assemblies based on heat-denatured β-Lg have been described for possible use as protective vehicles of bioactive compounds (Chen et al. 2006). Gels with a pH-dependent microstructure can be formed from β-Lg. Opaque particulate gels are formed near the isoelectric point (pI) of β-Lg (i.e., pH 4–6), while transparent fine-stranded gels are formed near neutral pH (Stading and Hermansson 1990).
Maiti et al. (2006) have studied the molecular interaction of EGCG with human serum albumin, and the binding capacity of EGCG to β-Lg has been reported recently (Riihimäki et al. 2008; Shpigelman et al. 2010). In order to gain better understanding of the mechanism of interaction and hence the potential of β-Lg as a carrier of EGCG, the interactions of this polyphenol with native and heat-denatured β-Lg were investigated at pH 7.0 and 4.0 using fluorescence, Fourier transform infrared (FTIR) spectroscopy, and the ferric reducing antioxidant power (FRAP) assay.
2 Materials and methods
2.1 Materials
β-Lactoglobulin (variant B from bovine milk, purity ≥90%), EGCG (purity 80%), D2O (99.9% deuterium), NaOD, and DCl were purchased from Sigma-Aldrich Chemical Co. (St. Louis, USA) and used without further purification.
β-Lg and EGCG solutions were freshly prepared previously each experiment in 20 mmol.L−1 phosphate buffer at pH 7.0 or in 20 mmol.L−1 acetate buffer at pH 4.0. For FTIR analyses, these buffers were prepared in D2O. The protein concentration and the EGCG concentration were determined spectrophotometrically (HP 8453 UV/vis spectrophotometer, Agilent Technologies Canada Inc., Mississauga, Canada) using ε 280 = 17,600 (mol.L−1)−1 cm−1 for β-Lg (Collini et al. 2000) and ε 270 = 11,920 (mol.L−1)−1 cm−1 for EGCG (Pelillo et al. 2004). Heat-denatured β-Lg (h-β-Lg) solutions were prepared by heating at 85 °C for 30 min in a 50:50 water/ethylene glycol bath and then cooling at room temperature. Solutions containing β-Lg (or h-β-Lg) and EGCG at molar ratios of 1:1, 1:2, and 1:10 were obtained by combining initial solutions.
2.2 FTIR spectroscopic measurements
Solutions of 2.7 mmol.L−1 β-Lg and 27.2 mmol.L−1 EGCG were prepared in D2O. Their pD was adjusted with NaOD or DCl to 7.4 and 4.4 using an Accumet pH meter 915 (Fisher Scientific, Ottawa, Canada), in view of the correction factor pD = pH + 0.4 (Boye et al. 1996). The β-Lg solution was kept for 48 h under a nitrogen atmosphere to achieve complete H/D exchange and h-β-Lg solution was prepared subsequently from this. These solutions were blended to obtain protein/EGCG ratios of 1:1, 1:2, and 1:10 relative to 1.4 mmol.L−1 β-Lg (or h-β-Lg). The samples were held for 2 h at room temperature before recording spectra. All procedures were performed under a nitrogen atmosphere.
FTIR measurements were conducted at 24 °C on a Magna 560 Nicolet spectrometer (Madison, USA) equipped with a mercury–cadmium–telluride detector. The spectrometer was continuously purged with dry air. Samples were placed between two CaF2 windows separated by 23-μm polyethylene terephthalate spacers. Each spectrum was the result of an average of 128 scans and apodized with a Happ–Genzel function. The contribution of D2O to the amide I′ region was eliminated by subtracting D2O spectra such that the baseline was flat between 2,000 and 1,700 cm−1. In order to identify each overlapped component present under the broad amide I’ band, Fourier self-deconvolution was applied to the spectra (Arrondo et al. 1993). The subtraction and deconvolution were obtained using the Omnic software provided with the spectrometer. Band narrowing was achieved with full width at a half-height of 18 cm−1 and a resolution enhancement factor of 2 cm−1.
2.3 Fluorescence spectroscopy
Protein–EGCG samples were prepared by mixing 40 μmol.L−1 β-Lg or h-β-Lg solution with an equal volume of EGCG solution at concentrations of 400, 80, and 40 μmol.L−1 and holding at room temperature for 2 h before analysis. Protein intrinsic fluorescence spectra were recorded on a Cary Eclipse Fluorescence Spectrophotometer (Varian Inc. Mississauga, Canada). Emission spectra were recorded from 300 to 500 nm with an excitation wavelength of 295 nm using a quartz optical cell with a 10 mm path length. The excitation and emission bandwidths were 5 nm. As the excitation and emission wavelength reported for EGCG is 331 and 455 nm, respectively (Nagaoka et al. 2002), the background contributed by EGCG absorbance was recorded and corrected during calculation of fluorescence parameters. For the calculation of the binding constant, we used seven different concentrations of EGCG (Figs. 3 and 4, insets plots) in order to enhance the experimental precision. The binding constant and stoichiometry were calculated using the following equation:

Fluorescence emission spectra (λ ex = 295 nm) of β-lactoglobulin at pH 7.0 in the presence of epigallocatechin-3-gallate (EGCG) at different protein/antioxidant molar ratios: A native protein; B heat-denatured protein; a β-Lactogobulin alone (40 μmol.L−1); b 1:1; c 1:2; d 1:10; e epigallocatechin-3-gallate alone (40 μmol.L−1). Insets Plot of log [(F 0 – F)/F] as a function of log [EGCG] as per Eq. 1

Fluorescence emission spectra (λ ex = 295 nm) of β-lactoglobulin at pH 4.0 in the presence of epigallocatechin-3-gallate (EGCG) at different protein/antioxidant molar ratios: a native protein; b heat-denatured protein; a β-lactoglobulin alone (40 μmol L−1); b 1:1; c 1:2; d 1:10; e epigallocatechin-3-gallate alone (40 μmol L−1). Insets Plot of log [(F 0 – F)/F] as a function of log [EGCG] as per Eq. 1
where F 0 is initial fluorescence intensity, F is the fluorescence intensity in the presence of the quenching agent (EGCG), K s is the static binding constant and n is the binding number(Ghosh et al. 2008).
2.4 FRAP assay
In order to study the effect of adduct formation on its antioxidant capacity, the ferric-reducing antioxidant power (FRAP) of EGCG was determined (Benzie and Strain 1996). This method is based on the reduction of a ferric complex of 2,4,6-tripyridil-s-triazine (Fe3+-TPTZ) to the ferrous form (Fe2+-TPTZ). Sample preparation was the same as for fluorescence measurement except that the final protein concentration was 10 μmol.L−1 and the EGCG concentrations were 10, 20, and 100 μmol.L−1. The FRAP reagent was prepared freshly by mixing 2.5 mL of 10 mmol.L−1 TPTZ solution and 2.5 mL of 20 mmol.L−1 FeCl3·6H2O in 25 mL of 0.3 mol.L−1 acetate buffer at pH 3.6. Sample solution (100 μL) was added to 3 mL of FRAP reagent, and after holding at room temperature for 15 min, absorbance was read at 593 nm on a HP 8453 UV/vis spectrophotometer (Palo Alto, USA). Absorbance measurements were done at 2, 3, 6, 24, 48, 72 h, and 7 days. The optical path length was 1 cm, and FRAP reagent was used as a blank. Aqueous solutions of known Fe2+ concentrations in the range of 100–2,000 μmol.L−1 were used for calibration. Data were expressed as micromoles of Fe2+ per liter (FRAP units).
3 Results and discussion
3.1 The β-Lg/EGCG interaction studied by fluorescence quenching
Intrinsic protein fluorescence is a valuable tool in the investigation of the structure, function, and reactivity of proteins and other biological molecules in solution. β-Lg contains two tryptophan residues (Trp19 and Trp61). Trp19 is in an apolar environment within the cavity of β-Lg, whereas Trp61 is adjacent to strand I located near the C terminus of the protein (Fig. 2). It has been shown that Trp19 is the major fluorophore in native β-Lg (Cho et al. 1994) and contributes about 80% of total fluorescence of the protein because Trp61 is probably quenched by the proximity of the Cys66–Cys160 disulfide bond (Chen and Barkley 1998).
Figure 3 shows fluorescence spectra of β-Lg (Fig. 3A) and h-β-Lg (Fig. 3B) in the presence of EGCG at different concentrations at pH 7.0. β-Lg produces a strong fluorescence emission with a λ max at 334 nm (Fig. 3A). Addition of EGCG shifts the λ max to longer wavelengths of 336, 338, and 345 nm, respectively at β-Lg/EGCG ratios of 1:1, 1:2, and 1:10, indicating that interaction with EGCG makes the environment of the β-Lg Trp residues more hydrophilic. Furthermore, the β-Lg fluorescence intensity gradually decreased to 10.2% that of the pure protein as the EGCG concentration increased to ten times the protein concentration. In comparison with β-Lg, h-β-Lg has a λ max of 340 nm with a red shift of 6 nm (Fig. 3B) due to the partial loss of the hydrophobicity of the Trp19 environment and stronger intensity because of the decreased quenching of Trp61 (Palazolo et al. 2000). Addition of EGCG to h-β-Lg induced changes in λ max somewhat analogous to those obtained with β-Lg but at longer wavelengths \( \left( {{\lambda_{{\max }}} = 352\;{\text{nm}}\;{\text{at}}\;{\text{a}}\;{\text{protein/antioxidant}}\;{\text{ratio}}\;{\text{of}}\;{1:10}} \right) \). This indicates that interaction with EGCG moves the Trp residues of h-β-Lg to a completely hydrophilic environment. EGCG also decreased the fluorescence intensity of h-β-Lg slightly more than it did with β-Lg, down to 8.7% that of the pure protein when the h-β-Lg/EGCG ratio was 1:10. The concentration dependence of the fluorescence intensity was analyzed using the linear plot of log [(F 0 – F)/F] as a function of log [EGCG] shown in the insets of Fig. 3A, B (Eq. 1). The number n of bound ligands per protein molecule is 0.94 ± 0.01 and 1.05 ± 0.02, respectively, for β-Lg and h-β-Lg, based on the slope of the straight line. The binding constant K s is 1.25 (±0.01) × 104 (mol.L−1)−1 for β-Lg and 3.70 (±0.24) × 104 (mol.L−1)−1 for h-β-Lg, as calculated from the intercept. The greater affinity of EGCG for heat-denatured protein is consistent with a recent study by Shpigelman et al. (2010) in which the pH was around neutral.
Figure 4 shows fluorescence spectra of β-Lg (Fig. 4A) and h-β-Lg (Fig. 4B) in the presence of various concentrations of EGCG at pH 4.0. The λ max for β-Lg was 336 nm and the fluorescence intensity about 72% of that obtained at pH 7.0. The lower intensity may be due to increased quenching caused by the formation of octamers near the pI of β-Lg (Sawyer et al. 1998). Adding EGCG did not change λ max, but fluorescence intensity dropped to 9.0% (at the 1:10 ratio) of that of the pure protein, which is a slightly greater decrease than at pH 7.0. In the case of h-β-Lg (Fig. 4B), the λ max was 338 nm and the intensity about 66% of that measured for native β-Lg at this pH, suggesting increased quenching due to greater aggregation as a result of denaturing. In the presence of the antioxidant at a molar ratio of 10, h-β-Lg fluorescence dropped to 10.8% that of the pure protein, without a significant shift in λ max. The change in fluorescence intensity was analyzed using the linear plot of log [(F 0 – F)/F] as a function of log [EGCG] shown in the insets of Fig. 4A, B. The number n of bound ligands per protein molecule is 0.94 ± 0.01 and 0.86 ± 0.01, respectively, for β-Lg and h-β-Lg. The binding constant K s is 1.05 (±0.06) × 104 (mol.L−1)−1 and 4.10 (±0.05) × 103 (mol.L−1)−1, respectively, for β-Lg and h-β-Lg, indicating that EGCG has a greater affinity for β-Lg than for h-β-Lg at pH 4.0. This might be due to increased aggregation of protein monomers and hence more shielding of functional groups. For the same reason, the affinity of EGCG for the proteins is lower at pH 4.0 than at pH 7.0. These results show that even though it decreased at acidic pH, the interaction of EGCG with both β-Lg and h-β-Lg can still be observed and characterized.
3.2 FTIR analysis of the influence of EGCG binding on β-Lg and h-β-Lg structure
Coupling between transition dipoles and hydrogen bonding play a major role in changes in β-Lg amide bond conformation as expressed in FTIR spectral bands. The 1,700–1,600 cm−1 spectral region corresponds mainly to the amide I′ C = O stretching mode and is related to protein secondary structure. Figure 5 presents the deconvoluted spectra of native β-Lg (Fig. 5A) and heat-denatured h-β-Lg (Fig. 5B) in the absence and presence of EGCG at pD 7.4. The deconvoluted spectrum of β-Lg (Fig. 5A) presents individual corresponding to particular secondary structures that are well documented (Lefèvre and Subirade 2000). The bands at 1,692 and 1,678 cm−1 are both related to β-sheets and turns. The component located at 1,660 cm−1 is related to turns, while that at 1,648 cm−1 is attributed to random coils and α-helix. The components at 1,634 and 1,623 cm−1 are attributed respectively to β-sheets and strongly bonded β-strands. Their simultaneous presence characterizes β-Lg in the dimer form (Lefèvre and Subirade 1999). The presence of EGCG at concentrations up to twice the protein concentration appears to have no significant effect on the secondary structure of β-Lg. Nevertheless, increasing the EGCG concentration to ten times that of β-Lg caused the components located at 1,678, 1,648, and 1,634 cm−1 to shift respectively to 1,677, 1,645, and 1,632 cm−1 with a decrease in their intensities, while the 1,692, 1,660, and 1,623 cm−1 components are nearly distinguishable. These results indicate a change in protein structure, the disappearance of the component at 1,623 cm−1, suggesting that EGCG at this concentration disrupted the intermolecular β-sheet hydrogen bonds of the protein dimers. Rawel et al. (2001) mentioned that the phenolic hydroxyl group is an excellent hydrogen bond donor and forms strong hydrogen bonds with the amide carbonyl group of the peptide backbone. EGCG has eight phenolic hydroxyl groups capable of forming hydrogen bonds with β-Lg by competition with intermolecular β-sheet hydrogen bonds. It thus appears likely that interaction with EGCG induced a change from the dimeric to the monomeric structure of β-Lg.

Amide I′ region of the de-convoluted Fourier transform infrared spectra of β-lactoglobulin in the presence of epigallocatechin-3-gallate at different protein/antioxidant molar ratios: A native protein and B heat-denatured protein; a β-Lactoglobulin alone; b 1:1; c 1:2; d 1:10. All samples were at pD 7.4 in D2O. Spectra were recorded at 24 °C
The deconvoluted spectrum of heat-denatured β-Lg (Fig. 5B) differs substantially from that of the native protein. Denaturing resulted in broadening of the spectral feature wavenumber range and the appearance of new components at 1,615 and 1,682 cm−1. These are common to numerous proteins and attributed to intermolecular hydrogen-bonded anti-parallel β-sheet structures resulting from aggregation (Lefèvre and Subirade 1999). The presence of EGCG at ten times the molar concentration of h-β-Lg induced a shift of the aggregation component located at 1,615 to 1,613 cm−1, indicating a strengthening of intermolecular hydrogen bonds in β-sheets (Lefèvre and Subirade 2000). This differs from the interaction of β-Lg with EGCG, which weakened the β-sheet intermolecular hydrogen bonds. Two possible explanations for this difference are different EGCG binding sites on h-β-Lg and β-Lg and the h-β-Lg/EGCG interaction being based on forces other than hydrogen bonds such as hydrophobic interactions.
Figure 6 shows the deconvoluted spectra of native β-Lg (Fig. 6A) and heat-denatured h-β-Lg (Fig. 6B) in the absence and presence of EGCG at pD 4.4. The β-Lg spectrum (Fig. 6A) is similar to that obtained at pD 7.4, but the six components mentioned above show red shifts of several nanometers, and a new and weak component appeared at 1,614 cm−1. This component persisted until the β-Lg/EGCG ratio reached 1:2. All spectral components decreased in intensity at the β-Lg/EGCG ratio of 1:10. The fading of the 1,626 and 1,636 cm−1 components characteristic of protein dimers and the 1,683 and 1,614 cm−1 components characteristic of protein aggregation suggests that EGCG may compete with intermolecular β-sheet hydrogen bonds at pD 4.4 as it does at pD 7.4 and that EGCG at this concentration affects the oligomeric form of the protein. In the case of h-β-Lg (Fig. 6B), the components located at 1,632, 1,647, and 1,658 cm−1 are still present, suggesting the presence of β-sheets and unordered structures or α-helix segments in the polypeptide chains at pD 4.4, since β-Lg is known to be more resistant to denaturing at acidic pH (Cayot and Lorient 1998). However, the broadening and decreased definition of the components and the disappearance of the 1,694 cm−1 component indicate the occurrence of partial protein denaturing at pD 4.4. The spectrum displays similar characteristics at an EGCG/h-β-Lg molar ratio of 1:10, suggesting that EGCG has little effect on the secondary structure of denatured protein at acidic pH.

Amide I′ region of the de-convoluted Fourier transform infrared spectra of β-lactoglobulin in the presence of epigallocatechin-3-gallate at different protein/antioxidant molar ratios: A native protein and B heat-denatured protein; a β-Lactoglobulin alone; b 1:1; c 1:2; d 1:10. All samples were at pD 4.4 in D2O. Spectra were recorded at 24 °C
3.3 Effect of complex formation on the antioxidant capacity of EGCG
The FRAP assay is a method of assessing antioxidant power by reduction of ferric to ferrous ion, which causes a colored ferrous-trypyridyltriazine complex to form at acidic pH (Benzie and Strain 1996). Figure 7 shows the FRAP value of EGCG at three different concentrations in the presence of 20 μmol.L−1 β-Lg or h-β-Lg at pH 7.0 and 4.0. The FRAP value was analyzed after 2 h of reaction time and obtained by subtracting the protein background from the protein–EGCG mixtures. At pH 7.0, the FRAP value of EGCG alone was about 153 at 20 μmol.L−1 and was proportional to concentration up to 200 μmol.L−1 (Fig. 7A). Adding β-Lg decreased the antioxidant activity to a FRAP value of 18 at 20 μmol.L−1 EGCG. This effect was of almost the same proportion at all EGCG concentrations. The mask effect on the antioxidant activity of flavonoids has been reported previously for plasma proteins (Arts et al. 2001). An explanation for this is that β-Lg forms hydrogen bonds with EGCG phenolic hydroxyl groups, which contribute to the antioxidant activity (Almajano et al. 2007b), in lieu of protein intermolecular β-sheet hydrogen bonds, as deduced from FTIR (Fig. 5A). It is noteworthy that h-β-Lg induced a very similar decrease in the antioxidant activity of EGCG, suggesting that h-β-Lg also interacts with EGCG phenolic hydroxyl groups by hydrogen bonding. However, no FTIR evidence was found for hydrogen bonding between EGCG and h-β-Lg (Fig. 5B). We therefore speculate that EGCG binds to different moieties of β-Lg and h-β-Lg at pH 7.0.

Antioxidant activity (ferric reducing antioxidant power) of 20, 40, and 200 μmol.L−1 epigallocatechin-3-gallate measured at pH 7.0 (A) and pH 4.0 (B); alone (blue square), with 20 μmol.L−1 β-lactoglobulin (pink square), or with 20 μmol.L−1 heat denatured-β-Lactoglobulin (green square)
At pH 4.0 (Fig. 7B), the antioxidant activity of EGCG was proportional to its concentration, but greater than at pH 7.0 (Fig. 7A). Adding either β-Lg or h-β-Lg again decreased the antioxidant value almost proportionately at all EGCG concentrations (Fig. 7B). These results suggest that both β-Lg and h-β-Lg interact with EGCG by hydrogen bonding with phenolic hydroxyl groups. However, the affinity of EGCG for β-Lg was about 2.6 times higher than for h-β-Lg at pH 4.0, based on fluorescence analysis. The decreases in FRAP values at pH 4.0 were smaller than those observed at pH 7.0 and almost identical for β-Lg and h-β-Lg, which is inconsistent with the observed differences in affinities from fluorescence results. We therefore speculate that forces other than hydrogen bonding, such as hydrophobic interaction, which has been reported for polyphenol interaction with proteins (Papadopoulou et al. 2005), occurs between EGCG and β-Lg in both states.
In order to study the possible protective effect of protein on EGCG stability, antioxidant activity as a function of time at a protein/EGCG ratio of 1:1 was measured at pH 7.0 and 4.0 (Fig. 8A, B). At pH 7.0, the antioxidant activity of EGCG gradually decreased over time, reaching a plateau after 72 h. However, activity did not change significantly in the presence of β-Lg, remaining about 1.2 times that of pure EGCG for 24 h and about 1.8 times that of pure EGCG for 48 h in the presence of h-β-Lg. The greater protection provided by h-β-Lg than by β-Lg is possibly due to the higher affinity of EGCG for h-β-Lg (Fig. 3). The activity decreased to 14 FRAP units at 168 h in the presence of h-β-Lg. This indicates that the protection provided by β-Lg in both its native and denatured states is time limited at pH 7.0. At pH 4.0, the antioxidant activity of EGCG remained constant until 72 h, after which a small decrease was observed, since EGCG is more stable at acidic pH. A similar change was observed in the presence of both β-Lg and h-β-Lg.

Antioxidant activity (ferric reducing antioxidant power) of pure epigallocatechin-3-gallate (blue diamond), β-lactoglobulin and epigallocatechin-3-gallate 1:1 (pink square), heat denatured-β-Lactoglobulin and epigallocatechin-3-gallate 1:1 (green triangle) measured over time at pH 7.0 (A) and pH 4.0 (B). The data are expressed as mean ± standard error of the mean of triplicate determinations
4 Conclusions
Based on our spectroscopic results, EGCG interacts with β-Lg and heat-denatured β-Lg at pH 7.0 and 4.0. The interactions changed the tertiary (Trp environment) and secondary structures of the protein. Hydrogen bonding occurs between EGCG and the protein in both its native and denatured states, which decreased the antioxidant activity of EGCG. Hydrophobic interaction may also be involved in the interaction. The affinity of EGCG for h-β-Lg at pH 7.0 is greater than for β-Lg and greater than for the protein in either state at pH 4.0. Complexing with β-Lg provides limited protection of EGCG against degradation at pH 7.0 but not at pH 4.0. This protection was slightly greater with h-β-Lg. The present research provides further insight into the characteristics of β-Lg binding with flavonoids such as EGCG and the impact of this interaction on their antioxidant activity.
References
Almajano MP, Delgado ME, Gordon MH (2007a) Albumin causes a synergistic increase in the antioxydant activity of green tea catechins in oil-in-water emulsions. Food Chem 10:1375–1382
Almajano MP, Delgado ME, Gordon MH (2007b) Changes in the antioxidant properties of protein solutions in the presence of epigallocatechin gallate. Food Chem 101:126–130
Arrondo JL, Muga A, Castresana J, Goñi FM (1993) Quantitative studies of the structure of proteins in solution by Fourier-transform infrared spectroscopy. Progr Biophys Mol Biol 59:23–56
Arts MJTJ, Haenen GRMM, Voss HP, Bast A (2001) Masking of antioxidant capacity by the interaction of flavonoid with protein. Food Chem Toxicol 39:787–791
Arts MJTJ, Haenen GRMM, Wilms LC, Beetstra SAJN, Heijnen CGM, Voss HP, Bast A (2002) Interactions between flavonoids and proteins: effect on the total antioxidant capacity. J Agric Food Chem 50:1184–1187
Baxter NJ, Lilley TH, Haslam E, Williamson MP (1997) Multiple interactions between polyphenols and a salivary proline-rich protein repeat result in complexation and precipitation. Biochemistry 36:5566–5577
Benzie IFF, Strain JJ (1996) The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem 239:70–76
Boye JI, Alli L, Ismail A (1996) Effects of physicochemical factors on the secondary structure of β-lactoglobulin. J Dairy Res 63:97–109
Brownlow S, Morais Cabral JH, Cooper R, Flower DR, Yewdall SJ, Polikarpov I, North ACT, Sawyer L (1997) Bovine β-lactoglobulin at 1.8 Å resolution-still an enigmatic lipocalin. Structure 5:481–495
Cayot P, Lorient D (1998) Structures et technofonctions des protéines du lait. [Structure and techno functions of milk proteins] Airlait recherches, France
Chen L, Remondetto GE, Subirade M (2006) Food protein-based materials as nutraceutical delivery systems. Trends Food Sci Technol 17:272–283
Chen Y, Barkley MD (1998) Towards understanding tryptophan fluorescence in proteins. Biochemistry 37:9976–9982
Cho Y, Batt CA, Sawyer L (1994) Probing the retinol-binding site of bovine β-lactoglobulin. J Biol Chem 259:11102–11107
Collini M, D’Alfonso L, Baldini G (2000) New insight on beta-lactoglobulin binding sites by 1-anilinonaphthalene-8-sulfonate fluorescence decay. Protein Sci 9:1968–1974
Dufour E, Haertlé T (1991) Binding of retinoids and β-carotene to β-lactoglobulin. Influence of protein modifications. Biochim Biophys Acta 1079:316–320
Fourneau C, Laurens A, Hocquemiller R, Cavé A (1998) Radical scavenging evaluation of green tea extracts. Phytotherapy Res 10:529–530
Ghosh KS, Sahoo BK, Dasgupta S (2008) Spectrophotometric studies on the interaction between (−)-epigallocatechin gallate and lysozyme. Chem Phys Lett 452:193–197
Harbowy ME, Balentine DA (1997) Tea Chemistry. Crit Rev Plant Sci 16:415–480
Kontopidis G, Holt C, Sawyer L (2002) The ligand-binding site of bovine β-lactoglobulin: evidence for a function. J Mol Biol 318:1043–1055
Lefèvre T, Subirade M (1999) Structural and interaction properties of β-Lactoglobulin as studied by FTIR spectroscopy. Int J Food Sci Technol 34:419–428
Lefèvre T, Subirade M (2000) Molecular differences in the formation and structure of fine-stranded and particulate β-lactoglobulin gels. Biopolymers 54:578–586
Lefèvre T, Subirade M (2001) Molecular structure and interaction of biopolymers as viewed by Fourier transformed infrared spectroscopy: model studies on β-lactoglobulin. Food Hydrocolloids 15:365–376
Liang L, Tajmir-Riahi HA, Subirade M (2008) Interaction of β-lactoglobulin with resveratrol and its biological implications. Biomacromolecules 9:50–56
Liang L, Subirade M (2010) β-Lactoglobulin/folic acid complexes: formation, characterization and biological implication. J Phys Chem B 114:6707–6712
Maiti TK, Ghosh KS, Dasgupta S (2006) Interaction of (−)-epigallocatechin-3-gallate with human serum albumin: fluorescence, Fourier transform infrared, circular dichroism and docking studies. Proteins: Struct Funct Bioinf 64:355–362
Nagaoka H, Toyoshima S, Takeda K (2002) Spectrofluorometric determination of catechins with 1,2-diphenylethylenediamine. Anal Sci 18(8):951–953
Nakao M, Takio S, Ono K (1998) Alkyl peroxyl radical-scavenging activity of catechins. Phytochemistry 49:2379–2382
Palazolo G, Rodriguez F, Farruggia B, Pico G, Delorenzi N (2000) Heat treatment of β-lactoglobulin: structural changes studied by partitioning and fluorescence. J Agric Food Chem 48:3817–3822
Papadopoulou A, Green RJ, Frazier RA (2005) Interaction of flavonoids with bovine serum albumin: a fluorescence quenching study. J Agric Food Chem 53:158–163
Papiz MZ, Sawyer L, Eliopoulos EE, North ACT, Findlay JBC, Sivaprasadarao R, Jones TA, Newcomer ME, Kraulis PJ (1986) The structure of [beta]-lactoglobulin and its similarity to plasma retinol-binding protein. Nature 324:383–385
Pelillo ME, Cuvelier B, Biguzzi T, Toschi G, Berset C, Lercker G (2004) Calculation of the molar absorptivity of polyphenols by using liquid chromatography with diode array detection: the case of carnosic acid. J Chromatogr A 1023:225–229
Rawel HM, Kroll J, Hohl UC (2001) Model studies on reactions of plant phenols with whey proteins. Nahrung/Food 45:72–81
Riihimäki LH, Vainio MJ, Heikura JMS, Valkonen KH, Virtanen VT, Vuorela PM (2008) Binding of phenolic compounds and their derivatives to bovine and reindeer β-lactoglobulin. J Agric Food Chem 56:7721–7729
Rohn S, Rawel H, Kroll J (2004) Antioxidant activity of protein-bound quercetin. J Agric Food Chem 52:4725–4729
Sawyer L, Brownlow S, Polikarpov I, Wu SY (1998) [beta]-Lactoglobulin: structural studies, biological clues. Int Dairy J 8:65–72
Sawyer L, Kontopidis G (2000) The core lipocalin, bovine β-lactoglobulin. Biochim Biophys Acta 1482:136–148
Shpigelman A, Israeli G, Livney YD (2010) Thermally-induced protein-polyphenol co-assemblies: beta Lactoglobulin-based nanocomplexes as protective nanovehicles for EGCG. Food Hydrocolloids 24:735–743
Stading M, Hermansson AM (1990) Viscoelastic behaviour of [beta]-lactoglobulin gel structures. Food Hydrocolloids 4:121–135
Wang Q, Allen JC, Swaisgood HE (1997) Binding of vitamin D and cholesterol to β-lactoglobulin. J Dairy Sci 80:1054–1059
Wang Q, Allen JC, Swaisgood HE (1998) Protein concentration dependence of palmitate binding to [beta]-lactoglobulin. J Dairy Sci 81:76–81
Wu SY, Pérez MD, Puyol P, Sawyer L (1999) β-Lactoglobulin binds palmitate within its central cavity. J Biol Chem 274:170–174
Zhu QY, Zhang A, Tsang D, Huang Y, Chen ZY (1997) Stability of green tea catechins. J Agric Food Chem 45:4624–4628
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Canada Research Chair in Proteins, Biosystems and Functional Foods.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Zorilla, R., Liang, L., Remondetto, G. et al. Interaction of epigallocatechin-3-gallate with β-lactoglobulin: molecular characterization and biological implication. Dairy Science & Technol. 91, 629–644 (2011). https://doi.org/10.1007/s13594-011-0036-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13594-011-0036-3