
Abstract
Gestational diabetes mellitus (GDM) is defined as a form of glucose intolerance. Evidences for late metabolic and behavioral consequences to offspring born from GDM are emerging. More recent and concerning evidences point to detrimental effects of GDM on the behavior and cognitive capacity of the offspring. We aimed to review what is known about the consequences of GDM to brain and behavior of the offspring. A research was made in PubMed using the words gestational diabetes, insulin resistance, memory, cognition, brain, and offspring. The most relevant papers according to citations and with results suggesting that hyperglycemia in pregnancy is associated with inflammation and some mechanisms which are potentially involved with the late brain insulin resistance and its consequences in the GDM’s offspring brain were selected. An increased risk of developing neuropsychiatric disorders to offspring from GDM mothers has been suggested. Transient intra-uterine exposure to high glucose levels can interfere with neuronal integrity, survival and connectivity to offspring’s brain. Fighting neuroinflammation during gestational period may avoid susceptibility for neurodegenerative disorders in the offspring.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gestational diabetes mellitus (GDM) is defined by the American Diabetic Association [1] as a glucose intolerance triggered during the second and third trimesters of pregnancy in previously normoglycemic women. Thus, hyperglycemia during pregnancy is a hallmark of GDM [2]. Diabetes in pregnancy is the most important metabolic condition occurring in approximately 10% of all pregnancies in developing countries like Brazil and India, and reaching greater incidence in developed countries such as USA [3, 4]. Family history of obesity or diabetes, non-white race, and maternal age are risk factors for developing GDM [5, 6].
GDM is related to several detrimental effects on the offspring, including an increased risk of congenital malformations and inflammation that affect several organs and tissues, including the central nervous system (CNS) [7,8,9]. The immediate consequences of GDM to infants include macrosomia, neonatal hypoglycemia, hypocalcemia, and respiratory distress syndrome at birth [10]. The most common morbidity is macrosomia occurring in 30% of infants who were exposed to fetal hyperglycemia [11]. Maternal factors that contribute to fetal macrosomia include obesity and elevated concentrations of lipids and amino acids [12].
There is also an increase of inflammation markers in GDM mothers such as higher plasma levels of tumor necrosis factor alpha (TNF-α) [13]. GDM contributes to offspring neuroinflammation and influences neuronal distribution in the brain, as well as neural stem cell (NSC) proliferation and apoptosis during embryogenesis, possibly contributing to cognitive deficit, behavioral changes, and memory loss in the adult life [9, 14, 15].
Learning and memory deficits are related to inflammation and changes in insulin signaling in the brain. Brain insulin resistance (IR) is present in diseases related to cognitive impairment, including neurodegenerative disorders, such as Alzheimer’s disease, and sepsis [16,17,18,19]. However, while the effect of brain IR is well documented in neurodegenerative diseases, the effect of insulin and its receptors on molecular mechanisms in inflammation, memory, and cognition in the offspring of GDM is poorly understood, and very few studies in literature are underlying the theme [7,8,9, 14, 15, 20,21,22,23,24,25,26].
This review aims to provide an overview about how intrauterine exposure to hyperglycemia during GDM influences the development of brain IR in the offspring, analyzing putative mechanisms or inflammation that could contribute to behavioral alterations and cognitive decline seen in adult offspring. A research was made in PubMed using the words gestational diabetes, insulin resistance, memory, cognition, brain, and offspring. The most relevant papers according to citations about GDM suggest hyperglycemia in pregnancy is associated with inflammation and putative mechanisms, which are potentially related to consequences to offspring brain.
Shared mechanisms between insulin resistance in type 2 diabetes and GDM
Insulin is a peptide hormone produced by β cells of the pancreas. It controls glucose metabolism and it is mainly secreted in response to the rise of circulating nutrients that occur immediately after food ingestion [27]. Thus, the elevation of the insulin level does not avoid the development of insulin resistance in muscle and adipose tissues [28, 29].
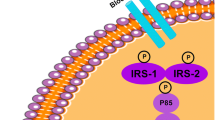
Physiological intracellular insulin signaling begins with the binding of insulin to its receptor in the surface of cells. Insulin receptors are formed by two alpha (α) and two beta (β) subunits. Insulin binding to its cell surface transmembrane receptor stimulates protein kinase activity of the β subunit. This leads to a conformational alteration of the molecule and to the insulin receptor phosphorylation in tyrosine residues [30]. Insulin receptors activate many intracellular substrates of which the most important are part of the insulin receptor substrate (IRS) family [31, 32]. IRS-1 and IRS-2 are directly related to glucose control in brain and peripheral tissues.
When insulin is bound to its receptor, the receptor phosphorylates IRS in tyrosine residues. Impaired kinase activity of insulin receptors appears to play an important role in insulin resistance in T2DM [32, 33]. The phosphatidylinositol-3-kinase (PI3-K) is an important compound in insulin signaling pathway. PI3-K is activated by IRS and consists of two subunits: p110 which has a catalytic subunit and the p85 regulatory subunit. The binding of IRS to the SH2 domains of the p85 subunit of PI3-K activates the associated catalytic domain [34].A number of receptors for growth factors and many oncoproteins work as intracellular binding sites for proteins containing SH2 domains such as PI3-K [35,36,37].
IR occurs when there is a failure in the activation or inhibition of IRS-1 and IRS-2 or if there is a reduced activation of other proteins such as activated kinase tyrosine (AKT) [36]. In peripheral tissues, inhibition of IRS-1 does not cause hyperglycemia but it leads to insulin resistance and growth retardation [32]. Reduced sensitivity to insulin induces β cells to increase the production of this hormone in order to maintain glycemia in the normal range. Besides the increasing of insulin, the tissue gets more resistant to it, favoring the return of the hyperglycemia [1, 38]. IRS-2 inhibition leads to hyperglycemia and hyperinsulinemia in the peripheral tissues.
There are differences in the IR mechanisms described in T2DM and GDM. During gestation, there are compensatory mechanisms such as hypertrophy and hyperplasia of β pancreatic cells to keep normoglycemic levels for the mother and the offspring [39, 40]. In GDM, these compensatory mechanisms tend to fail, leading to hyperglycemia, IR, and DNA damage to β pancreatic cells of the mother, rendering them more susceptible to develop obesity, diabetes, and neurodegenerative disorders later in life. Although, these events are well established in rodents, the compensatory alterations that happen in human β pancreatic cell during pregnancy remain controversial [39, 40].
Hyperglycemia in GDM is responsible for causing modifications in genes involved in proliferation and differentiation of NSC of the offspring, and these changes may favor in adult life oxidative stress and metabolic disturbance [14, 41].
Long-term consequences of GDM
For many years, GDM was considered to be a transient condition, which ruled out investigation of whether it was related to long-lasting consequences to either the mother or the fetus [7, 9]. It is known that mothers who develop diabetes during gestation are at increased risk of developing T2DM later in life due to the induction of metabolic changes and DNA damage to β pancreatic cells [42]. Risk of adverse maternal, fetal, and neonatal outcomes is increased as a function of maternal hyperglycemia [2, 42]. A study made by Cheung and Byte [42] with females who developed diabetes during pregnancy reviewed earlier studies examining the prevalence of GDM and risk of subsequent diabetes through an epidemiological tool known as population etiological fraction. They found over 31% of women who had GDM developed T2DM later in life and concluded that effective measures to prevent this situation are necessary and could have a significant impact over population health.
Infants born from diabetic mothers also show an increased risk of becoming obese and developing T2DM later in life [2]. The magnitude of fetal-neonatal risks and the reduction on neuronal activity and neurogenesis are proportional to the severity of maternal hyperglycemia [10, 43]. Besides changes in metabolism in the offspring of diabetic mothers, there are also morphological and functional alterations in hypothalamic structures caused by abnormal hormone levels [44]. Catecholaminergic system contributes to changes in the hypothalamus of the offspring affecting the neurodevelopment and promoting metabolic alterations such as an impaired leptin sensitivity [45, 46]. These changes can happen during pregnancy or lactation and are associated with obesity in adulthood, and also to brain changes [44, 45, 47]. More recent and concerning evidences point to detrimental effects of GDM on the behavior and cognitive capacity of the offspring [9, 20]. An increased risk of developing neuropsychiatric disorders in children from GDM mothers has also been suggested [9]. Due to the need of new researches in this field and the difficulty of doing human biochemical research in children’s brain, studies using experimental models are necessary to indicate possible ways to avoid or inhibit offspring brain damage or metabolic alterations in the offspring.
GDM impairs the development of CNS and causes cognitive and behavioral abnormalities in the offspring [48]. Studies using animal models have shown that GDM influences the behavior and fetal phenotype [9, 11]. An original research made with rats induced to diabetic pregnancy by a single intraperitoneal streptozotocin (STZ) injection (30–35 mg/Kg) showed that GDM’s offspring at 60 days of age presented increased anxiety levels when exposed to challenging situations, including the social interaction test and the elevated plus maze, and also revealed hyperactivity in the open-field test [49]. Identical results were seen when evaluating the offspring of a transgenic model for metabolic syndrome and correlating these behavior changes with hippocampal inflammation [50].
Changes in brain structure can occur in fetuses from GDM mothers resulting in increased susceptibility to autism and schizophrenia later in life [51]. More recently, a study made with humans analyzed children born from multiethnic mothers diagnosed with maternal diabetes at 26 weeks of gestation revealed a strong association between GDM and increased risks of developing autism in the offspring [23].
A review also pointed to an increased risk of schizophrenia in GDM offspring due to the classic mechanisms of maternal diabetes such as hyperglycemia, oxidative stress, alterations in lipid metabolism, and mitochondrial structure affecting memory and cognition [52].
There are a few studies about how hyperglycemia and hyperinsulinemia effect the developing brain in humans [31, 53, 54]. A research conducted by Linder and colleagues (2015) assessed the influence of maternal metabolism in the development of the fetal brain through fetal magneto encephalography, a modern technique to evaluate fetal brain activity. They determined fetal brain postprandial activity to be slower in the offspring of women with GDM. Their results indicated that maternal diabetes affects brain development and leads to CNS IR in the fetuses [53,54,55].
Evidence for interference of GDM in insulin signaling pathways in the offspring’s brain
Insulin has an important neuroprotective role and also modulates synapse plasticity mechanisms [56]. Insulin signaling also controls synapse density with consequent regulation of circuit function and plasticity [57,58,59]. Insulin receptors have been described in several brain structures, but those with the highest density include the hippocampus and cerebral cortex, where they were found to regulate memory and cognition [60].
Interestingly, for a long period of time, the brain was considered insensitive to insulin, and the CNS would not be insulin-dependent within this old concept [61]. However, it has been demonstrated that systemic metabolic expenditure and energetic homeostasis in the CNS are insulin-dependent and that neurons in the hippocampus, hypothalamus, and other brain regions that are sensitive to the presence of insulin are directly related to activities such as ingested food control and the energy expended for example [61]. Subjects with diabetes have a pre-disposition to develop cognitive deficits and show a greater chance of dementia, Alzheimer’s disease, and Schizophrenia [17, 18, 61].
The reduced activation of insulin pathway is proposed to have negative effects on GDM’s offspring cognition and memory [62]. Insulin and insulin-like growth factor-1 (IGF-1) are important in the development of these functions, and diabetes during pregnancy strongly influences the regulation of the receptors of insulin and IGF-1 [20, 22]. Alterations in IGF-1 gene expression and epigenetic regulation in hippocampus increase the risk for dementia to the offspring of maternal diabetes [22]. Recent studies in rodents revealed that retardation of fetal dendritic development in GDM is associated with IGF-1 signals [63].
Evidences suggest that the inhibition or malfunctioning of the IRS-1 is the main cause of memory loss and cognition failure due to brain IR [11, 22]. The reduction of the expression of the insulin receptors is related to memory loss and cognitive deficits at the brain [64]. Unfortunately, these molecular mechanisms of brain IR are not described in the GDM offspring, and most of what is known about brain IR comes from researches involving chronic neurological disorders such as Alzheimer’s or Parkinson’s diseases, among others [18, 48, 65].
Hyperinsulinemia and hyperglycemia in several brain areas could not be associated with changes in glucose transporter (GLUT) expression in the past decade [66]. Most of the past studies analyzing glucose transporters evaluated GLUT4 expressions which are present in peripheral tissues, but not in the brain [66] which expresses mostly GLUT1 transporting glucose across the blood-brain barrier and into the astrocytes and GLUT3 that carries glucose into neurons [67, 68]. However, a recent study evaluating middle-aged rats under a high-fat diet revealed that an increase in GLUT1 is associated with tissue damage in the hippocampus [69]. Expression levels of GLUT1 in cell membranes are normally increased by diminished glucose levels in hypoxia and reduced by increased glucose levels being widely distributed in fetal tissues [70]. It has been hypothesized that higher levels of GLUT1 expression are correlated with hypoxia, blood-brain barrier disruption, and may actually be a compensatory response to energy demands by the cells [71]. GLUT1 expression has also been described as a measure of how glucose enters endothelial cells and in neural stem cells when in a hypoxia situation [69]. Although these studies were not done under GDM conditions, they provide molecular evidences of the role of hyperglycemia, hypoxia, and inflammation in the brain, and there are a few studies showing evidence of their effect over maternal diabetes [69].
A recent meta-analysis [54] evaluated maternal diabetes and the cognitive performance in the offspring. Exclusion criteria were no control group population, the presence of any pathology in the offspring, and pre-clinical researches. The studies analyzed were made in humans and involved 6140 infants, and they found that children born from GDM and between 1 and 2 years old had lower scores in psychomotor and cognitive development.
Putative mechanisms involved in long-term consequences to brain function in GDM offspring
Morphological changes in the brain of the GDM offspring occur, most of the times, in the embryonic state and may have long-term consequences [51]. Hyperglycemia present in GDM pregnant is thought to generate a choric hypoxia state in the fetus [9]. These metabolic maternal changes affect neurodevelopment in many different ways such as a reduced myelination [72], neuron distribution [73] and connectivity at cortex [9], neurogenesis in hippocampus and cortex [74], and neuron apoptosis [56].
In the presence of hypoxia, the fetus needs to maintain the iron plasma level, and its supply does not attend its demand. Human babies born from mothers with GDM possess iron content estimated in just 40% of normal [25, 56]. Iron plays an important role in neurotransmitter synthesis, neurogenesis, and myelination [75, 76]. Iron deficiency leads to hippocampal changes which compromise its normal functions and result in behavioral alterations similar to those seen in schizophrenic patients [77]. Other neurodegenerative disorders that developed in fetuses originated from maternal diabetes which may be related to iron deficiency and hypoxia as results from the hyperglycemia and hyperinsulinemia present in this condition [77]. It is also common in fetal iron deficiency to the offspring manifesting higher levels of irritability and a depressive behavior [78].
Hypoxia and iron deficiency can also contribute to an increase in the inflammatory burden incurred by the fetus [79]. Iron deficiency, hypoxia, and higher TNF-α levels are factors which can be triggered for the changes in glycemia leading to possible neurocognitive sequelae in offspring of diabetic mothers [25].
Metabolic alterations occur in GDM [25] such as higher plasma TNF-α levels [13] and increase in total blood glucose in the dams [20] and can influence neuron development of the offspring [80].Recently, a study showed that alterations in glyoxalase 1, a detoxifying enzyme and its pathway, lead to increased levels of circulating methylglyoxal, which causes a reduction in embryonic mice cortical neural precursor cells causing long-lasting alterations in adult neurons, such as a diminished number of mature neurons and the reduction of neurogenesis, affecting behavior and cognition during the animal’s whole life [9]. The inflammation of the CNS may begin due to infection, toxic metabolites, or brain injury, and the immune response depends mainly on glial cells [9].
A maternal pro-inflammatory state can influence the brain development in the offspring
Microglia and astrocytes are glial cells directly related to neuroinflammation influencing memory and cognition [81, 82]. Usually, when having an exacerbated activation of microglia and astrocytes, a diminished activity of PI3-K insulin pathway and consequently a higher inhibition of insulin receptors and substrates are observed [81, 83, 84]. Even knowing the importance of microglia and astrocytes in inflammation, there are no studies evaluating their roles during all brain stages of development, starting from the embrionary period until adult life in GDM offspring. Hyperglycemia during pregnancy can contribute to a maternal pro-inflammatory state that can influence the brain development in the offspring [20, 24].
Microglia and astrocytes are responsible for producing and increasing the TNF-α levels having a harmful impact in the developing brain [82, 85].It has been shown that increased brain levels of TNF-α lead to peripheral and brain IR [64]. Higher plasma levels of TNF-α might cross placenta and could cause morphological alterations in the offspring’s brain [64]. A study revealed that circulating TNF-α concentrations may inhibit the insulin production [13, 86].
It is already known that inflammation plays an important role in generating morphological changes in offspring and/or worsening the IR consequences in offspring from GDM leading to severe brain alterations such as an abnormal distribution of neurons in the cortical areas and a reduced neurogenesis [7, 9]. A study published recently by Vuong and colleagues [87] provides evidence that in rats, maternal obesity associated with GDM influences microglial activation and neuroinflammation in newborn offspring. GDM was induced in female Sprague-Dawley rats using a high-fat and sucrose diet for 6 weeks prior mating, throughout gestation and lactation, while lean control dams were fed a low-fat diet. The dams experienced increased weight gain during pregnancy and starting mid-gestation developed hyperinsulinemia and moderate hyperglycemia. Offspring of GDM dams presented impaired recognition memory in the object recognition test, associated with reduced synaptophysin expression in CA1 and dentate gyrus (hippocampus areas). Increased levels of pro-inflammatory cytokines interferon gamma, IL-1α, IL-4, and TNF-α were also observed in the brain of the neonatal offspring of GDM dams. Finally, the microglial morphological transformation and astrogliosis observed in the GDM offspring persisted into young adulthood. GDM appears to condition microglia to a neuroinflammatory environment such that resulted in derangement of the hippocampal CA1 pyramidal neurons and cognitive impairments in young adulthood [87].
Serum maternal oxidative stress canals can induce or contribute to the development of a pro-inflammatory state and is associated with increased rates of perinatal problems [88,89,90,91]. Oxidative stress happens when the levels of reactive oxygen species (ROS) present within a tissue are greater than can be counteracted by the antioxidants present at the same tissue [88]. Pregnant women with GDM have increased level of DNA damage. A recent study showed that markers of lipid peroxidation and DNA oxidation are significantly increased in GDM women compared to healthy group of patients which indicates increased oxidative stress in GDM patients [89]. The human placenta is susceptible to oxidative stress and oxidative damage but methods of predicting pregnancy complications are limited [88]. The usage of biomarkers of oxidative stress in pregnancy for clinical applications must be detectable in biological fluids and be highly stable [92]. Some multiple markers of oxidative stress are known such as protein carbonyls and superoxide; ROS production provokes oxidation of lipids such as polyunsaturated fatty acids (PUFAs) and advanced glycation end-products (AGEs) [88].
Changes in the antioxidant levels of enzymes contribute to the development of oxidative stress in maternal diabetes [93]. An antioxidant is a molecule that inhibits the oxidation of other molecules. Oxidation is a chemical reaction that produces free radicals and leads to chain reactions that might damage cells [94]. Diet [95], medicine intake [96], and physical exercise influence the antioxidant levels, and cells are protected from oxidative stress by an interacting network between antioxidant enzymes [94]. A study made by Aziz and colleagues [93] with rats induced to maternal diabetes through a combination of different methods such as being fed with high-fat sucrose diet, and receiving a single intraperitoneal injection of streptozotocin (35 mg/Kg) and nicotinamide (120 mg/Kg) on gestational day 0 showed reduced activity of some antioxidant enzymes. The authors analyzed the superoxide dismutase, which catalyzes the breakdown of the superoxide anion into oxygen and hydrogen peroxide [97]; catalase, which catalyzes the conversion of hydrogen peroxide to water and oxygen [98]; and glutathione peroxidase, whose main role is to prevent the organism from oxidative damage [99], and their levels were significantly reduced in the dams induced to maternal diabetes [93]. This study highlight that impaired antioxidant condition can be linked with maternal diabetes stress.
Another marker of oxidative stress can be the receptor for AGEs (RAGE), which can trigger a pro-inflammatory pathway, and is associated with the risk of developing psychiatric disorders [13]. RAGE alterations have been described as important for GDM-induced offspring changes in the CNS [20]. Maternal diabetes was induced in rats during mid-pregnancy by STZ creating a pro-inflammatory state, which was triggered by RAGE signaling. Primary hippocampal neurons in culture obtained from the offspring showed an altered excitability and a hyperpolarized membrane-resting potential. As NF-kB, a pro-inflammatory transcription factor, is increased by RAGE, Western blots were performed with hippocampus samples, and its levels were found to be significantly higher in the offspring of STZ-treated dams. In early adulthood, offspring of GDM animals had reduced anxiety-like behavior, and altered object place preference revealed in behavior tests. These results suggest a disturbance in hippocampal development and alteration in behavior mediated by increasing levels of RAGE in fetal brain caused by hyperglycemia during pregnancy [20].
A recent study investigated another possibility of influencing the CNS development of the offspring through changes in the expression of genes responsible for regulating apoptosis in the hippocampus of neonate Wistar rats born to mothers who were induced to diabetes with a single intraperitoneal injection of STZ [56]. The study evaluated male offspring at P0, P7, and P14 and revealed that maternal hyperglycemia may cause disturbances in the expression of Bcl-2 and Bax genes, two extremely important genes in apoptosis regulation. The authors suggest that these disturbances may be the reason for the anomalies in cognition and behavior observed in offspring born to diabetic mothers.
Conclusions
The embryo exposure to GDM induces neuroinflammation [87], derangement of hippocampal neurons [9], and cognitive changes [20]. The offspring brains can suffer the consequences of inflammatory mechanisms during gestation leading to brain IR [87]. Failure in brain insulin signaling in the GDM offspring might explain in part the delay in cognitive development, and this hypothesis has never been evaluated properly (Fig. 1). Cognitive decline and memory loss in adults, especially at mild age or third age, might be a consequence of an embryo exposition to inflammation during a gestational period.

GDM consequences in the offspring’s brain. GDM is caused by hyperglycemia during pregnancy that leads to maternal serum inflammation and embrionary inflammation, which can influence the offspring’s brain development. One of the possible mechanisms that could be responsible for these changes in the embryo, and our hypothesis is that the microgliosis and astrogliosis during embrionary period would lead to an elevation of the TNF-α expression. In adult life, offspring born from diabetic mothers would be more susceptible to develop brain IR and consequently neurodegeneration to be revealed by the early cognition impairment and memory loss
Lifestyle intervention is necessary and requires a scientific basis for applying these possible interventions in order to achieve and deliver results from an economic and effective perspective in the fight against IR [54].An effective strategy to prevent and combat hyperglycemia and IR during GDM is the adoption of a balanced diet associated with physical exercises, which will favor the better functioning of insulin-dependent and non-insulin-dependent pathways, contributing to increased insulin sensitivity and glucose uptake [21]. Lifestyle intervention in pregnant women who were obese has shown to reduce plasma levels of inflammation markers [100].
Hyperglycemia that occurs in GDM stimulates an inflammatory state that can lead the fetus to brain IR driving them to develop memory loss and cognition impairment [101]. The data reviewed suggests therapies using inhibitors of inflammation pathways could be used in order to reduce inflammation avoiding these embryo malformations during GDM. This would contribute to the inhibition of memory loss and cognitive deficit in the offspring diminishing the long-lasting effects of the GDM. Finally, we suggest that blocking TNF-α signaling at different stages of embryo development could be an effective approach, as this pathway seems to mediate several alterations that affect cognition [9, 20]. The development of effective anti-inflammatory therapies is of great relevance to prevent the long-lasting effects of GDM over the offspring’s brain.
References
American Diabetes Association (ADA). Diagnosis and classification of diabetes mellitus. Diabetes Care [Internet]. 2014 [cited 2016 Dec 4];37 Suppl 1(January):S81–90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24357215 .
American Diabetes Association (ADA). Standards of medical care in diabetes—2014. Diabetes Care [Internet]. 2014 [cited 2014 Jul 10];37 Suppl 1(October 2013):S14–80. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24357209 .
Desisto CL, Kim SY, Sharma AJ. Prevalence estimates of gestational diabetes mellitus in the United States, pregnancy risk assessment monitoring system ( PRAMS ), 2007–2010. Prev Chronic Dis. 2014;11(12):1–9.
IDF. International Diabetes Federation. Diabetes Atlas [Internet]. Seventh Ed. 2015. Available from: http://www.diabetesatlas.org
Kim SY, England L, Sappenfield W, Hoyt G, Bish CL, Salihu HM, et al. Racial/ethnic differences in the percentage of gestational diabetes mellitus cases attributable to overweight and obesity, Florida, 2004-2007. Prevention. 2012;9(6):2012.
Cypryk K, Szymczak W, Czupryniak L, Sobczak M. Gestational diabetes mellitus—an analysis of risk factors Cukrzyca ciążowa—analiza czynników ryzyka. Endokrynol Pol. 2008;59(5):393–7.
Mills JL, Baker L, Goldman AS. Malformations in infants of diabetic mothers occur before the seventh gestational week implications for treatment. Diabetes. 1979;28:292–3.
Sharpe PB, Chan A, Haan E A, Hiller JE. Maternal diabetes and congenital anomalies in South Australia 1986-2000: a population-based cohort study. Birth Defects Res A Clin Mol Teratol [Internet]. 2005 [cited 2016 Dec 18];73(9):605–11. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16007590 .
Yang G, Cancino GI, Zahr SK, Guskjolen A, Voronova A, Gallagher D, et al. A glo1-methylglyoxal pathway that is perturbed in maternal diabetes regulates embryonic and adult neural stem cell pools in murine offspring. Cell Rep [Internet]. ElsevierCompany.; 2016 Oct [cited 2016 Oct 21];17(4):1022–36. Available from: http://linkinghub.elsevier.com/retrieve/pii/S2211124716313237
Frías JL, Frías JP, Frías PA, Frías MLM. Infrequently studied congenital anomalies as clues to the diagnosis of maternal diabetes mellitus. Am J Med Genet Part A. 2007;2909:2904–9.
Jing Y, Song Y, Yao Y, Yin J, Wang D, Gao L. Retardation of fetal dendritic development induced by gestational hyperglycemia is associated with brain insulin/IGF-I signals. Int J Dev Neurosci. 2014;37:15–20.
Boulet SL, Alexander GR, Salihu HM, Pass M. Macrosomic births in the United States: determinants, outcomes, and proposed grades of risk. Am J Obstet Gynecol. 2003;188:1372–8.
McLachlan KA, O’Neal D, Jenkins A, Alford FP. Do adiponectin, TNFalpha, leptin and CRP relate to insulin resistance in pregnancy? Studies in women with and without gestational diabetes, during and after pregnancy. Diabetes Metab Res Rev [Internet]. 2006 [cited 2017 Feb 5];22(2):131–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16170833 .
Fu J, Tay SSW, Ling E A, Dheen ST. High glucose alters the expression of genes involved in proliferation and cell-fate specification of embryonic neural stem cells. Diabetologia [Internet]. 2006 [cited 2016 Dec 18];49(5):1027–38. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16508779 .
Liao DM, Ng YK, Tay SSW, Ling E A, Dheen ST. Altered gene expression with abnormal patterning of the telencephalon in embryos of diabetic Albino Swiss mice. Diabetologia [Internet]. 2004 [cited 2016 Dec 18];47(3):523–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14963649 .
Bomfim TR, Forny-germano L, Sathler LB, Brito-moreira J, Houzel J, Decker H, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. J Clin Invest. 2012;122(4):1339–53.
Clarke JR, Lyra NM, Figueiredo CP, Frozza RL, Ledo JH, Beckman D, et al. Alzheimer-associated Aβ oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol Med. 2015;7(2):190–211.
Ferreira ST, Clarke JR, Bomfim TR, De Felice FG. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement [Internet]. Elsevier; 2014 [cited 2015 Mar 28];10(1 Suppl):S76–83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24529528 .
Lee C-C, Kuo Y-M, Huang C-C, Hsu K-S. Insulin rescues amyloid beta-induced impairment of hippocampal long-term potentiation. Neurobiol Aging [Internet]. 2009 [cited 2015 Apr 3];30(3):377–87. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17692997 .
Chandna AR, Kuhlmann N, Bryce C A, Greba Q, Campanucci V A, Howland JG. Chronic maternal hyperglycemia induced during mid-pregnancy in rats increases RAGE expression, augments hippocampal excitability, and alters behavior of the offspring. Neuroscience [Internet]. IBRO; 2015 [cited 2016 Dec 20];303:241–60. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26151680 .
El Hajj N, Schneider E, Lehnen H, Haaf T. Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment. Reproduction [Internet]. 2014 Dec [cited 2016 Oct 17];148(6):R111–20. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4241689&tool=pmcentrez&rendertype=abstract
Hami J, Sadr-nabavi A, Sankian M, Baladi-Mood M, Haghir H. The effects of maternal diabetes on expression of insulin-like growth factor-1 and insulin receptors in male developing rat hippocampus. Brain Strcture Funct. 2013;218:73–84.
Linder K, Schleger F, Kiefer-Schmidt I, Fritsche L, Kümmel S, Heni M, et al. Gestational diabetes impairs human fetal postprandial brain activity. J Clin Endocrinol Metab [Internet]. 2015 [cited 2016 Oct 22];100(11):4029–36. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26465393 .
Liu S, Guo Y, Yuan Q, Pan Y, Wang L, Liu Q, et al. Melatonin prevents neural tube defects in the offspring of diabetic pregnancy. J Pineal Res [Internet]. 2015 [cited 2015 Oct 27];508–17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26475080 .
Nelson CA, Wewerka S, Thomas KM, Tribby-walbridge S, Georgieff M. Neurocognitive sequelae of infants of diabetic mothers. Behav Neurosci. 2000;114(5):950–6.
Tanigawa K, Kawaguchi M, Tanaka O, Kato Y. Skeletal malformations in rat offspring long-term effect of maternal insulin-induced hypoglycemia during organogenesis. Diabetes. 1991;40:1115–21.
Vind BF, Birk JB, Vienberg SG, Andersen B, Beck-Nielsen H, Wojtaszewski JFP, et al. Hyperglycaemia normalises insulin action on glucose metabolism but not the impaired activation of AKT and glycogen synthase in the skeletal muscle of patients with type 2 diabetes. Diabetologia [Internet]. 2012 [cited 2014 Jul 18];55(5):1435–45. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22322917 .
Turner N, Cooney GJ, Kraegen EW, Bruce CR. Fatty acid metabolism, energy expenditure and insulin resistance in muscle. J Endocrinol [Internet]. 2014 [cited 2014 Nov 27];220(2):T61–79. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24323910 .
Fröjdö S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: a review of the current evidence from humans. Biochim Biophys Acta [Internet]. Elsevier B.V.; 2009 [cited 2014 Jul 18];1792(2):83–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19041393 .
Carvalho CRO, Carvalheira JBC, Lima MHM, Zimmerman SF, Caperuto LC, Amanso A, et al. Novel signal transduction pathway for luteinizing hormone and its interaction with insulin: activation of Janus kinase/signal transducer and activator of transcription and phosphoinositol 3-kinase/Akt pathways. Endocrinology [Internet]. 2003 [cited 2015 Jan 2];144(2):638–47. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12538627 .
Cai D, Dhe-Paganon S, Melendez P A, Lee J, Shoelson SE. Two new substrates in insulin signaling, IRS5/DOK4 and IRS6/DOK5. J Biol Chem [Internet]. 2003 [cited 2017 Feb 6];278(28):25323–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12730241 .
Folli F, Saad MJ, Backer JM, Kahn CR. Insulin stimulation of phosphatidylinositol 3-kinase activity and association with insulin receptor substrate 1 in liver and muscle of the intact rat *. J Biol Chem. 1992;267:22171–7.
Chowdhury KK, Legare DJ, Lautt WW. Exercise enhancement of hepatic insulin-sensitising substance-mediated glucose uptake in diet-induced prediabetic rats. Br J Nutr [Internet]. 2013 [cited 2014 Jul 18];109(5):844–52. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23021417 .
Backer JM, Myers MG Jr, Shoelson SE, Chin DJ, Sun X, Hu P, et al. Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 1992;11(9):3469–79.
Fantin VR, Wang Q, Lienhard GE, Keller SR, Valeria R. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am J Physiol Endocrinol Metab. 2000;278:127–33.
Saad MJA, Araki E, Rothenberg PL, White MF, Kahn CR. Regulation of insulin receptor substrate-1 in liver and muscle of animal models of insulin resistance. J Clin Invest. 1992;90:1839–49.
Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren J, Previs S, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–4.
Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet [Internet]. Elsevier Ltd; 2011 [cited 2014 Jul 11];378(9786):169–81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21705072 .
Parsons A, Sorenson L. Dwarf mice: effect of lactogenic hormones * of. Endocr Soc. 2014;136(5):2013–22.
Pasek RC, Gannon M. Advancements and challenges in generating accurate animal models of gestational diabetes mellitus. Am J Physiol Endocrinol Metab [Internet]. 2013 1 [cited 2015 Nov 30];305(11):E1327–38. Available from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4073988&tool=pmcentrez&rendertype=abstract
Wentzel P, Eriksson UJ. Antioxidants diminish developmental damage induced by high glucose and cyclooxygenase inhibitors in rat embryos in vitro. Diabetes [Internet]. 1998 1 [cited 2016 Dec 18];47(4):677–84. Available from http://diabetes.diabetesjournals.org/cgi/doi/10.2337/diabetes.47.4.677
Cheung NW, Byth K. Population health significance of gestational diabetes. Diabetes Care. 2005;26(7):2005–9.
Perkins JM, Dunn JP, Jagasia SM. Perspectives in gestational diabetes mellitus: a review of screening, diagnosis, and treatment. Clin Diabetes. 2007;25(2):57–62.
Plagemann A, Dorner G. Alterations of hypothalamic catecholamines in the newborn offspring of gestational diabetic mother rats. Dev Brain Res. 1998;109:201–9.
Melo A, Benatti R, Ignacio-Souza L, Okino C, Torsoni AS, Milanski M, et al. pregnancy and lactation. Metabolism [Internet]. Elsevier Inc. 2014;63(5):682–92. Available from. https://doi.org/10.1016/j.metabol.2014.02.002.
Steculorum SM, Bouret SG. Maternal diabetes compromises the organization of hypothalamic feeding circuits and impairs leptin sensitivity in offspring. Endocrinology. 2015;152:4171–9.
Steculorum SM, Bouret SG. Maternal diabetes compromises the Organization of Hypothalamic Feeding Circuits and Impairs Leptin Sensitivity in offspring. Endocrinology. 2011;152(11):4171–9.
De la Monte SM. Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer’s disease. Drugs [Internet]. Springer International Publishing; 2017 [cited 2017 Feb 9];77(1):47–65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27988872 .
Kinney BA, Rabe MB, Jensen RA, Steger RW. Maternal hyperglycemia leads to gender-dependent deficits in learning and memory in offspring. Exp Biol Med. 2003;228(2):152–9.
Dinel A-L, André C, Aubert A, Ferreira G, Layé S, Castanon N. Cognitive and emotional alterations are related to hippocampal inflammation in a mouse model of metabolic syndrome. PLoS One [Internet]. 2011 Jan [cited 2017 Feb 7];6(9):e24325. Available from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3174932&tool=pmcentrez&rendertype=abstract
Xiang AH, Wang X, Martinez MP, Walthall JC, Curry ES, Page K, et al. Association of maternal diabetes with autism in offspring. Jama [Internet]. 2015 [cited 2017 Feb 17];313(14):1425–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25871668 .
Van Lieshout RJ, Voruganti LP. Examen critique Diabetes mellitus during pregnancy and increased risk of schizophrenia in offspring: a review of the evidence and putative mechanisms. J Psychiatry Neurosci. 2008;33(5):395–404.
Bolaños L, Matute E, Ramírez-Dueñas MDL, Zarabozo D. Neuropsychological Impairment in School-Aged Children Born to Mothers With Gestational Diabetes. J Child Neurol [Internet]. 2015 [cited 2017 Feb 6];30(12):1616–24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25814475 .
Camprubi Robles M, Campoy C, Garcia Fernandez L, Lopez-Pedrosa JM, Rueda R, Martin MJ. Maternal diabetes and cognitive performance in the offspring: a systematic review and meta-analysis. PLoS One [Internet]. 2015 Jan [cited 2017 Feb 6];10(11):e0142583. Available from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4643884&tool=pmcentrez&rendertype=abstract
Cai S, Qiu A, Broekman BFP, Wong EQ, Gluckman PD, Godfrey KM, et al. The influence of gestational diabetes on neurodevelopment of children in the first two years of life: a prospective study. PLoS One [Internet]. 2016 Jan [cited 2017 Feb 6];11(9):e0162113. Available from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=5014336&tool=pmcentrez&rendertype=abstract
Haghir H, Hami J, Lotf N, Peyvandi M, Ghasemi S, Hosseini M. Expression of apoptosis-regulatory genes in the hippocampus of rat neonates born to mothers with diabetes. Metab Brain Dis. 2017:1–12.
Plum L, Schubert M, Brüning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol Metab [Internet]. 2005 [cited 2016 Nov 15];16(2):59–65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15734146 .
Wan Q, Xiong ZG, Man HY, Ackerley CA. Recruitment of functional GABA A receptors to postsynaptic domains by insulin. Nature. 1997;388(August):686–90.
Xie L, Sovari AA, Tran DX, Morita N, Xie F, Karagueuzian H, et al. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of AB oligomers. Proc Natl Acad Sci [Internet]. 2009 22 [cited 2016 Dec 20];106(18):7678–7678. Available from http://www.pnas.org/cgi/doi/10.1073/pnas.0901917106
Chiu S-L, Chen C-M, Cline HT. Insulin receptor signaling regulates synapse number, dendritic plasticity and circuit function in vivo. Neuron. 2011;58(5):708–19.
Park C, Seeley R, Craft S, Woods S. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol Behav [Internet]. 2000;68(4):509–14. Available from http://linkinghub.elsevier.com/retrieve/pii/S0031938499002206
Silverman BL, Metzger BE, Cho NH, Loeb CA. Impaired glucose tolerance in adolescent offspring of diabetic mothers. Diabetes Care. 1995;18(5):611–7.
Ross MG, Desai M, Khorram O, Mcknight RA, Lane RH, Torday J. Gestational programming of offspring obesity: a potential contributor to Alzheimer’s disease. Curr Alzheimer Reasearch. 2007;4:213–7.
Lourenco M V, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab [Internet]. 2013 [cited 2015 Feb 4];18(6):831–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24315369 .
Arnold SE, Lucki I, Brookshire BR, Carlson GC, Browne C A, Kazi H, et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis [Internet]. Elsevier Inc.; 2014 Jul [cited 2017 Jan 15];67:79–87. Available from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4083060&tool=pmcentrez&rendertype=abstract
Leloup C, Magnan C, Alquier T, Mistry S. Intrauterine hyperglycemia increases insulin binding sites but not glucose transporter expression in discrete brain areas in term rat fetuses. Pediatr Res. 2004;56(2):263–7.
Adams OP. The impact of brief high-intensity exercise on blood glucose levels. Diabetes Metab Syndr Obes [Internet]. 2013 Jan [cited 2014 Dec 15];6:113–22. Available from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3587394&tool=pmcentrez&rendertype=abstract
Nicholas LM, Morrison JL, Rattanatray L, Ozanne SE, Kleemann DO, Walker SK, et al. Differential effects of exposure to maternal obesity or maternal weight loss during the periconceptional period in the sheep on insulin signalling molecules in skeletal muscle of the offspring at 4 months of age. PLoS One [Internet]. 2013 Jan [cited 2016 Nov 12];8(12):e84594. Available from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3873457&tool=pmcentrez&rendertype=abstract
Maurer MH, Geomor HK, Bürgers HF, Schelshorn DW, Kuschinsky W. Adult neural stem cells express glucose transporters GLUT1 and GLUT3 and regulate GLUT3 expression. FEBS Lett [Internet]. 2006 [cited 2017 Feb 9];580(18):4430–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16854415 .
Freeman L, Zitlin V, Stevens C, Granholm A-C. Diet induced effects on neuronal and glial elements in the middle-aged rat hippocampus. Nutr Neurosci. 2011;14(1):32–44.
Harik N, Harik S, Kuo N-T, Sakai K, Przybylski R, LaManna J. Time-course and reversibility of the hypoxia-induced alterations in cerebral vascularity and cerebral capillary glucose transporter density. Brain Res. 1996;737:335–8.
Eidelman AL, Samueloff A. The pathophysiology of the fetus of the diabetic mother. Semin Perinatol. 2002;26(3):232–6.
Curristin SM, Cao A, Stewart WB, Zhang H, Madri JA, Morrow JS, et al. Disrupted synaptic development in the hypoxic newborn brain. Proc Natl Acad Sci U S A [Internet]. 2002 26 [cited 2017 Feb 5];99(24):15729–34. Available from http://www.pnas.org/cgi/content/short/99/24/15729
Mcquillen PS, Sheldon RA, Shatz CJ, Ferriero DM. Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci. 2003;23(8):3308–15.
Buzina-suboticanec K, Buzina R, Stavljenic A, Tadinac-babic M. Effects of iron supplementation on iron nutrition status and cognitive functions in children. Food Nutr Bull. 1998;19(4):298–306.
Petry C, Eaton M, Wobken J, Mills MM, Johnson D, Georgieff M. Iron deficiency of liver, heart, and brain in newborn infants of diabetic mothers. J Pediatr. 1992;121:109–14.
Pritchard WS. Cognitive event-related potential correlates of schizophrenia. Psychol Bull. 1986;100(1):43–66.
Beard JL, Felt B, Schallert T, Burhans M, Connor JR, Georgieff MK. Moderate iron deficiency in infancy: biology and behavior in young rats. Behav Brain Res [Internet]. 2006 [cited 2017 Feb 5];170(2):224–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16569441 .
Vaughn J, Brown J, Carter JP. The effects of maternal anemia on infant behavior. J Natl Med Assoc. 1986;78(10):963–8.
Kinalski M, Telejko B, Kuz M, Kre A, Kinalska I. Tumor necrosis factor alpha system and plasma adiponectin concentration in women with gestational diabetes. Horm Metab Res. 2005;37:450–4.
Perry VH, Nicoll J A R, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol [Internet]. Nature Publishing Group; 2010 [cited 2016 Sep 22];6(4):193–201. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20234358 .
Barker AJ, Ullian EM. New roles for astrocytes in developing synaptic circuits. Commun Integr Biol 2008;207–11, 1, 211.
Tomassoni D, Nwankwo IE, Gabrielli MG, Bhatt S, Muhammad AB, Lokhandwala MF, et al. Astrogliosis in the brain of obese Zucker rat: a model of metabolic syndrome. Neurosci Lett [Internet]. Elsevier Ireland Ltd. 2013;543:136–41. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23545209
Gehrmann J, Matsumoto Y, Kreutzberg GW. REVIEWS Microglia: intrinsic immuneffector cell of the brain. Brain Res Rev. 1995;20:269–87.
Paolicelli RC, Gross CT. Microglia in development: linking brain wiring to brain environment. Neuron Glia Biol [Internet]. 2011;7(1):77–83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22857738
Fein A, Kostina E, Savion S, Orenstein H, Shepshelovich J, Torchinsky A, et al. Expression of tumor necrosis factor-a in the pregnant uterus of diabetic mice: effect of maternal immunopotentiation. Am J Reprod Immunol. 2001;46:161–8.
Vuong B, Odero G, Rozbacher S, Stevenson M, Kereliuk SM, Pereira TJ, et al. Exposure to gestational diabetes mellitus induces neuroinflammation, derangement of hippocampal neurons, and cognitive changes in rat offspring. J Neuroinflammation. J Neuroinflammation. 2017:1–13.
Cuffe JSM, Xu C, Anthony V. Biomarkers of oxidative stress in pregnancy complications. Biomark Med. 2017;11:295–306.
Toljic M, Egic A, Munjas J, Orlic NK, Milovanovic Z, Radenkovic A, et al. Increased oxidative stress and cytokinesis-block micronucleus cytome assay parameters in pregnant women with gestational diabetes mellitus and gestational arterial hypertension. Reprod Toxicol [Internet]. Elsevier Inc.; 2017; Available from https://doi.org/10.1016/j.reprotox.2017.04.002
Usluoğullari B, Usluogullari CA, Balkan F, Orkmez M. Role of serum levels of irisin and oxidative stress markers in pregnant women with and without gestational diabetes Role of serum levels of irisin and oxidative stress markers in pregnant women with and without gestational diabetes. Gynecol Endocrinol [Internet]. Informa UK Limited, trading as Taylor 8 Francis Group; 2017;0(0):-000. Available from https://doi.org/10.1080/09513590.2017.1284789
Li H, Yin Q, Li N, Ouyang Z, Zhong M. Plasma markers of oxidative stress in patients with gestational diabetes mellitus in the second and third trimester. Obstet Gynecol Int Hindawi Publishing Corporation. 2016;2016:1–8.
Ho E, Galougahi K, Liu C, Bhindi R, Figtree GA. Biological markers of oxidative stress: applications to cardiovascular research and practice. Redox Biol [Internet]. Elsevier. 2013;1(1):483–91. https://doi.org/10.1016/j.redox.2013.07.006.
Aziz HSA, John CM, Yusof NIMS, Nordin M, Ramasamy R, Adam A, et al. Animal model of gestational diabetes mellitus with pathophysiological resemblance to the human condition induced by multiple factors ( nutritional, pharmacological, and stress ) in rats. Biomed Hindawi Publishing Corporation; 2016;2016.
Davies KJA. Oxidative stress: the paradox of aerobic life. Buochem Soc Symp. 1995;61:1–31.
Stanner SA, Hughes J, Kelly CNM, Buttriss J. A review of the epidemiological evidence for the “antioxidant hypothesis.”. Public Health Nutr. 2017;7:407–22.
Sena E, Wheble P, Sandercock P, Macleod M. Systematic review and meta-analysis of the efficacy of tirilazad in experimental stroke. Stroke. 2007:388–95.
Bannister J, Bannister W, Rotilio G. Aspects of the structure, function, and applications of superoxide dismutase. Crit Rev Biomed. 1987;22:111–80.
Chelikani P, Fita I, Loewen P. Diversity of structures and properties among catalases. Cell. 2009;61:192–208.
Sedighi O, Makhlough A, Shokrzadeh M, Hoorshad S. Association between plasma selenium and glutathione peroxidase levels and severity of diabetic nephropathy in patients with type two diabetes mellitus. Nephro Urol Mon. 2014;6(5):10–3.
Renault KM, Carlsen EM, Nilas L, Secher NJ, Cortes D, Olsen SF, et al. Accepted article preview: published ahead of advance online publication. Int J Obes 2017;
Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia [Internet]. 2012 [cited 2014 Jul 12];55(6):1577–96. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22526604 .
Acknowledgments
Thanks to CAPES, CNPq, and FAPERJ for the support of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All procedures did not involve any human participants and/or animals, because it is a review article. This study is in accordance with the ethical standards of the national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
De Sousa, R.A.L. Gestational diabetes is associated to the development of brain insulin resistance in the offspring. Int J Diabetes Dev Ctries 39, 408–416 (2019). https://doi.org/10.1007/s13410-018-0618-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13410-018-0618-1