
Abstract
Phytic acid or phytate (myo-inositol hexakisphosphate) is the principal storage indigestible form of phosphorus in different crops. It is considered as an antinutrient in human as well as animal (including fish, poultry, pig, chicken etc.) diet due to its chelating behavior of certain essential divalent minerals (Fe2+, Mg2+, Zn2+, Ca2+ etc.). The unabsorbed, indigested form of phosphorus also causes phosphate pollution in the soil by animal wastes. Phytate degrading enzymes like phytases (myo-inositol hexakisphosphate phosphohydrolase) in this regard can be very useful and also economically feasible to reduce the risk of phosphate pollution and increase the nutrient value in animal feeds at the same time. The Klebsiella phytases are suitable to use in the food industries of plant origin for their excellent thermal stability and high pH tolerance. From the present in silico investigation, it was found that Klebsiella phytases were 46–47 kDa molecular weight protein of histidine phosphatase superfamily having thermostability and alkalinity nature. This thermostability can be achieved due to possession of higher percentage of α helices and β sheets at the same time; the presence of higher aliphatic indices (range in between 88 and 91) etc. Interestingly, a strong correlation was found to be pertinent from phylogenetic studies of proteins with their cDNA among both species and strain level. Hence, the present study would be beneficial for future researchers (3D model available in Protein Model Database with acc. no.: PM0080562) to meet the demand of agricultural and industrial production of bacterial phytases particularly for agricultural farming.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phytase is a subclass of phosphatase enzyme, which primarily breaks down the phytic acid into myo-inositol and inorganic phosphorus (Wyss et al. 1999; Mullaney and Ullah 2003). Its molecular mass ranges between 40 and 70 kDa (Mullaney and Ullah 2003; Kumar et al. 2012). The enzyme phytase mediates the hydrolytic degradation by cleaving off the phosphate moieties from the phytic acid in a sequential manner. There are several classes of Phytase viz. the 3-phytase (EC 3.1.3.8) and 6-phytase (EC 3.1.3.26) that liberates P moiety at position C3 and C6 respectively from the hexaphosphate ring, histidine acid phosphatases (HAPhy) (EC 3.1.3.2) which show optimum activity at pH 5.0 and alkaline phosphatases (EC 3.1.3.1). It exhibited optimum activity at pH 8.0, β-propeller phytase (EC 3.1.3.8) (BPPhy) (Huang et al. 2009) which play an important role in phytate-phosphorus cycling (Mullaney and Ullah 2003). Besides, purple acid phosphatases (EC 3.1.3.2), (PAPhy) and protein tyrosine phosphatase-like phytases are recently discovered classes of phytase (Nakashima et al. 2007; Kumar et al. 2012).
Phytate is the main stored form of phosphorus in various seeds, brans, cereals, grains etc. (Nakashima et al. 2007; Gupta et al. 2015). Phytate phosphorus is not bioavailable to monogastric animals for the lack of digestive enzyme phytase (Gupta et al. 2015). So, unutilized phytate passes intact through the gastrointestinal tract which ultimately elevates the level of organic phosphorus in manure causing P pollution (Gupta et al. 2015). Excess phosphorus excretion ultimately leads to phosphorus accumulation in the soil, eutrophication of water bodies, and great increase of phytoplankton in a water body for increased levels of nutrient (Nakashima et al. 2007; El-Toukhy et al. 2013). This, in turn, results in the depletion of oxygen in the water (hypoxia) which may cause death to aquatic animals. On the other hand, ruminant animals can utilize phytate as a nutritional factor because of the presence of phytases secreted by ruminant microbes (Bravo et al. 2003; Nakashima et al. 2007). However, phytate phosphorus bioavailability can be enhanced by supplementation of the fodder with the enzyme phytase to meet the phosphorus requirement of the non-ruminants (Bravo et al. 2003; Nakashima et al. 2007). Phytic acid is considered as an antinutrient or mineral absorption inhibitor as it has the ability to bind certain dietary minerals like iron, zinc, and manganese, calcium etc. causing impairment of the respective mineral absorption and hence, promoting mineral deficiencies (Escobin-Mopera et al. 2012; El-Toukhy et al. 2013). On the other hand, phytate also has a number of potential benefits, such as, it acts as phosphorus store in sprouting seeds, as a source of myo-inositol (a cell wall precursor), can act as potent antioxidant particularly in regards to iron, exhibits anticancer properties and exhibits positive impact on cholesterol and blood sugar control (Gontia-Mishra and Tiwari 2013).
Moreover, Phytase enzyme is also acquiring a great demand day by day in various industries including in the field of agricultural biotechnology (Escobin-Mopera et al. 2012; Dahiya 2016). The estimated sales value of this enzyme is about 500 million USD per annum (Abelson 1999) and phytase supplementation was recorded approximately 70% of global monogastric feed (Escobin-Mopera et al. 2012). Although, few commercially produced phytases from fungi and bacteria sold in the market (Escobin-Mopera et al. 2012), bacterial phytases have proved more advantageous over fungal ones due to better catalytic capability, higher substrate specificity and increased resistance to proteolysis (Konietzny and Greiner 2004). However, there is an intense interest to isolate and characterize different classes of phytase to meet the diverse needs for wider industrial applications because it is nearly impossible to cover all the commercial needs by a single phytase (Konietzny and Greiner 2004).
Enzymatic activity of phytase protein was previously observed in plants (rice, maize, soybean, legume seeds, barley etc.) animals (rats, chickens etc.), a number of Phytase producing microbes including fungi (Aspergillus sp., Candida sp., Saccharomyces sp., Rhizopus sp., Penicillium sp.) and bacteria (Escherichia coli, Klebsiella pneumoniae, Bacillus sp., Pseudomonas sp.) have been studied (Mullaney and Ullah 2003; Elkhalil et al. 2007; Escobin-Mopera et al. 2012; El-Toukhy et al. 2013).
However, in silico analysis of Klebsiella phytase protein not previously investigated to elucidate their structure–function as well as phylogenetic aspects. Hence, the present study was undertaken to computationally assess the detailed physicochemical characteristics, phylogenetic relatedness, structural and functional characterization of phytase proteins among Klebsiella spp.
Materials and methods
Data extraction and phylogenetic analysis
UniProt knowledgebase (www.uniprot.org/help/uniprotkb) (Apweiler et al. 2004) was used for retrieval of amino acid sequences of Klebsiella species having specific phytase activity. The amino acid sequences, specific for phytase enzyme of Klebsiella pneumoniae subsp. pneumoniae was first retrieved from UniProtKB database with UniProtKB entry A0A0M4JZ39_KLEPN (currently available in UniParc of UniProtKB—http://www.uniprot.org/uniparc/) and treated as a target/query sequence to obtain template sequences by using the BLAST tool of UniProtKB database. The aligned amino acid sequences were selected on the basis of lowest E-value, highest sequence identity, maximum query coverage and bit score. In this way, a total of 15 different protein sequences of Klebsiella species were downloaded in FASTA format for in silico analyses. The coding sequences (CDSs) or genes of the respective protein sequences were downloaded in FASTA format from the public nucleic acid database, the EMBL-EBI bank. The retrieved protein, as well as the respective coding sequences of different Klebsiella species and Klebsiella pneumoniae strains, were evolutionary analyzed and the corresponding phylogenetic trees were built by using MEGA7 (Molecular Evolutionary Genetics analysis) software (Kumar et al. 2016).
Structural and functional analyses of Klebsiella phytases
The primary sequence analysis was done by calculating the physiochemical characteristics of retrieved protein sequences which include isoelectric point (pI), molecular weight (MW), extinction coefficient, instability index (II), aliphatic index (AI), GRAVY or Grand Average of Hydropathicities by using ExPASY-ProtParam tool (http://web.expasy.org/protparam/) (Gasteiger et al. 2005). The secondary structural features i.e. secondary elements (like helix, turn, sheet, coil etc.) were predicted by two web-based servers—PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) and CFSSP: Chou and Fasman Secondary Structure Prediction Server (http://cho-fas.sourceforge.net/) (Chou and Fasman 1974a, b). The comparative protein model of Klebsiella pneumoniae phytase was built through the SWISS-Model Workspace (http://swissmodel.expasy.org/) (Biasini et al. 2014) using the selected template. The predicted protein model was evaluated through both QMEAN and UCLA—DOE LAB SAVES (The Structure Analysis and Verification Server version4) server (http://services.mbi.ucla.edu/SAVES/), which runs six programs for checking and validating protein structures during and after model refinement. Finally, the authenticated model in specified (.pdb) format deposited to Protein Model Database (PMDB—https://bioinformatics.cineca.it/PMDB/) and PMDB ID was obtained. Motif finder (http://www.genome.jp/tools/motif/), Interpro scan (http://www.ebi.ac.uk/Tools/pfa/iprscan5/), ScanProsite (http://prosite.expasy.org/scanprosite/); all these analytical databases were used for the functional analysis of the retrieved sequences.
Results
Phylogenetic analysis of protein and gene sequences of phytase
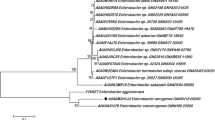
From the retrieved primary amino acid sequences and their respective gene sequences of UniProtKB, total four different phylograms (Figs. 1a, b, S1a, b) were constructed. Two phylograms consisted of evolutionary distance analysis by Phytase amino acid sequences of 15 species of Klebsiella (Fig. 1a) and 13 strains of K. pneumoniae (Fig. 1b). Another couple of trees (Fig. S1a, b) were of respective CDSs i.e. phytase gene sequences, also inferring evolutionary relatedness among the said taxa. Comparative phylogenetic analysis revealed that the selected target sequence K. pneumoniae subsp. pneumoniae (A0A0M4JZ39_KLEPN) clustered with K. variicola (A0A0M3RP91_KLEVA) and showed 97% similarity between them (Fig. 1a). This cluster again clustered with K. quasipneumoniae subsp. similipneumoniae (W8XLI4_9ENTR) with 95% similarity (Fig. 1a). Moreover, Fig. 1b shows K. pneumoniae subsp. pneumoniae (A0A0M4JZ39_KLEPN) clustered with K. pneumoniae MGH44 (V3KA25_KLEPN) with 98% similarity in strain level (Fig. 1b). Interestingly, a similar observation was noted about clustering in case of trees made with the respective CDSs (Fig. S1a, b). Here also, K. pneumoniae subsp. pneumoniae (ALD57067.1) found in the same cluster with K. variicola (ALD07427.1) and showed 99% similarity (Fig. S1a) while K. pneumoniae subsp. pneumoniae (ALD57067.1) also clustered with K. pneumoniae MGH44 (ESM59273.1) showing 100% similarity. The nearest neighbour, K. quasipneumoniae subsp. similipneumoniae (CDN05508.1) also found clustered similarly with 99% similarity with the first cluster.

Phylogenetic tree of phytase proteins of a different Klebsiella spp. and b different strains of Klebsiella pneumoniae
Physicochemical characterization
Characterization of theoretical physiochemical features was done for every sequence and presented separately (Tables 1, 2). The amino acid residue and molecular weight of this protein among selected strains were 422 (except Q7WSY1_RAOTE) and 46 kDa respectively. Isoelectric points were in the range of 8–10. The molar extinction coefficient (assuming all Cys residues are reduced) in case of different species of Klebsiella ranging from 61,420 to 68,410 M−1 cm−1 (Table 1) but it is fixed to 68,410 M−1 cm−1 in case of different strains of K. pneumoniae (Table 2). All of the instability indices calculated from Expasy ProtParam tool were above 40. Aliphatic indices of the strains were considerably higher, range in between 88 and 91 and calculated GRAVY was very low (in negative terms) in all cases. Graphical representations of the comparison of the composition of different amino acid residues of the 15 species of Klebsiella and the same among 13 different strains of K. pneumoniae have shown in Fig. 2a, b respectively. In both cases, the X-axis and Y-axis represent the percentage of amino acid composition and the amino acid residues respectively while the colour bars represent selected sequences. The red circles indicated the regions of dissimilarities of amino acid composition among the strains.

Composition comparison of different amino acid residues of a 15 species of Klebsiella and b 13 strains of Klebsiella pneumoniae subsp. pneumoniae (red circles showing difference in composition from each other) (color figure online)
Prediction of secondary structure
Predicted secondary arrangements of the target sequence [phytase of K. pneumoniae subsp. pneumoniae (A0A0M4JZ39_KLEPN)] are shown in Fig.3 and S2. Disordered protein binding sites were not observed in the secondary structure map (Fig. S2). Helix, sheet and turn, the three main types of secondary arrangements predicted through Chou–Fasman web server were 68.5, 68.2 and 10.4% respectively (Fig. 3).

Built secondary structure of Klebsiella pneumoniae subsp. pneumoniae (A0A0M4JZ39_KLEPN) phytase from Chou and Fasman server showing the percentage of different secondary arrangements
3D modeling of protein and quality analysis
The 3D models of Phytase protein of K. pneumoniae subsp. pneumoniae (A0A0M4JZ39_KLEPN) as viewed by PyMol (Fig 4a–d). From this model, it was noted that the selected target sequence was actually a tetramer i.e. it consisted of four monomeric chains labeled as Chain A, B, C and D (Fig. 4d). Helices, sheets and loops were coloured in red, yellow and green respectively (Fig. 4d). Besides, distinct disulfides were observed in this protein shown as green sphericals. The compared evaluation study (Fig. 5a–e) from both QMEAN and SAVES server revealed the overall quality of the predicted model. Z scores gained from QMEAN4 and SAVES were < 1 in both cases. The overall quality factor determined from SAVES ERRAT was 98.164 and the average 3D-1D score of 99.49% amino acid residue was ≥ 0.2 as revealed from Verify3D.

Tertiary 3D modelled structure of phytase of Klebsiella pneumoniae subsp. pneumoniae (A0A0M4JZ39_KLEPN) viewed by PyMol: a, b four distinct chains of the protein, c surface view of the phytase protein showing 4 chains in 4 different colours and d tertiary structure showing prominent secondary arrangements and disulfides (red = helix, yellow = sheet, green = loop, green balls = disulfides) (color figure online)

Quality analysis of predicted protein from different servers: a local quality estimate from QMEAN server, b comparison of built model with non-redundant set of structures from QMEAN server, c QMEAN4 score of predicted protein, d Ramachandran plot from RAMPAGE and e overall quality check by ERRAT (SAVES server)
Functional analysis
There were two motifs found (Fig. S3a, b) in the target sequence by the motif finder. Both belong to the Histidine phosphatase superfamily. His_Phos_2 found in the position of 32–357 and His_Phos_1 found in the 107–127 position in the amino acid sequence of the said strain (A0A0M4JZ39_KLEPN). Therefore the consensus sequences of the motifs were 325 and 20 amino acids in length respectively.
Discussion
Phytase is a subclass of phosphatase enzyme, which primarily catalyzes the hydrolysis of phytic acid into myo-inositol and inorganic phosphorus (Wyss et al. 1999). Till date, a number of phytase producing microbes have been isolated, characterized to fulfill the growing demand of this enzyme in various industries (Abelson 1999; Konietzny and Greiner 2004; Escobin-Mopera et al. 2012; Gontia-Mishra and Tiwari 2013; Dahiya 2016). In silico analysis in this regard found very important to characterize them more accurately. Computational investigation of Bacillus β-propeller phytase has examined recently (Kumar et al. 2014; Verma et al. 2016) but Klebsiella phytases have not analyzed earlier in details by in silico tools. However, a detailed account of computational identification and homology modeling of phytase protein of Yersinia mollaretii (Shivange et al. 2012) and Fusarium oxysporum (Gontia-Mishra et al. 2014) were done. Besides, specifically the structural and functional analyses of histidine phosphatase superfamily (Rigden 2008) and histidine acid phytase (Kumar et al. 2012) were studied earlier. In silico analysis of biological proteins has been shown immense contribution since few years (Verma et al. 2016; Pramanik et al. 2017) in the field of computational biology illustrating the structural and functional aspects of the proteins.
The present study covers the overall phylogenetic, structural and functional analysis of Klebsiella phytase enzyme. From, the phylogenetic analysis the only interpretation which can be made is that amino acid sequence and their respective CDSs contribute parallel to the evolutionary relatedness of the selected taxa indicating a positive correlation between the protein and gene sequences.
However, apart from the wet lab studies, in silico analysis of the physicochemical features of a protein is very important to get a theoretical overview of the proteins. This study demonstrates a number of physicochemical features which included isoelectric point, molecular weight, aliphatic index, instability index, extinction coefficient and grand average of hydropathicities. The molecular weight of a monomeric Phytase is generally around 14 kDa (Zhang et al. 2013); here, it is around 46 kDa for the tetrameric phytase protein. The isoelectric points were in the range of 8–10 which indicated that the proteins were alkaline in nature. Although previous in silico analysis of Bacillus phytase was reported to be acidic in nature (Verma et al. 2016), Zhang et al. (2011) reported the presence of alkaline phytases (sometimes called basic phytases) in Serratia sp. TN49 from the gut of Batocera horsfieldi larvae. A protein can be said to be unstable if the instability index rises above 40. In this case, the protein seemed to be unstable as all the calculated values of the instability index for the selected taxa just crossed 40. But there were many other factors which indicate strongly that the protein was stable. Aliphatic indices of the strains were considerably higher (range in between 88 and 91) which indicated that the proteins were thermostable in nature, and calculated GRAVY was very low which implied that the proteins have better interaction with water as also shown by Verma et al. (2016) and Pramanik et al. (2017). Moreover, amino acid composition at species level showed considerable variation (Fig. 2a) whereas the variation was almost negligible at strain level (Fig. 2b).
Based on the predicted secondary arrangement, it was found that Klebsiella phytase proteins can be classified in two major groups: helix and sheet (Fig. 3) and almost similar percentages of helices and sheets were present. This demonstration supported by a number of structural findings of different microbial phytases. The crystal structure assessments of phytases have revealed a variety of structural parameters (Kostrewa et al. 1997; Lim et al. 2000).
The 3D model prepared from the selected target sequence revealed that it was actually a tetramer i.e. it consisted of four monomeric chains labeled as Chain A-D (Fig. 4a). Tetrameric phytases have been reported previously both in fungi—Debaryomyces castellii CBS 2923 (Ragon et al. 2009) and bacteria—Yersinia mollaretii (Shivange et al. 2012). Helices, sheets and loops are coloured in red, yellow and green respectively (Fig. 4a). According to SAVES ERRAT, a good high resolution (> 3 Å) structure generally produce values around 95% or higher. As the overall quality factor determined from SAVES ERRAT was 98.164 (Fig. 5e), the resolution of present built 3D model of Klebsiella phytase is greater than 3 Å which is desirable. According to SAVES Verify3D, at least 80% of the amino acids should score ≥ 0.2 in the 3D/1D profile to get pass for being a standard good quality structure, which is here 99.49%. Disulfide bonds were frequently found in the modeled protein (Fig. 4d) which also indicated about the stability of the protein. Trivedi et al. (2009) showed that disulfide bonds in proteins were formed by oxidation of thiol groups of cystine residues and were advantageous to provide the stability to the proteins (Cheng et al. 2007). However, the built protein was deposited in PMDB database and the accession number of the model is PM0080562.
From the functional analysis, there were two motifs found (Fig. S3a, b) in the target sequence, both of which belong to the histidine phosphatase superfamily. This superfamily is a broad functionally diverse group of proteins that share a conserved catalytic core centered on a histidine which gets phosphorylated during the course of the reaction (Rigden 2008). The superfamily can be classified into two major branches. Although, the relationship between the two branches is not so distinct by (PSI-)BLAST but is evident from more sensitive sequence searches and structural comparisons (Rigden 2008). The branch-2 composed mainly of acid phosphatases and phytases and the branch-1 contains a wide range of catalytic functions, fructose 2, 6-bisphosphatase and cofactor-dependent phosphoglycerate mutase. In this study, His_Phos_2 and His_Phos_1 belong to Histidine phosphatase superfamily branch-2 and branch-1 respectively. A similar observation about the identification of motif superfamily was recorded by Kumar et al. (2012). The consensus sequences of these motifs can be very useful for designing primers for the isolation and identification of these types of phytases (Kumar et al. 2012).
Phytase is one of the important subclasses of phosphatase class of enzyme. It plays important roles in solubilization of insoluble phosphate to soluble one to increase bioavailable phosphorus to plants and exhibits a major role in ameliorating phosphate pollution from the soil. Several food industries use bacterial phytases to enhance nutritional value in the diet as phytases cleave the phytates. In order to elucidate a comprehensive account of Klebsiella phytase enzyme, this computational based study was made considering its physicochemical, structural, functional characters and phylogenetic relationship. The selected Klebsiella phytase was found to be thermostable, alkaline protein of histidine phosphatase superfamily having a molecular weight of 46 kDa and the 3D high-resolution model predicted to be a tetrameric protein. It can also be concluded that Klebsiella phytase may be very suitable, eco-friendly and cost-effective in various food industries for its excellent thermal stability and high pH tolerance. Thus, the present investigation will be helpful to the future researchers particularly in the field of computational proteomic studies of this phytase protein and wet laboratory studies as well.
Abbreviations
- UniProt KB:
-
UniProt knowledgebase
- UniParc:
-
UniProt archive
- BLAST:
-
Basic local alignment search tool
- MEGA:
-
Molecular evolutionary genetics analysis
- EMBL:
-
The European Molecular Biology Laboratory
- QMEAN:
-
Qualitative Model Energy ANalysis
References
Abelson PH (1999) A potential phosphate crisis. Science 283:2015
Apweiler R, Bairoch A, Wu CH et al (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res 32:D115–D119
Biasini M, Bienert S, Waterhouse A et al (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. https://doi.org/10.1093/nar/gku340
Bravo D, Sauvant D, Bogaert C, Meschy F (2003) Quantitative aspects of phosphorus absorption in ruminants. Reprod Nutr Dev 43:271–284
Cheng C, Wong K, Lim B (2007) The effect of disulfide bond on the conformational stability and catalytic activity of beta-propeller phytase. Protein Pept Lett 14:175–183
Chou PY, Fasman GD (1974a) Conformational parameters for amino acids in helical, β-376 sheet, and random coil regions calculated from proteins. Biochemistry 13:211–222. https://doi.org/10.1021/bi00699a001
Chou PY, Fasman GD (1974b) Prediction of protein conformation. Biochemistry 13:222–379. https://doi.org/10.1021/bi00699a002
Dahiya S (2016) Industrial application of phytases. Int J Appl Res 2:95–98
Elkhalil EAI, Männer K, Borriss R, Simon O (2007) In vitro and in vivo characteristics of bacterial phytases and their efficacy in broiler chickens. Br Poult Sci 48:64–70
El-Toukhy NMK, Youssef AS, Mikhail MGM (2013) Isolation, purification and characterization of phytase from Bacillus subtilis MJA. Afr J Biotechnol 12:2957–2967. https://doi.org/10.5897/ajb2013.12304
Escobin-Mopera L, Ohtani M, Sekiguchi S et al (2012) Purification and characterization of phytase from Klebsiella pneumoniae 9-3B. J Biosci Bioeng 113:562–567
Gasteiger E, Hoogland C, Gattiker A et al (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press, New York, pp 571–607
Gontia-Mishra I, Tiwari S (2013) Phylogenetic analysis of fungal phytases. Food Technol Biotechnol 51:313–326
Gontia-Mishra I, Singh VK, Tripathi N et al (2014) Computational identification, homology modelling and docking analysis of phytase protein from Fusarium oxysporum. Biologia 69:1283–1294
Gupta KR, Gangoliya SS, Singh NK (2015) Reduction of phytic acid and enhancement of bioavailable micronutrients in food grains. J Food Sci Technol 52:676–684. https://doi.org/10.1007/s13197-013-0978-y
Huang H, Shi P, Wang Y et al (2009) Diversity of beta-propeller phytase genes in the intestinal contents of grass carp provides insight into the release of major phosphorus from phytate in nature. Appl Environ Microbiol 75:1508–1516. https://doi.org/10.1128/aem.02188-08
Konietzny U, Greiner R (2004) Bacterial phytase: potential application, in vivo function and regulation of its synthesis. Braz J Microbiol 35:11–18
Kostrewa D, Grüninger-Leitch F, D’Arcy A et al (1997) Crystal structure of phytase from Aspergillus ficuum at 2.5A resolution. Nat Struct Biol 4:185–190
Kumar V, Singh G, Verma AK (2012) In silico characterization of histidine acid phytase sequences. Enzyme Res 2012:1–8
Kumar V, Singh G, Sangwan P et al (2014) Cloning, sequencing, and in silico analysis of β-propeller phytase Bacillus licheniformis strain PB-13. Biotechnol Res Int 2014:1–11. https://doi.org/10.1155/2014/841353
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol. https://doi.org/10.1093/molbev/msw054
Lim D, Golovan S, Forsberg CW et al (2000) Crystal structures of E. coli phytase and its complex with phytate. Nat Struct Biol 7:108–113
Mullaney EJ, Ullah AHJ (2003) The term phytase comprises several different classes of enzymes. Biochem Biophys Res Commun 312:179–184
Nakashima BA, McAllister TA, Sharma R, Selinger LB (2007) Diversity of phytases in the rumen. Microb Ecol 53:82–88. https://doi.org/10.1007/s00248-006-9147-4
Pramanik K, Soren T, Mitra S, Maiti TK (2017) In silico structural and functional analysis of Mesorhizobium ACC deaminase. Comput Biol Chem 68:12–21. https://doi.org/10.1016/j.compbiolchem.2017.02.005
Ragon M, Hoh F, Aumelas A et al (2009) Structure of Debaryomyces castellii CBS 2923 phytase. Struct Biol Cryst Commun Acta Cryst F65:321–326. https://doi.org/10.1107/s1744309109008653
Rigden DJ (2008) The histidine phosphatase superfamily: structure and function. Biochem J 409:333–348. https://doi.org/10.1042/bj20071097
Shivange AV, Serwe A, Dennig A et al (2012) Directed evolution of a highly active Yersinia mollaretii phytase. Appl Microbiol Biotechnol 95:405–418. https://doi.org/10.1007/s00253-011-3756-7
Trivedi MV, Laurence JS, Siahaan TJ (2009) The role of thiols and disulfides on protein stability. Curr Protein Pept Sci 10:614–625
Verma A, Singh VK, Gaur S (2016) Computational based functional analysis of Bacillus phytases. Comput Biol Chem 60:53–58
Wyss M, Brugger R, Kronenberger A et al (1999) Biochemical characterization of fungal phytases (myo-inositol hexakisphosphate phosphohydrolases): catalytic properties. Appl Environ Microbiol 65:367–373
Zhang R, Yang P, Huang H et al (2011) Two types of phytases (histidine acid phytase and β-propeller phytase) in Serratia sp. TN49 from the gut of Batocera horsfieldi (Coleoptera) larvae. Curr Microbiol 63:408–415. https://doi.org/10.1007/s00284-011-9995-0
Zhang G-Q, Wu Y-Y, Ng T-B et al (2013) A phytase characterized by relatively high pH tolerance and thermostability from the shiitake mushroom Lentinus edodes. Biomed Res Int 2013:7. https://doi.org/10.1155/2013/540239
Acknowledgements
Authors are thankful to Department of Science and Technology, New Delhi, Government of India for financial assistance to the first author (INSPIRE Fellow Reg. No. IF150197).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pramanik, K., Pal, P., Soren, T. et al. In silico structural, functional and phylogenetic analysis of Klebsiella phytases. J. Plant Biochem. Biotechnol. 27, 362–372 (2018). https://doi.org/10.1007/s13562-018-0445-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-018-0445-y