
Abstract
We recently demonstrated that interleukin-6 (IL-6)- and vascular endothelial growth factor (VEGF)-induced osteosarcoma (OS) cell proliferation and migration are parallel to significant increased expression of SH3GL1 and the phosphorylation level of P130cas. The expression level of SH3GL1 was widely upregulated in human OS tissues, and their overexpression was significantly correlated with more aggressive clinicopathological features. Conversely, depletion of SH3GL1 by adenovirus shRNA abrogates P130cas phosphorylation and IL-6- and VEGF-induced OS cell proliferation and migration. To further pinpoint the mechanism how SH3GL1 was responsible for cell proliferation and migration, we deleted SH3GL1 in vitro and in vivo. In vitro, depletion of SH3GL1 abrogates P130cas phosphorylation and IL-6- and VEGF-induced OS cell proliferation and migration. SH3GL1 knockdown caused cell cycle arrest in G0/G1 phase via downregulation of cyclin D1, caused activation of p27KIP, and attenuated the activation of p-Rb. Interestingly, SH3GL1 knockdown also markedly attenuated the phosphorylation level of Akt/GSK-3β/FAK. In vivo, depletion of SH3GL1 by shRNA inhibited the tumor tissue growth and the expression of p-P130cas. Collectively, our results strongly suggest that SH3GL1 is a novel target for anti-osteosarcoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteosarcoma (OS) is a familiar malignancy which takes over approximately 35% of all bone malignancies currently [1]. Extensive studies about osteosarcoma have been made recently, but the understanding cellular mechanisms remain incomplete. Although, the therapeutic receptors expressions have been successfully employed in the treatment of estrogen receptor-positive osteosarcoma, osteosarcoma still remains an incurable disease. So, identification of novel targets and practical targets for the clinically more aggressive forms of osteosarcoma are urgently needed. For this idea, a series of molecules have been identified, including MicroRNA-34c [2], Cyr61 [3], and Orphan receptor tyrosine kinase (ROR2) [4] which are involved in tumorigenesis and progression of osteosarcoma.
SH3GL1 is a member of a unique SH3 family, which expresses ubiquitously mammalian tissues. SH3GL1 as a membrane-binding protein which is essential for clathrin-mediated endocytosis [5,6,7]. During clathrin-mediated endocytosis, SH3GL1 together with dynamin, CIN85 and amphiphysin facilitates to shape endocytic vesicle and endosomes. Some investigations showed that SH3GLB1 was a Bax-binding protein [8,9,10,11,12,13] and interacts with Beclin 1 through UVRAG to regulate cells autophagy and tumorigenesis [11]. Besides, SH3GLB1 was required for the maintenance of mitochondrial morphology [12]. Other studies showed that SH3GL1 was a fusion partner to the mixed lineage leukemia (MLL) gene [14]. The two binding partners’ fusion resulted from chromosome translocation and leukemia [15]. And, SH3GL1 plays an important role in tumorigenesis [16, 17], development and invasion [8,9,10]; however, the direct evidences of SH3GL1 in osteosarcoma have not been well clarified.
Based on other investigations about the functions of SH3GL1, and in the study, we further clarify the mechanisms of which deficiency of SH3GL1 mediates its anti-proliferative and anti-migration effects in OS cells. Specifically, we investigated the effects of SH3GL1 deletion on cell-cycle progression and migration.
Materials and methods
Reagents
Antibody targeting SH3GL1, Cyclin D1, p27 and GAPDH were purchased from Santa Cruz Biotech (Santa Cruz, CA). Antibodies targeting P130cas, phospho-P130cas, Akt, phospho-Akt, GSK-3β and phospho-GSK-3β were purchased from Cell Signaling Technology (Beverly, MA). Interlukin-6 (IL-6) and vascular endothelial growth factor (VEGF) were purchased from Sigma (Sigma-Aldrich, USA). Cell Counting Assay Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies (Japan). Fetal bovine serum (FBS) and MEM medium were purchased from Gibco (Gibco, USA). DAPI was purchased from Ladder Molecular Biomedical Research Center (Guangzhou, China).
Tissues and patients
80 paired tumor tissues and adjacent normal tissues were collected from patients with osteosarcoma who underwent surgical resection at Department of Orthopaedics of Tianjin Hospital from 2013 to 2015. The study was approved by the Ethics Committee of Tianjin Hospital (Tianjin, China). The clinicopathologic characteristics of all the patients including age, gender, focus type, tumor size, grade, T stage, lung metastasis, and histological type were collected from their medical records.
Cell culture
Osteosarcoma cell lines MG-63 and K7M2-WT were purchased from the American Tissue Culture Collection (ATCC, USA). Cells were cultured in MEM medium supplemented with 10% fetal bovine serum (FBS, HyClone, USA), 5 U/ml heparin, 100 U/ml penicillin, and 100 U/ml streptomycin in a humidified atmosphere of 5% CO2 at 37 °C.
Infection of cells with SH3GL1 shRNA
SH3GL1-Human, 4 unique 29mer shRNA constructs in adenovirus vector (Gene ID = 6455) were purchased from OriGene Technologies Inc. The shRNA constructs were designed against multiple splice variants at SH3GL1 gene locus (GenBank No. NM_001199943, NM_001199944,NM_003025 and XM_050099). To knockdown of SH3GL1, cells were transfection with the adenovirus vector combined with SH3GL1 shRNA (20 MOI). Briefly, the cells were cultured in medium without serum and antibiotics, then added SH3GL1 shRNA vector to the cells in quiescent state and swirled gently to ensure uniform distribution. After incubation for 8 h at 37 °C, cells were transferred into fresh medium with 10% FBS and cultured for 48 h. The expression of SH3GL1 protein was detected by western blot analysis, and the cells were used for the following study.
Cell viability assay
Cell proliferation was determined using Cell Counting Assay Kit-8 (Dojindo Molecular Technologies, Japan) incorporation as previously described [18], and according to the manufacturer’s instructions. Briefly, the experiment was performed after a cell synchronization, 100 µL of already processed cells were plated on 96-well plates at a density of 1 × 105 cells/ml for 48 h, the media contain different concentrations of IL-6 and VEGF, respectively. The quiescent cells were incubated with 10 µL of CCK-8 for 2 h, and then measured the absorbance at 450 nm using a microplate reader (Bio-Tek, Winooski, VT, USA).
Cell cycle analysis
Cell cycle status was evaluated by flow cytometry as described previously [19]. Cells were plated in 60-mm dishes with 10% FBS for 24 h, then resuspended with PBS three times and switched into serum-free MEM medium containing 2 mg/ml of BSA for 2 h, and then the cells were transfected with the recombinant Ad-SH3GL1 shRNA, 48 h later, the cells were collected and fixed with ice-cold 75% alcohol for 24 h at 4 °C, and then treated for cell cycle analysis using propidium iodide-staining method, and DNA content was analyzed by flow cytometry.
Transwell assay
Briefly, cells were cultured in MEM medium with 0.1% BSA for 12 h, and washed with PBS twice. Then cells were resuspended in MEM medium with 0.1% BSA. 1 × 105 cells and the same amount of control cells was plated in the upper chamber, and the lower chamber contained MEM medium with 0.1% BSA. Cells were allowed to migrate for 8 h in a humidified atmosphere of 5% CO2 at 37 °C. The cells migrated into the lower chamber were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet solution, and then observed under inverted microscope.
Western blot analysis
Western blot analysis was performed as described previously [19]. Briefly, cells were collected, rinsed with ice-cold PBS, lysis buffer (P0013, Beyotime, Jiangsu, China), and 1% protease inhibitor (cocktail: CalbioChem, Darmstadt, Germany) for 30 min. The protein was collected by centrifugation at 12,000×g for 12 min at 4 °C. Protein concentration was determined by BCA assay (Beyotime, Jiangsu, China). Equal amounts of total protein (40 μg) were separated by 8–12% SDS-PAGE and transferred to PVDF membranes (Immobilon-P, Millipore). The membranes were blocked at room temperature for 45 min in 5% non-fat milk (10 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween 20, 5% non-fat milk), incubated 2 h at room temperature initially with primary antibodies, and then with the appropriate secondary peroxidase-conjugated antibodies. Blots were developing with enhanced chemiluminescence substrates (Beyotime, Jiangsu, China) and then were visualized by exposure to Kodak X-ray film. Integrated optical density of target bands was accurately determined by the Image-J gel analysis system. SH3GL1, β-actin, cyclin D1, Akt and phospho-Akt (Ser473), P27, Rb and phospho-Rb, P130cas, phospho-P130cas, GSK-3β and phospho-GSK-3β were tested in this study.
Immunoprecipitation
The potential interaction between SH3GL1 and P130cas were confirmed by co-immunoprecipitation in OS cells. Cell lysates were centrifuged at 12,000×g for 15 min at 4 °C. Protein concentrations were determined by BCA protein assay, with bovine serum albumin (BSA) as the standard. Then samples containing 150 µg of protein were incubated with limiting amounts of anti-P130cas (1:50, Santa Cruz) at 4 °C overnight and then co-incubated with protein A/G agarose beads for 4 h at 4 °C. Screw cap was removed and the column was placed into a new tube. Beads were washed with PBS and resuspended in radio-immunoprecipitation assay for western blot analysis using anti-SH3GL1 antibody. The IgG antibody was used as the internal control.
Xenograft formation and in vivo research
BALB/c nude mice (Male, 4 weeks old) were purchased from Vital River Laboratories (Beijing, China). 1.5 × 107 K7M2-WT cells suspended in 1 ml of antibiotic-free PBS were injected subcutaneously into the neck of the mice. After 5–6 days, there was an obvious tumor at the injection site. When the tumors reached a size of 50 mm3, mice were randomly divided into three groups: the treatment group (injected with 100 MOI Ad-SH3GL1 shRNA vector via tail vein route every 3 days), the control group (injected with an equal volume of Ad-lacz via the same route at the same time), and the normal group (injected with an equal volume of PBS via the same route at the same time). There are six mice in each group. Every time before injection we measured the tumor dimensions with digital vernier caliper [the tumor size = π/6 × length × (width)2]. All the animal experiments were approved by Tianjin Hospital (Tianjin, China) Committee for Animal Research and performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (issued by the Ministry of Science and Technology of China, Beijing).
Immunofluorescence staining
The xenograft-formed tumor tissues were isolated from tumor-bearing mice, sliced into 8 μM and then incubated with antibodies of SH3GL1 (1:200, Santa Cruz, CA, USA) and p-P130cas (1:200, CST, Beverly, MA, USA). Cell nucleuses were stained with DAPI. The fluorescence images were detected using a fluorescent microscope (IX71, Olympus, Japan) and the fluorescence integrated density was examined using Image J Software.
Immunohistochemistry
The expression levels of SH3GL1 in paraffin sections were analyzed by immunohistochemical staining SP method. The human tumor tissue sections and human adjacent normal tissue sections were deparaffinized at 60 °C for 5 min, and then rehydrated through graded alcohol. Endogenous peroxidase activity was blocked by incubation in 3% hydrogen peroxide for 5 min. The sections were immunostained with SH3GL1 antibody (1:100, Santa Cruz, CA, USA) for 2 h at room temperature, then the sections were incubated with horseradish peroxidase-conjugated secondary antibody (1:100, Ladder Molecular Biomedical Research Center, Guangzhou, China) for 1 h. The sections were observed under a light microscope (Olympus, Tokyo, Japan).
Statistical analysis
All statistical analyses were performed using the SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). Measurement data were represented as mean ± SD, and determined using Student’s t test or ANOVA test. The correlation between SH3GL1 expression and clinicopathological characteristics was assessed by the Pearson χ 2 test. The p values less than 0.05 was considered significant.
Results
Upregulation of SH3GL1 correlates with more aggressive clinicopathological features in OS
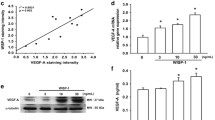
SH3GL1 expression in human tumor tissues and adjacent normal tissues was analyzed by immunohistochemistry. The data showed that SH3GL1 expression levels were significantly higher in the primary OS tumor tissues than in the adjacent normal tissues. Representative results are shown in Fig. 1. Then, 80 paired tissues were used to investigate the association between SH3GL1 expression and clinicopathological features. As shown in Table 1, SH3GL1 expression levels were concurrently highly related to bigger tumor size, lung metastasis, high grade and advanced T stage; however, there was no significant association between SH3GL1 expression with age, gender, focus type, and histological type.

SH3GL1 expression in human osteosarcoma tissues. a, b Representative immunohistochemistry images (×400) and statistical analysis of SH3GL1 expression in human osteosarcoma tissues. Data represent mean ± SD (n = 80), *p < 0.05 vs. adjacent normal tissue
SH3GL1 expression parallels with rate of OS cells proliferation
We first examined whether SH3GL1 expression correlated with the rate of cell proliferation. MG-63 cells were stimulated with various concentrations (5, 10, 20 ng/ml) of VEGF and various concentrations (10, 30, 100 ng/ml) of IL-6 for 24 h, then cell viability was detected using CCK-8 kit and the expression of endogenous SH3GL1 was examined by western blot analysis. As shown in Fig. 2a–d, after treatment with VEGF or IL-6 for 4 h, the cell proliferation was significantly faster and parallel to a significant increase in the endogenous SH3GL1 protein expression. Given that VEGF and IL-6 affect SH3GL1 expression in MG-63 cells, we speculated that SH3GL1 may be involved in the proliferation of OS cells. To prove this viewpoint, reproducibility of the experiments was tested in K7M2-WT cells. We showed that those results were reproducible, as shown in Fig. 2e–h. It may demonstrate that SH3GL1 was a necessity for proliferation. To further investigate the potential interaction between SH3GL1 and proliferation, we determined the effects of SH3GL1 knockdown on MG-63 cells proliferation. The transfection efficiency was detected by western blot analysis. As shown in Fig. 2i, after transfection with the recombinant adenovirus vector Ad-SH3GL1 shRNA for 48 h, the protein expression of SH3GL1 was decreased. However, adenovirus empty vector transfection had no significant effect on SH3GL1 expression. The effect of SH3GL1 knockdown on cell proliferation was determined using CCK-8 kit (Fig. 2j). Infection of SH3GL1 shRNA could inhibit VEGF- and IL-6-induced cell growth to 85.7 ± 7.2 and 92.9 ± 8.2%, respectively (Fig. 2j, l), whereas the infection of adenovirus empty vector had no significant effect on IL-6- and VEGF-induced cell growth.

SH3GL1 expression parallels with OS cell proliferation. a–d Dose effect of VEGF or IL-6 treatment for 48 h on the expression of SH3GL1 and cell viability in MG-63 cells. Data represent mean ± SD (n = 4), *p < 0.05 vs. ctrl. e–h Dose effect of VEGF or IL-6 treatment for 24 h on the expression of SH3GL1 and cell viability in K7M2-WT cells. Data represent mean ± SD (n = 4), *p < 0.05 vs. ctrl. i, j Knockdown of SH3GL1 reduces the IL-6- and VEGF-induced cell viability in MG-63 cells. Data represent mean ± SD (n = 4), *p < 0.05 vs. ctrl; # p < 0.05 vs. ctrl; & p < 0.05 vs. VEGF only. k, l Knockdown of SH3GL1 reduces the IL-6- and VEGF-induced cell viability in K7M2-WT cells. Data represent mean ± SD (n = 4), *p < 0.05 vs. ctrl; # p < 0.05 vs. ctrl; & p < 0.05 vs. IL-6 only
SH3GL1 shRNA arrests cell cycle in G0/G1 phase
To figure out the regulating effect of SH3GL1 on OS cell proliferation, we investigate whether SH3GL1 knockdown affects the cell cycle distribution of the highly malignant OS cell line MG-63, and found that silencing of SH3GL1 increases the cell population in G0/G1 phase and decreases the cell population in S phase and G2/M phase (Fig. 3a, b). To pinpoint the molecular mechanisms by which SH3GL1 shRNA induced G0/G1 arrest, we analyzed cyclin D1, p27KIP, and p-Rb expression levels in the model. As shown in (Fig. 3c, d), SH3GL1 shRNA treatment downregulated the expression of cyclin D1 and the phosphorylation levels of Rb but p27KIP expression was elevated in OS cell line MG-63.

SH3GL1 knockdown arrests cell cycle in G0/G1 phase. a, b Representative images and statistical analysis of cell cycle distribution of MG-63 cells after SH3GL1 knockdown. a Stimulated by IL-6. b Stimulated by VEGF. Data represent mean ± SD (n = 4), *p < 0.01 vs. ctrl. c, d Changes of G1/S phase transition-regulating proteins in MG-63 cells after being transfected with SH3GL1 shRNA. Data represent mean ± SD (n = 4), *p < 0.01 vs. ctrl
SH3GL1 is indispensable for cell migration
Cancer cell migration plays a key role in tumor invasion and metastasis, thus we explored the effect of SH3GL1 knockdown on the migration of OS cell line MG-63. Our data showed that SH3GL1 knockdown attenuated the migration ability of MG-63 cells (Fig. 4a, b).

SH3GL1 knockdown attenuates cell migration in MG-63 cells. a, b SH3GL1 knockdown attenuates the migration ability of MG-63 cells detected by transwell assay. a Stimulated by IL-6. B Stimulated by VEGF. Data represent mean ± SD (n = 4), *p < 0.01 vs. ctrl. c, d Representative western blot images and Statistical analysis of SH3GL1 and phospho-P130cas in MG-63 cells after transfected with SH3GL1 shRNA. Data represent mean ± SD (n = 4), *p < 0.01 vs. ctrl. e Co-immunoprecipitation of SH3GL1 with P130cas from MG-63 cell lysates. Cell lysate was immunoprecipitated (IP) by anti-P130cas antibody (IP) or not (input), and analyzed by western blot (WB) with anti-SH3GL1 antibody
Interaction between SH3GL1 and P130cas
In Fig. 4c, d, western blot results showed that phospho-P130cas protein expression was significantly decreased in SH3GL1 shRNA group, prompting that they have co-localization in cells. The interaction between endogenous SH3GL1 with P130cas in MG-63 cells was confirmed by co-immunoprecipitation (Fig. 4e).
SH3GL1 shRNA attenuates Akt/GSK-3β/FAK signaling
The phosphorylation of Akt is well known to regulate the progression of cell cycle G1 to S in many types of cells including OS cells [18]. Therefore, we investigated the phosphorylation status of Akt after silencing SH3GL1 by SH3GL1 shRNA. As shown in Fig. 5, SH3GL1 knockdown significantly attenuated the phosphorylations of Akt, GSK-3β, and FAK, yet SH3GL1 shRNA transfection did not significantly alter the protein expression levels of Akt, GSK-3β, and FAK.

SH3GL1 knockdown attenuates Akt/GSK-3β/FAK signaling. a, b SH3GL1 knockdown significantly attenuates the phosphorylations of Akt/GSK-3β/FAK without or with IL-6 treatment. Data represent mean ± SD (n = 4), *p < 0.01 vs. ctrl; # p < 0.05 vs. ctrl; & p < 0.05 vs. IL-6 only. c, d SH3GL1 knockdown significantly attenuates the phosphorylations of Akt/GSK-3β/FAK without or with VEGF treatment. Data represent mean ± SD (n = 4), *p < 0.01 vs. ctrl; # p < 0.05 vs. ctrl; & p < 0.05 vs. VEGF only
Depletion of SH3GL1 by shRNA inhibited the tumor tissue growth in vivo
To study whether SH3GL1 knockdown is effective in inhibiting growth of osteosarcoma in vivo, we established the xenograft model of osteosarcoma in nude mice. 6 days later, the nude mice were treated with 100 MOI Ad-shRNA vector and the expression of SH3GL1 was significantly decreased in the treated group, suggesting that the recombinant adenovirus vector Ad-SH3GL1 shRNA was effective in vivo (Fig. 6d, e, **p < 0.01 vs. ctrl). As it is shown in Fig. 6a, b, injection with 100 MOI shRNA could suppress the tumor growth remarkably on >15 days of treatment (*p < 0.05, n = 6). During the course of treatment, no significant changes were observed in mice body weight among all groups (Fig. 6c), which suggests that at the treatment dosage there was no significant toxicity to mice. In line with our research in vitro, the phosphorylation of p130cas was abrogated with the depletion of SH3GL1 (Fig. 6f, g).

Depletion of SH3GL1 by Ad-shRNA inhibited the tumor tissue growth in vivo. a Representative images of tumors at day 30 in control, Ad-lacz, and Ad-shRNA group. b Changes of tumor size after the first time of systemic dosage with Ad-shRNA vector. c Changes of mice body weight during the dosage. d, e Effect of Ad-shRNA on the expression of SH3GL1 in tumor tissue. Data represent mean ± SD (n = 6), *p < 0.01 vs. ctrl. f, g Depletion of SH3GL1 abrogates the phosphorylation of P130cas in vivo. Data represent mean ± SD (n = 6), *p < 0.01 vs. ctrl
Discussion
Several reports have indicated that SH3GL1 may be involved in malignant biological behavior of tumor cells [16, 17]. In this study, we found that the expression level of SH3GL1 was widely upregulated in human OS tissues, and their overexpression was significantly correlated with more aggressive clinicopathological features. However, the direct evidence of the molecular mechanisms of SH3GL1 for regulation of cell growth in OS cells has not been well understood. In the present study, we found that knockdown of SH3GL1 arrested cell cycle in G0/G1 phase in MG-63 cells and K7M2-WT cells. To explore the molecular mechanisms, we analyzed the expression of proteins regulating the cell cycle progression at this phase. Our results showed that knockdown of SH3GL1 decreased the expression of cyclin D1. Moreover, we examined the effects of SH3GL1 knockdown on the activation of p27KIP and p-Rb. The two cyclin-dependent kinase inhibitors have been reported to arrest cell cycle in G1/S transition by reversibly inhibiting several cyclin–CDK complexes [18,19,20,21,22]. Our results revealed that SH3GL1 knockdown caused the activation of p27KIP, and attenuated the activation of p-Rb. All these results provided the direct evidence that SH3GL1 is indispensable for cell proliferation. Akt/GSK-3β/FAK signaling pathway has been demonstrated to be involved in cell survival and apoptosis [17, 22, 23]. In this study, we found that SH3GL1 knockdown significantly attenuated the phosphorylations of Akt, GSK-3β and FAK in MG-63 cells.
In summary, attenuation of SH3GL1 expression effectively suppressed the phosphorylation activation of Akt/GSK-3β/FAK signaling pathway, this inactivation leads to the downregulation of cyclin D1, caused activation of p27KIP, attenuated the activation of p-Rb, and arrests cell cycle in G0/G1 phase, thus regulating cell proliferation. Besides, migration of cancer cells plays a key role in tumor invasion and metastasis. In this study, we observed that SH3GL1 knockdown markedly impaired the migration of OS cells. Results in vitro and in vivo both showed that SH3GL1 shRNA treatment inhibited the phosphorylation of p130cas. As is known to all, p130cas is the key molecule in integrin-mediated intracellular signaling processes [24,25,26,27,28]. Our results hint that SH3GL1 contributes to OS cell adhesion and tumor metastasis.
In short, all these findings contribute to our further understanding of the role SH3GL1 played in osteosarcoma. Moreover, SH3GL1 is a novel target for anti-OS.
References
Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3–13.
van der Deen M, Taipaleenmäki H, Zhang Y, et al. MicroRNA-34c inversely couples the biological functions of the runt-related transcription factor RUNX2 and the tumor suppressor p53 in osteosarcoma. J Biol Chem. 2013;288:21307–19.
Sabile AA, Arlt MJ, Muff R, et al. Cyr61 expression in osteosarcoma indicates poor prognosis and promotes intratibial growth and lung metastasis in mice. J Bone Miner Res. 2012;27:58–67.
Morioka K, Tanikawa C, Ochi K, et al. Orphan receptor tyrosine kinase ROR2 as a potential therapeutic target for osteosarcoma. Cancer Sci. 2009;100:1227–33.
Lua BL, Low BC. Activation of EGF receptor endocytosis and ERK1/2 signaling by BPGAP1 requires direct interaction with EEN/endophilin II and a functional RhoGAP domain. J Cell Sci. 2005;118:2707–21.
Lynch DK, Winata SC, Lyons RJ, et al. A Cortactin-CD2-associated protein (CD2AP) complex provides a novel link between epidermal growth factor receptor endocytosis and the actin cytoskeleton. J Biol Chem. 2003;278:21805–13.
Davis Steffen Runz, Daniel A, et al. Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron. 2003;40:749–62.
Etxebarria A, Terrones O, Yamaguchi H, et al. Endophilin B1/Bif-1 stimulates BAX activation independently from its capacity to produce large scale membrane morphological rearrangements. J Biol Chem. 2009;284:4200–12.
Takahashi Y, Meyerkord CL. Wang HBif-1/endophilin B1: a candidate for crescent driving force in autophagy. Cell Death Differ. 2009;16:947–55.
Rostovtseva TK, Boukari H, Antignani A, et al. Bax activates endophilin B1 oligomerization and lipid membrane vesiculation. J Biol Chem. 2009;284:34390–9.
Takahashi Y, Coppola D, Matsushita N, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51.
Karbowski M, Jeong SY, Youle RJ. Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol. 2004;166:1027–39.
Wong AS, Lee RH, Cheung AY, et al. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat Cell Biol. 2011;13:568–79.
Yam JW, Jin DY, So CW, et al. Identification and characterization of EBP, a novel EEN binding protein that inhibits Ras signaling and is recruited into the nucleus by the MLL-EEN fusion protein. Blood. 2004;103:1445–53.
Liu Han, Chen Bing, Xiong Hui, et al. Functional contribution of EEN to leukemogenic transformation by MLL-EEN fusion protein. Oncogene. 2004;23:3385–94.
Baldassarre T, Watt K, Truesdell P, et al. Endophilin A2 promotes TNBC cell invasion and tumor metastasis. Mol Cancer Res. 2015;13:1044–55.
Guan H, Zhao P, Dai Z, et al. SH3GL1 inhibition reverses multidrug resistance in colorectal cancer cells by downregulation of MDR1/P-glycoprotein via EGFR/ERK/AP-1 pathway. Tumour Biol. 2016;37:12153–60.
Zhao H, Li M, Li L, et al. MiR-133b is down-regulated in human osteosarcoma and inhibits osteosarcoma cells proliferation, migration and invasion, and promotes apoptosis. PLoS One. 2013;8:e83571.
Tang YB, Liu YJ, Zhou JG, et al. Silence of ClC-3 chloride channel inhibits cell proliferation and the cell cycle via G/S phase arrest in rat basilar arterial smooth muscle cells. Cell Prolif. 2008;41:775–85.
Zhang YH, Zhao CQ, Jiang LS, et al. Lentiviral shRNA silencing of CHOP inhibits apoptosis induced by cyclic stretch in rat annular cells and attenuates disc degeneration in the rats. Apoptosis. 2011;16:594–605.
Jain AK, Raina K, Agarwal R. Deletion of p21/Cdkn1a confers protective effect against prostate tumorigenesis in transgenic adenocarcinoma of the mouse prostate model. Cell Cycle. 2013;12:1598–604.
Raju U, Ariga H, Koto M, et al. Improvement of esophageal adenocarcinoma cell and xenograft responses to radiation by targeting cyclin-dependent kinases. Radiother Oncol. 2006;80:185–91.
Badr G, Al-Sadoon MK, Abdel-Maksoud MA, et al. Cellular and molecular mechanisms underlie the anti-tumor activities exerted by Walterinnesia aegyptia venom combined with silica nanoparticles against multiple myeloma cancer cell types. PLoS One. 2012;7:e51661.
Crompton BD, Carlton AL, Thorner AR, et al. High-throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in Ewing sarcoma. Cancer Res. 2013;73:2873–83.
Kang YS, da Jeong E, Lee EK, et al. p130Cas controls the susceptibility of cancer cells to TGF-β-induced growth inhibition. Biochem Biophys Res Commun. 2013;438:116–21.
Zhao Y, Kumbrink J, Lin BT, et al. Expression of a phosphorylated substrate domain of p130Cas promotes PyMT-induced c-Src-dependent murine breast cancer progression. Carcinogenesis. 2013;34:2880–90.
Tornillo G, Elia AR, Castellano I, et al. p130Cas alters the differentiation potential of mammary luminal progenitors by deregulating c-Kit activity. Stem Cells. 2013;31:1422–33.
Zeng F, Luo F, Lv S, et al. A monoclonal antibody targeting neuropilin-1 inhibits adhesion of MCF7 breast cancer cells to fibronectin by suppressing the FAK/p130cas signaling pathway. Anticancer Drugs. 2014;25:663–72.
Author information
Authors and Affiliations
Contributions
En-qi Li carried out the studies and the data statistics, and En-qi Li and Jin-li Zhang drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
Human experiments were approved by the Ethics Committee of Tianjin Hospital (Tianjin, China). Animal experiments were approved by Tianjin Hospital (Tianjin, China) Committee for Animal Research and performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (issued by the Ministry of Science and Technology of China, Beijing).
Rights and permissions
About this article

Cite this article
Li, Eq., Zhang, Jl. Essential role of SH3GL1 in interleukin-6(IL-6)- and vascular endothelial growth factor (VEGF)-triggered p130cas-mediated proliferation and migration of osteosarcoma cells. Human Cell 30, 300–310 (2017). https://doi.org/10.1007/s13577-017-0178-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-017-0178-6