
Abstract
MicroRNAs (miRNAs) are single-stranded, non-coding, small (~ 22 nt) RNAs that regulate mRNA targets in plants and animals. Gymnema sylvestre (Retz.) is an important medicinal plant that lacks genomic as well as transcriptomic information. Here, to identify homologous miRNAs, we screened 6028 unique known plant miRNAs against 272,161 unigenes of G. sylvestre (Retz.) generated by paired-end deep transcriptome sequencing. We utilized 76 aligned unigenes for extracting the precursor sequences in G. sylvestre (Retz.) and identified 16 potential candidate miRNAs belonging to 12 miRNA families. The mean minimal folding free energy index (MFEI) value of these 16 predicted miRNAs was − 0.966, ranging from − 0.653 to − 2.37, while the G + C% content varied between 27.3 and 61.9%. Nucleotide composition analysis revealed that cytosine was the dominant nucleotide in mature miRNAs (26.3%), while uracil was the next most prevalent nucleotide (25.7%), followed by guanine (24.3%) and adenine (23.7%). In most cases (81%), the first position of the 5′ end was occupied by uracil in the identified mature G. sylvestre (Retz.) miRNAs. Differential expression of target unigenes revealed that, as compared to leaf tissue, a total 16 and 12 target unigenes were differentially expressed in flower and fruit tissues, respectively. Among the differentially expressed target unigenes were Auxin Response Factors 17-like, Copia-type polyprotein Phytoene synthase 2, as well as several encoding unnamed, predicted, uncharacterized, and/or hypothetical proteins. After detailed prediction of miRNAs and their corresponding target unigenes, we report for the first time a total of 13 miRNA and 213 corresponding target unigenes in G. sylvestre (Retz.).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The microRNAs of any organism represent a set of single-stranded, non-coding, small (~ 22 nt) RNAs that play an important role in regulating mRNA targets. They have been reported in plants as well as animals. Their modes of action in influencing target mRNAs may be either cleavage and/or translational repression (Chen 2004; Jones-Rhoades et al. 2006). miRNAs regulate the development of plant aerial (Palatnik et al. 2003) and underground parts (Boualem et al. 2008), phenophase transitions (Aukerman and Sakai 2003; Lauter et al. 2005), reproductive development of male and female organs (Wu et al. 2006), and floral development (Cartolano et al. 2007). They are also reported to be involved in responses to biotic and abiotic stress (Shukla et al. 2008; Zhang et al. 2008b; Ding et al. 2009). Through genetic screening in the nematode Caenorhabditis elegans, the first small RNA (lin-4) was discovered in 1993 (Lee et al. 1993; Wightman et al. 1993). The regulatory function of small RNA was demonstrated through the regulation of lin-14 by lin-4. This lin-4 RNA is now considered to be the origin of many miRNAs (Lau et al. 2001; Lee and Ambros 2001; Lagos-Quintana et al. 2001).
Next generation sequencing (NGS) techniques coupled to sophisticated computational and bioinformatic prediction methods have revolutionized molecular biology and made it possible to predict miRNAs as well as their targets with different functionality (Lai et al. 2003; Nam et al. 2005; Li et al. 2006; Huang et al. 2007). However, only a small subset of the total miRNA system can be captured through computational techniques and bioinformatic algorithms.
In contrast to animals, in which the processing of primary micro RNA (pri-miRNA) takes place in the nucleus and cytoplasm, processing of pri-miRNA into mature miRNA in plants is a two-step process carried out by processed by a double-stranded (ds) RNA specific RNaseIII enzyme Dicerlike 1 (DCL1) into a mature miRNA and antisense strand to the small RNAs (miRNA*) duplex that occurs exclusively in the nucleus (Starega-Roslan et al. 2015; Jones-Rhoades et al. 2006). Mature miRNAs are then bound by Argonaute (Ago) subfamily proteins, whose mRNA-targeting activity results in post-transcriptional regulation of genes (Kim et al. 2009). Discoveries of miRNAs are rapidly increasing; the total number of miRNAs in the miRBase database was 10,883 in 2009, 28,625 in 2014, and 38,589 in the most recent release (release v22, March 12, 2018).
Gymnema sylvestre (Retz.), locally known as “Madhunashini” in India, is a tropical medicinal herb. Due to its anti-diabetic properties, leaves of this plant are used in various traditional medicines. Asclepias geminate Roxb., Periploca sylvestris Retz., Marsdenia sylvestris (Retz.) are botanical synonyms of this climber species. In Indian Ayurvedic medicinal system, Madhunashini holds a long history. The first report on use of this plant to treat diabetes is nearly 2000 years old. G. sylvestre (Retz.) contains triterpine saponins, and pregnane and its derivatives like cardiac glycosides that have high medicinal value. Unfortunately, genomic and transcriptomic data are not available for this important medicinal plant. Gene regulation mediated by miRNA has become one of the most active areas in molecular biology. In the present study, we generated transcriptomic data and attempted to identify miRNAs as well as their probable targets in G. sylvestre (Retz.).
2 Materials and methods
2.1 RNA isolation, cDNA library preparation, and quality check (QC)
Gymnema leaf and flower samples were collected during the last week of November, and the developing fruits were collected during the second week of December in 2016. Fresh samples were used for total RNA isolation using the Norgen Total RNA isolation kit (NORGEN Biotek, 1720, 37500) following the manufacturer’s instructions. An Agilent RNA 6000 Nano chip was used to test the quality and calculate the RNA Integrity Number of the total RNA on a Bioanalyzer. To deplete the ribosomal RNA and fragment, all three samples were treated with the Illumina make TruSeq Stranded Total RNA-Ribo-Zero kit. The fragmented mRNA was converted into first-strand cDNA, followed by second-strand generation, A-tailing, adapter ligation and finally ended by limited number of PCR amplification of the adaptor-ligated libraries. And the quantity and quality were checked using a Agilent make High Sensitivity DNA Reagents Kit. A Bioanalyzer 2100 (Agilent Technologies) was used to analyse amplified libraries using a High Sensitivity (HS) DNA chip as per the manufacturer’s instructions.
2.2 Transcriptome sequencing
The qualitatively-screened cDNA library was loaded into an Illumina HiSeq 2500 platform for cluster generation and sequencing through 2 × 150 paired-end sequencing. Complementary adapter oligos were used to bind library molecules on a paired-end flow cell. Designed adapters were used for selective cleavage of the forward strands after re-synthesis of the reverse strand during sequencing. The copied reverse strand was then used to sequence from the opposite end of the fragment.
2.3 De novo assembly and unigene prediction from transcripts
Keeping a minimum Phred Score (QV) of 20, the raw data was filtered and processed through Trimmomatic-0.36 (Bolger et al. 2014). Trinity software (Haas et al. 2013), with default parameters, was used to prepare the de novo assembly of high-quality reads without any reference sequence. Reads were further assembled into contigs and minimally-overlapping contigs were clustered into connected components. A CD-HIT package using CD-HIT-EST (Li and Adams 2006) was used to remove short, redundant transcripts and to predict unigenes. A master assembly was prepared by pooling a total of 157.39 million reads from the three different samples using Trinity software at a kmer value of 25. The statistical elements of the assembly were calculated using in-house Perl scripts.
2.4 Criteria for orthologous miRNA annotation
Identification and characterization of miRNAs from transcriptome data of G. sylvestre (Retz.) is summarized in Fig. 1.

Workflow for identification of miRNAs from unigene sequences
2.5 Overview of unigenes and known miRNAs used for miRNA identification
Unigenes generated after clustering the transcript sequences obtained from combo assembly of G. sylvestre (Retz.) were used along with known plant miRNAs for identification of miRNA precursors. Very recent release of miRBase (http://www.mirbase.org, release v22, March 12, 2018), which consists of 48,885 mature and 8589 hairpin sequences, was used for this study. Sequences belonging to Viridiplantae were separated and redundant sequences were removed using an in-house script to generate a non-redundant set of potential miRNAs that were used for further analysis. Thus, a total of 6028 unique known plant miRNAs were screened against G. sylvestre (Retz.) unigenes for identification of homologous miRNAs. These known plant miRNAs were used as query for homology search against G. sylvestre (Retz.) unigenes using the standalone BLAST+ 2.2.30 program with a word size of 7.
The criteria for conserved plant miRNA annotation established by Blake et al. (2008) were used for orthologous miRNA identification in this study. These criteria include conservation of the miRNA precursor hairpin and the mature miRNA sequence. For filtering of the stem–loop structure and the mature miRNA sequence conservation, specific criteria were as follows: miRNAs should not align with unigenes in reverse complementarity, alignment should not include any gaps, maximum mismatch allowed was 3, the difference between length of mature miRNAs and alignment length should not be more than 3, the expected value (E) defining the random background noise was kept ≤ 0.01 to describe the number of hits we can expect to see by chance while searching the NCBI database. A lower cut-off E value was used to ensure significant match of our query sequences with the NCBI database. Unigenes with miRNA hits should not show any similarity against the NCBI non-redundant protein database. The sliding window approach was used for the extraction process, considering ~ 80 nt upstream and 80 nt downstream of the position at which the mature miRNA aligned, in increments of ~ 20 nt.
2.6 miRNA hairpin prediction
All sequences that had three or fewer mismatches with previously identified mature miRNAs were then filtered using their predicted secondary structures. RNAfold webserver (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) was used for prediction of hairpin-like secondary structure of pre-miRNAs. Based on earlier studies, the criteria used for selecting the pre-miRNA structures are as follows: the sequence could fold into an apparent stem-loop hairpin secondary structure, predicted mature miRNA should be located in the stem region of the hairpin structure, predicted mature miRNAs should have no more than 3 nt mismatches with the known miRNAs and no more than 6 mismatches with the corresponding active miRNA* sequence, there should not be any loop or break in the active miRNA* sequence, and the minimal folding free energy (MFE) of the predicted secondary structure should be lower than − 20 kcal/mol. The predicted miRNAs were named according to the miRBase database. The mature miRNAs were labelled as “miR” with the prefix “gsy” for G. sylvestre (Retz.).
2.7 Minimal folding free energy (MFE), adjusted minimal folding free energy (AMFE), and minimal folding free energy index (MFEI)
The minimal folding free energy index (MFEI) of miRNA precursors in G. sylvestre (Retz.) was calculated as follows: MFEI = [(AMFE) × 100]/(G% + C%) (Zhang et al. 2006b). Adjusted minimal folding free energy (AMFE) was calculated as follows: AMFE = [(MFE/length of RNA sequence) × 100] (Zhang et al. 2006b).
2.8 miRNA target prediction
To identify the potential target genes of the predicted G. sylvestre (Retz.) miRNAs from the unigene database the plant miRNA target finder program psRNATarget (http://plantgrn.noble.org/psRNATarget/) was used, selecting the “small RNAs and targets” option. The predicted mature miRNA sequences were used as query for finding the complementary sequences in the G. sylvestre (Retz.) unigene set using the following parameters: maximum expectation value of 3, hsp size (length of complementary scoring) of 19, range of central mismatch for translational inhibition 9–11 nt, calculated target accessibility using Max UPE (maximum energy to unpair the target site) of 25, flank length around the target site of 17 nt upstream and 13 nt downstream, and number of top targets of 50.
2.9 KOG and transcription factor analysis for miRNA targets
KOG analysis and transcription factor identification was carried for the identified target unigenes. Target unigene sequences were searched for similarity against the KOG database and the Plant Transcription Factor Database (PlantTFDB) (Guo et al. 2008) using BLASTX with an E-value threshold of 1e−5.
2.10 Functional annotation of miRNA targets
The predicted target unigene sequences were subjected to similarity search against the NCBI non-redundant (nr) database using the BLASTP algorithm. Protein sequence similarity searches against Uniprot, KOG, and Pfam databases were carried out for functional annotation, followed by gene ontology (GO) mapping and annotation using Blast2GO pro. Target unigenes were searched against all transcription factor protein sequences in the PlantTFDB (Guo et al. 2008) using BLASTP with an E-value cut-off of < 1e−10. GO mapping (Young et al. 2010) provides ontology of defined terms representing gene products.
2.11 Differential expression of target unigenes
To calculate the amount of gene expression, reads were mapped onto the 213 unigene sequences individually to determine the raw read counts using BWA-MEM (Li et al. 2013). Differential analysis was carried using the DeSeq2 R package, which generates normalized values in terms of “basemean”. Basemean values are used for log fold change (FC) and p value evaluations. Unigenes were considered upregulated if log2FC > 0 and downregulated if log2FC < 0. Unigenes having log2FC > 0 and p value < 0.05 were considered as significantly upregulated, whereas unigenes with log2FC < 0 and p value < 0.05 were considered as significantly downregulated (Wang et al. 2010).
3 Results
3.1 Transcripts clustering and prediction of hairpin structures of the pre-miRNAs
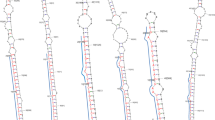
Transcripts were clustered based on nucleotide sequences and the longest transcripts in a given cluster were considered as unigenes. Statistics of the unigenes are given in Table 1. After application of all criteria (see materials and methods), a total of 76 mature and unigene alignments were utilized for extracting the precursor sequences in G. sylvestre (Retz.). Predicted hairpin structures of the pre-miRNAs using the RNAfold webserver (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) are given in Figs. 2, 3, 4 and 5. A total of 16 candidate miRNAs were identified. These 16 miRNAs, belonging to 12 miRNA families, are shown in Table 2.

Predicted hairpin stem loop secondary structures of 5 miRNAs identified in Gymnema sylvestre (Retz.). a gsy-miR160a b gsy-miR319b c gsy-miR169g d gsy-miR398b e gsy-miR162a-5p. Sequence highlighted in yellow represents the mature miRNA. (Color figure online)

Predicted hairpin stem loop secondary structures of 5 miRNAs identified in Gymnema sylvestre (Retz.). f gsy-miR162a-3p g gsy-miR167b-3p h gsy-miR2111a-5p i gsy-miR166g-5p j gsy-miR399f. Sequence highlighted in yellow represents the mature miRNA. (Color figure online)

Predicted hairpin stem loop secondary structures of 3 miRNAs identified in Gymnema sylvestre (Retz.). k gsy-miR159a l gsy-miR160 m gsy-miR167h. Sequence highlighted in yellow represents the mature miRNA. (Color figure online)

Predicted hairpin stem loop secondary structures of 3 optional miRNAs identified in Gymnema sylvestre (Retz.). n gsy-miR4238 o gsy-miR319c and p gsy-miR171c. Sequence highlighted in yellow represents the mature miRNA. (Color figure online)
3.2 MFEI value and refinement of miRNAs families and annotation and differential expression of targets
The mean MFEI value of the 16 predicted miRNAs was − 0.966, ranging between − 0.653 and − 2.37. A high MFEI value is indicative of an actual miRNA, however, lower values do not rule out a sequence as a true miRNA (Zhang et al. 2006b, 2007). The G + C% ranged from 27.27 to 61.9%, with a mean value of 50.18%. The 16 precursor sequences were further checked using the iMcRNA webserver (Liu et al. 2015), which helped identify the real microRNA precursors from the false microRNA precursors, with results showing that all precursors were real. A screenshot of the prediction results obtained from iMcRNA for all 16 miRNAs precursors is shown in Supplementary Fig. S1. However, from these 16 families, two miRNAs from family gsy-miR4238 and gsy-miR319c lacked the minimum two nucleotide mismatch required for dicer activity, as mentioned by Taylor et al. (2017), and one miRNA from family gsy-miR171c did not have a corresponding target annotated in the unigene database. Thus, we were left with 13 miRNAs with corresponding targets. A total of 271 targets were identified (265 unique sequences) with respect to these 13 miRNAs (Fig. 6). From KOG analysis of the predicted 265 targets, a total of 114 targets had a hit in the KOG database. KOG analysis (Fig. 7) showed that the most enriched KOG categories were “Posttranslational modification, protein turnover, chaperones (O)” and “Carbohydrate transport and metabolism (G)”, followed by “RNA processing and modification (A)”. Transcription factor analysis showed that a total of 131 targets had hits against the PlantTFDB. The most enriched transcription factor families were GATA, followed by HSF and bHLH (Fig. 8). GO mapping (Young et al. 2010) provides ontology of defined gene products. Functional annotation of the 265 target unigene sequences against the NR database was carried out, followed by GO analysis. From a total of 265 unigenes, NR hits were obtained for 213 unigenes. Further GO annotation of these unigenes resulted in assignment of GO to 84 unigene sequences. GO category distribution is shown in Table 3.

Number of identified targets for each of the 13 miRNAs

KOG classification for targets sequences of 13 miRNAs

Transcription factor distribution for targets sequences of 13 miRNAs
KEGG automatic annotation server (KAAS) was used for ortholog assignment and mapping of targets to biological pathways. All unigenes were compared against the KEGG database using BLASTX with a threshold bit-score value of 60 (default). The unigenes were enriched in 5 level-1 categories and 13 level-2 functional pathway categories. The mapped unigenes represented metabolic pathways of major biomolecules such as carbohydrates, lipids, amino acids, glycans, cofactors, vitamins, terpenoids, polyketides, etc. The mapped unigenes also represented genes involved in genetic information processing, environmental information processing, cellular processes, and organismal systems. Categorical unigene distribution is shown in Table 4.
Based on the annotation of targets against 13 miRNAs, a total of 119 unique target pairs were obtained and used to generate Circos plots. Among the identified pairs, the mode of action was cleavage and translation inhibition in 110 and 9 pairs, respectively. A Circos plot for 13 predicted miRNAs and their respective targets is shown in Fig. 9. In the plot, the upper right-hand tracks on the circle represent the 13 miRNAs starting from gsy-miR159a to gsy-miR399f. The 119 target accessions are displayed on the rest of the circle. The targets were labelled according to their BLASTX annotations.

Circos plot between the 13 predicted miRNAs and their respective targets. The upper right-hand side tracks of the circle represent the 13 miRNAs starting from gsy-miR153a to gsy-miR399f, while the 119 targets accessions are displayed on the rest of the circle. The targets are labelled according to their BLASTX annotations. Inner, coloured lines connect miRNAs to their respective targets. (Color figure online)
As compared to the leaf tissue, the number of significantly differentially expressed target unigenes were 16 and 12 in flower and fruit tissues, respectively. For flower tissue, 10 target unigenes were downregulated and 6 were upregulated, as compared to the leaf tissue. In fruit tissue, five target unigenes were upregulated and seven were downregulated. There were seven target unigenes in common among those differentially expressed in flower and fruit tissues, as compared to the leaf tissue. The DGEs of the target unigenes are presented in Fig. 10. Differentially expressed target unigenes encoded Auxin Response Factor 17-like, Copia-type Polyprotein, Phytoene synthase 2, along with other unnamed, predicted, uncharacterized, and/or hypothetical proteins.

Heat map showing top 16 significantly expressed genes in flower tissue as compared to leaf tissue (a) and 14 significantly expressed genes in fruit tissue as compared to leaf tissue (b). Differential analysis was carried using DeSeq2 R package. Basemean values were used for log fold change and p value evaluation. Unigenes were considered upregulated if log2FC > 0 and downregulated if log2FC < 0. Unigenes having log2FC > 0 and p value < 0.05 were considered as significantly upregulated, whereas unigenes with log2FC < 0 and p value < 0.05 were considered as significantly downregulated. Colour chart gradient from red to yellow indicates significant downregulation and significantly upregulation of genes, respectively. (Color figure online)
4 Discussion
From the nucleotide composition, cytosine was found to be the dominant nucleotide (26.3%) in mature miRNAs; uracil was the next most prevalent one (25.7%), followed by guanine (24.3%) and adenine (23.7%). In most (81%) cases, the first position of the 5′ end was occupied by uracil in the mature G. sylvestre (Retz.) miRNAs. The highest number of targets (40%) were annotated as being involved in genetic information processing, followed by metabolism (18.5%). Environmental processing and cellular processing both represented 14.8% in each category, whereas the smallest number of targets (11.1%) were annotated as being involved in organismal systems.
Utmost care was taken during the prediction of miRNAs and their targets and for the first time in G. sylvestre (Retz.), we report a total of 13 miRNA families in this study. Thermodynamic stability of the secondary structure of RNA or DNA was measured in terms of the MFE, the unit of which was expressed in kcal/mol (Mathews et al. 1999; Zuker 2003). Structural stability of a molecule increases with a decrease in its MFE value. AMFE was calculated to normalize the MFE, as these values are strongly correlated with the length of the sequence (Zhang et al. 2008a, b). MFEI is an index developed by Zhang et al. (2006a, b), and is used as a criterion to differentiate between miRNAs and other RNAs based on MFE, sequence length, and G + C nucleotide composition (Zhang et al. 2006b, 2008a). To improve accuracy in predicting miRNA targets, near-perfect complementarity of plant miRNAs for their targets is desirable (Rhoades et al. 2002; Jones-Rhoades and Bartel 2004; Schwab et al. 2005, 2006). Based on earlier reports, uracil at the first 5′ nucleotide position of a mature sequence is associated with its important role in the recognition of a miRNA by Argonaute1 (Mi et al. 2008; Montgomery et al. 2008; Takeda et al. 2008; Zhang et al. 2008a) and in G. sylvestre, we report that uracil (81.25%) was the dominant nucleotide at the first position of the 5′ end of mature miRNAs.
Availability of data and materials
The transcriptome raw data are available at NCBI under Project SUB2977090 as SAMN07528738 (leaf), SAMN07528739 (flower) and SAMN07528740 (fruit).
References
Aukerman MJ, Sakai H (2003) Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15:2730–2741
Blake C, Meyers MJ, Axtell BB, David PB, David B, John LB, Xiaofeng C, James CC, Xuemei C et al (2008) Criteria for annotation of plant microRNAs. Plant Cell 20:3186–3190
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Boualem A, Laporte P, Jovanovic M, Laffont C, Plet J, Combier JP, Niebel A, Crespi M, Frugier F (2008) MicroRNA166 controls root and nodule development in Medicago truncatula. Plant J 54:876–887
Cartolano M, Castillo R, Efremova N, Kuckenberg M, Zethof J, Gerats T, Schwarz-Sommer Z, Vandenbussche M (2007) A conserved microRNA module exerts homeotic control over Petunia hybrida and Antirrhinum majus floral organ identity. Nat Genet 39:901–905
Chen X (2004) A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303:2022–2025
Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y (2009) Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot 103:29–38
Guo AY, Chen X, Gap G, Zhang QH, Liu XC, Zhong YF, Gu X, He K, Luo J (2008) Plant TFDB: a comprehensive plant transcription factor database. Nucleic Acids Res 36:966–969
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B et al (2013) De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat Protoc 8:1494–1512
Huang TH, Fan B, Rothschild FM, Hu ZL, Li K, Zhao SH (2007) MiRFinder: an improved approach and software implementation for genome-wide fast microRNA precursor scans. BMC Bioinform 8:341
Jones-Rhoades MW, Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 14:787–799
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10:126–139
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T (2001) Identification of novel genes coding for small expressed RNAs. Science 294:853–858
Lai EC, Tomancak P, Williams RW, Rubin GM (2003) Computational identification of Drosophila microRNA genes. Genome Biol 4:R42. https://doi.org/10.1186/gb-2003-4-7-r42
Lau NC, Lim PL, Weinstein EG, Bartel DP (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294:858–862
Lauter N, Kampani A, Carlson S, Goebel M, Moose SP (2005) microRNA172 downregulates glossy15 to promote vegetative phase change in maize. Proc Natl Acad Sci USA 102:9412–9417
Lee RC, Ambros V (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science 294:862–864
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Li W, Adam G (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659
Li SC, Pan CY, Lin WC (2006) Bioinformatics discovery of microRNA precursor from human ESTs and introns. BMC Genom 7:164
Li JW, Wan R, Yu CS, Co NN, Wong N, Chan TF (2013) ViralFusionSeq: accurately discover viral integration events and reconstruct fusion transcripts at single-base resolution. Bioinformatics 29:649–651
Liu B, Fang L, Liu F, Wang X, Chen J, Chou KC (2015) Identification of real microRNA precursors with a pseudo structure status composition approach. PloS One 10:e0121501
Mathews DH, Sabina J, Zuker M, Turner DH (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 288:911–940
Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H et al (2008) Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133:116–127
Montgomery TA, Howell MD, Cuperus JT, Li D, Hansen JE, Alexander AL, Chapman EJ, Fahlgren N, Allen E et al (2008) Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 133:128–141
Nam JW, Shin KR, Han J, Lee Y, Kim VN, Zhang BT (2005) Human microRNA prediction through a probabilistic co-learning model of sequence and structure. Nucleic Acids Res 33:3570–3581
Palatnik JF, Edwards A, Wu X, Schommer C, Schwab R, Carrington JC, Weigel D (2003) Control of leaf morphogenesis by microRNAs. Nature 425:257–263
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP (2002) Prediction of plant microRNA targets. Cell 110:513–520
Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D (2005) Specific effects of microRNAs on the plant transcriptome. Dev Cell 8:517–527
Schwab R, Ossowski S, Riester M, Warthmann N, Weigel D (2006) Highly specific gene silencing by artificial microRNAs in Arabidopsis. Plant Cell 18:1121–1133
Shukla LI, Chinnusamy V, Sunkar R (2008) The role of microRNAs and other endogenous small RNAs in plant stress responses. Biochim Biophys Acta 1779:743–748
Starega-Roslan J, Galka-Marciniak P, Wlodzimierz JK (2015) Nucleotide sequence of miRNA precursor contributes to cleavage site selection by Dicer. Nucleic Acids Res 43:10939–10951. https://doi.org/10.1093/nar/gkv968
Takeda A, Iwasaki S, Watanabe T, Utsumi M, Watanabe Y (2008) The mechanism selecting the guide strand from small RNA duplexes is different among argonaute proteins. Plant Cell Physiol 49:493–500
Taylor RS, Tarver JE, Foroozani A, Donoghue PCJ (2017) MicroRNA annotation of plant genomes—do it right or not at all. BioEssays 39:1–6
Wang L, Feng Z, Wang X, Xi Wang, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Wightman B, Ha I, Ruvkun G (1993) Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75:855–862
Wu MF, Tian Q, Reed JW (2006) Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression and regulates both female and male reproduction. Development 133:4211–4218
Young MD, Wakefield MJ, Smyth GK, Oshlack A (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11:1–12
Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA (2006a) Conservation and divergence of plant microRNA genes. Plant J 46:243–259
Zhang BH, Pan XP, Cox SB, Cobb GP, Anderson TA (2006b) Evidence that miRNAs are different from other RNAs. Cell Mol Life Sci 63:246–254
Zhang B, Wang Q, Wang K, Pan X, Liu F, Guo T, Cobb GP, Anderson TA (2007) Identification of cotton microRNAs and their targets. Gene 397:26–37
Zhang B, Pan X, Stellwag EJ (2008a) Identification of soybean microRNAs and their targets. Planta 229:161–182
Zhang JF, Yuan LJ, Shao Y, Du W, Yan DW, Lu YT (2008b) The disturbance of small RNA pathways enhanced abscisic acid response and multiple stress responses in Arabidopsis. Plant Cell Environ 31:562–574
Zuker M (2003) M fold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgements
We acknowledge funding through Financial Assistance Programme (FAP) Scheme 2015-16 from Gujarat State Biotechnology Mission (GSBTM), Govt. of Gujarat, Gujarat, India, the ICAR-DMAPR, Anand, and the Indian Council of Agricultural Research, New Delhi for providing the basic facilities for this research, the germplasm explorer and the curators of the genotype used in this study, Dr. Jitendra Kumar, the Ex-Director of the ICAR-DMAPR for his moral support and guidance while preparing the research proposal for external funding.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
About this article

{kind=link}
Cite this article
Kalariya, K.A., Meena, R.P., Saran, P.L. et al. Identification of microRNAs from transcriptome data in gurmar (Gymnema sylvestre). Hortic. Environ. Biotechnol. 60, 383–397 (2019). https://doi.org/10.1007/s13580-019-00135-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13580-019-00135-7