
Abstract
Background
MicroRNAs (miRNAs) are a class of non-coding, endogenous, small RNAs that negatively regulate gene expression by inducing degradation or translational inhibition of target mRNAs. Aberrant expression of miRNAs appears to be a common characteristic of hematological malignancies including leukemias.
Aim
Here we review the available data supporting a role of aberrant expression of miRNAs in the pathogenesis of leukemias including acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), chronic myeloid leukemia (CML), and chronic lymphocytic leukemia (CLL).
Conclusions
The expression signatures of miRNAs provide exciting opportunities in the diagnosis, prognosis, and therapy of leukemia. Since miRNAs can function as either oncogenes or tumor suppressor genes in leukemogenesis, the potential of using these small RNAs as therapeutic targets opens up new opportunities for leukemia therapy by either inhibiting or augmenting their activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
MicroRNAs (miRNAs or miRs) are a class of non-coding small RNAs of ~22 nucleotides that regulate expression of target genes at the post-transcriptional level [1, 2]. Since the discovery of the first miRNAs [3, 4], these small genes have added a new layer of complexity to the regulation of normal and pathological cell functions [5]. It is currently estimated that the human genome encodes more than 1,000 unique mature miRNAs, each of which may control several hundred target genes to regulate up to one-third of human transcripts (http://www.mirbase.org, Release 18.0, November 2011).
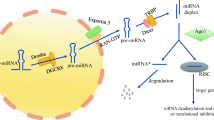
The biogenesis of miRNAs begins in the nucleus and is completed in the cytoplasm [6–8] (Fig. 1). MiRNAs recognize and target the 3’-untranslated region (3’-UTR) of specific mRNAs bearing a complementary target site. Depending on the degree of complementarity between mRNA target sites and the nucleotide sequence from position 2–8 at the 5’ end of miRNAs (the seed region), regulation of target gene expression occurs by one of two possible mechanisms: (i) Ago-catalyzed cleavage of target mRNA when the miRNA has a perfect (or near perfect) complementarity to that mRNA or (ii) repression of translation when the miRNA shows an imperfect complementarity to the target mRNA [9].

Schematic illustration of biogenesis, processing, and function of microRNA: The biogenesis of miRNAs begins in the nucleus with the synthesis of a relatively long double-stranded RNA molecule, known as pri-miRNA. The resulting pri-miRNA transcript is cleaved by Drosha and its interacting partner DGCR8, producing a characteristic stem loop precursor, pre-miRNA. The pre-miRNA is then transported to the cytoplasm by a protein complex consisting of Exportin-5 and Ran-GTPase. In the cytoplasm, the pre-miRNA undergoes its final processing step, which involves cleavage by Dicer and TRBP below the stem-loop. This produces a duplex molecule containing the mature miRNA (often designated miR) and its complementary miRNA*. The miRNA: miRNA* duplex is separated into two single-stranded nucleotide molecules by an RNA helicase; the miRNA* fragment is released and rapidly degraded in the cytoplasm, whereas the mature miRNA molecule binds to an Argonaute (Ago) protein and incorporates into the RNA-induced silencing complex (RISC), which is the active form that affects mRNA and its translation. Primary miRNA, pri-miRNA; precursor miRNA, pre-miRNA; Drosha, RNase III endonuclease; DGCR8, DiGeorge syndrome critical region 8; Dicer, RNase III endonuclease; Ago, Argonaute; RISC, RNA-induced silencing complex
MiRNAs are involved in the control of several cellular processes altered in cancer, such as proliferation, differentiation, and apoptosis [2, 10, 11]. Given this wide variety of functions, several miRNAs have emerged as candidate oncogenes and tumor suppressors involved in the networks specifically altered during cancer development and progression [12–14]. Since dysregulation of miRNAs has been shown in many types of solid tumors and leukemias [15–19], miRNAs may serve as novel clinically useful cancer biomarkers. We here summarize the current data from miRNA expression profiles in leukemia studies and the current knowledge on the role of aberrant expression of miRNAs in the pathogenesis of leukemias. In addition, the potential for therapy is discussed.
2 MicroRNAs in leukemias
2.1 MicroRNA expression in acute myeloid leukemia
Acute myeloid leukemia (AML) is a heterogeneous group of neoplastic hematopoietic diseases characterized by the accumulation of primitive myeloid cells arrested at early stages of differentiation. The hallmark of AML is the presence of one of many specific cytogenetic abnormalities being found in about 55 % of adult AML patients [20, 21]. The miRNA signatures were reported to be associated with specific cytogenetic translocations such as t(15;17), t(8;21), inv(16) or molecular abnormalities such as nucleophosmin (NPM1), CCAAT/enhancer binding protein alpha (C/EBPα) or internal tandem duplication of FMS-like tyrosine kinase 3 (FLT3-ITD) mutations [22] (Table 1).
Specific alterations in miRNA expression distinguish subtypes of AML and deregulation of specific miRNAs may play a role in the development of leukemias with specific genetic rearrangements. A quantitative expression profiling of 157 miRNAs in a cohort of 100 primary AML patients points toward a distinctive signature of AML bearing a t(15;17) translocation, including the up-regulation of miRNAs located in the human 14q32 imprinted domain. The set of miRNAs that was differentially expressed compared with normal hematopoietic tissue included miR-127, miR-154, miR-154*, miR-299, miR-323, miR-368, and miR-370, providing molecular signatures characteristic of the major translocation-mediated gene fusion events in AML [23]. Jongen-Lavrencic et al [24] identified the miRNA signatures characteristic of AMLs with the cytogenetic abnormalities t(15;17), t(8;21), and inv(16). A prominent signature identified in AML patients with t(15;17) displayed a strong up-regulation of miR-382, miR-134, miR-376a, miR-127, miR-299-5p, and miR-323. Two members of a known tumor suppressor miRNA family, let-7b and let-7c, were down-regulated in AML with t(8;21) and also in AML with inv(16). MiR-127 is another known tumor suppressor miRNA which was shown to be significantly down-regulated in AML associated with inv(16).
In a large-scale genome-wide miRNA profiling study of AML patients, Li et al [20] found that miR-126/126* was specifically overexpressed in both t(8;21) and inv(16) AMLs, rearrangements resulting in the disruption of Core Binding Factors (CBF), whereas miR-224, miR-368, and miR-382 were almost exclusively overexpressed in t(15;17) AMLs. They found that the overexpression of miR-126/126* in CBF AMLs was associated with partial promoter demethylation of the CpG island in which miR-126/126* is embedded, but not with amplification or mutation of the genomic locus. Also, this study revealed that miR-126 inhibited apoptosis and increased the viability of AML cells, and enhanced the proliferation of mouse normal bone marrow (BM) progenitor cells alone and in cooperation with the t(8;21) fusion gene, likely through targeting Polo-like kinase 2 (PLK2), a tumor suppressor.
Since the expression levels of miR-223 steadily increase during myelopoiesis and the suppression of this increase blocks granulocytic maturation, it seems that miR-223 is an important player in myeloid differentiation [25]. A regulatory circuitry comprised of miR-223 and two transcription factors NFI-A and C/EBPα has been proposed to regulate human granulopoiesis. The two transcription factors modulate the expression levels of miR-223 through competitive binding to the pre-miR-223 promoter. NFI-A maintains miR-223 at low levels, whereas its replacement by retinoic acid (RA)-induced C/EBPα results in miR-223 up-regulation and enhanced granulocytic differentiation of leukemic cells. Interestingly, miR-223 represses NFI-A translation, creating a negative feedback loop that favors granulocytic differentiation [25, 26]. To investigate the possibility that leukemia fusion proteins could epigenetically inhibit the expression of miR-223, Fazi et al [27] analyzed patient’s primary leukemia blasts and demonstrated a down-regulation of miR-223 in AML blasts harboring the chromosomal translocation t(8;21) generating the AML1/ETO (AE) fusion product. This study identified miR-223 as a direct transcriptional target of the AE fusion protein and showed that the expression of AE induces epigenetic silencing of the myelopoiesis regulator miR-223 through the recruitment of chromatin remodeling enzymes. Ectopic expression of miR-223, down-regulation of AE protein levels, or the use of demethylating agents enhanced miR-223 expression levels and restored differentiation of leukemic blasts. These findings support the possibility that the epigenetic silencing of miR-223 may be associated with a differentiation block of myeloid precursors underlying leukemogenesis.
It was also shown that when miR-223 was overexpressed, acute promyelocytic leukemia (APL) patient-derived NB4 cells carrying the t(15:17) chromosomal translocation and expressing the PML/RARα fusion product, were able to undergo granulocytic differentiation [26]. Interestingly, ectopic expression of miR-223 induced granulocytic differentiation in the human myeloblastic HL60 cell line, which does not carry oncogenic fusion products [27]. Overall, these findings indicate that miR-223 plays an important role during granulopoiesis and point to the ability of miR-223 to reprogram myeloid differentiation in distinct leukemia subtypes independently from the presence of a specific genetic lesion.
To assess the function of miR-223 in an in vivo context, Johnnidis et al [28] engineered a loss of function allele by excising the miR-223 gene in mice and found, rather surprisingly, that these miR-223-deficient mice exhibit granulocytosis and a hyperinflammatory state. They also showed that MEF2C, a transcription factor that promotes myeloid progenitor proliferation, is a target of miR-223, and that genetic ablation of MEF2C suppresses progenitor expansion and corrects the neutrophilic phenotype in miR-223 null mice. Therefore, miR-223 seems to act as a negative regulator of progenitor cell proliferation and granulocyte differentiation and activation. These data are not consistent with the previous findings indicating that miR-223 is a positive regulator of granulocytic differentiation [26]. This apparent contradiction could be explained on the basis of different strategies used in each study, i.e., overexpression strategies used by ref. 26 could have different effects compared with those that are extrapolated from the knockout studies [28]. In ref. 26 the expression levels of miR-223 was manipulated in a leukemic cell line resembling an early granulocyte progenitor, whereas the complete abrogation of miR-223 expression in ref. 28 led to an extreme experimental situation in which miR-233 was absent in the entire granulocytic lineage in knockout mice. Additionally, it should be noted that, in real nature, genes are rather expressed in tightly controlled graded levels and small changes in their expression may trigger distinct biological functions [29]. This is supported by the observation that different miR-223 functions during different stages of myeloid cell development are attributed to different concentrations of miR-223 [28].
The t(2;11)(p21;q23) chromosomal translocation, specifically observed in patients with AML and myelodysplastic syndrome, entails an elevated expression of miR-125b (from 6- to 90-fold). Bousquet et al [30] reported that in vitro transfection of miR-125b prevented primary human CD34+ cell differentiation and also blocked the myelomonocytic differentiation of HL60 and NB4 leukemic cell lines upon chemical treatment. Therefore, miR-125b up-regulation may account for the differentiation block observed in leukemic cells in vivo.
The miR-17-92 cluster was confirmed to be involved in the development of MLL-rearrangement AMLs (M4/M5, with the majority of leukemic cells being monoblasts) [20]. Fontana et al [31] showed that three miRNAs (miR-17-5p, miR-20a, and miR-106a) control monocytopoiesis through AML1 targeting, leading to M-CSF receptor down-regulation, enhanced blast proliferation and inhibition of monocytic differentiation and maturation. Mi et al [32] showed that the miR-17-92 cluster was aberrantly overexpressed in MLL-rearranged acute leukemias. They observed that forced expression of this miRNA cluster could significantly enhance the viability and inhibit the apoptosis of human HeLa and 293 T cells, and more importantly, increase proliferation and inhibit differentiation of mouse normal BM progenitor cells alone and in cooperation with MLL fusions, leading to transformation of the cells. They identified 363 potential miR-17-92 target genes whose expression was inversely correlated with this miRNA expression. Gene Ontology revealed that these potential target genes are significantly enriched in pathways/networks related to cell differentiation, particularly hematopoiesis (including both myeloid and B-cell differentiation), cell cycle, and apoptosis.
Wong et al [33] showed that the miR-17-92 cluster regulates leukemia stem cell (LSC) potential in a mouse model of MLL-associated AML through direct suppression of the cyclin-dependent kinase inhibitor p21. Cluster expression was significantly reduced upon the exit of LSCs from the self-renewing compartment, whereas forced expression blocked myeloid leukemia cell differentiation, enhanced proliferation, and significantly decreased the latency for MLL leukemia development. Knockdown of p21 in MLL-transformed cells phenocopied the expression of the miR-17-92 cluster, validating p21 as a physiologic and direct in vivo target of the miR-17-92 cluster in regulating LSC frequency and accelerating MLL leukemia.
MiR-196b, located between the homeobox A9 (HOXA9) and HOXA10 genes, is another miRNA which has been found to be specifically overexpressed in AML patients with MLL rearrangements [20, 23, 24, 34, 35]. Popovic et al [35] demonstrated that expression of miR-196b is induced by leukemogenic MLL fusion proteins, and that overexpression of miR-196b in normal BM hematopoietic progenitor cells leads to an increase in proliferation and survival capacity, as well as a partial block of myeloid cell differentiation. Thus, miR-196 overexpression seems to be an important event in the development of leukemias caused by MLL fusion proteins.
In a follow-up study, Marcucci et al [36] reported a miRNA signature associated with the presence of the C/EBPα mutation in cytogenetically normal AML (CN-AML) patients. Up-regulated miR-181a and miR-181b were confirmed to be a part of a miRNA expression signature associated with C/EBPα mutations that predicted a favorable outcome in CN-AML. Since increased expression levels of miR-181a and miR-181b have been reported during erythroid differentiation [37], high expression levels of the members of the miR-181 family may contribute to the partial erythroid differentiation reported in leukemic blasts harboring C/EBPα mutations [36].
The expression signature of some miRNAs has been found to be associated with clinical outcome and survival of patients with leukemia [38]. In a high-risk subgroup of CN-AMLs (younger than 60 years with molecular features such as FLT3-ITD, NPM1 or both), miRNA expression profiling revealed a miRNA signature that was associated with event-free survival. The prognostic signature included miR-181a and miR-181b, which were inversely associated with the risk of an event (failure to achieve complete remission, relapse, or death) and miR-124, miR-128-1, miR-194, miR-219-5p, miR-220a, and miR-320, which were positively associated with the risk of an event. There was an inverse correlation between expression levels of the miR-181 family and expression levels of predicted target genes involved in mechanisms of innate immunity including genes encoding toll-like receptors and interleukin-1β. It is thought that down-regulation of miRNAs of the miR-181 family contributes to an aggressive leukemia phenotype through mechanisms associated with the activation of specific innate immunity pathways. Taken together, these findings point toward a miRNA signature in molecularly defined, high-risk CN-AML that is associated with clinical outcome and with target genes encoding proteins involved in specific innate immunity pathways [39]. By analyzing a large set of AML patients with predominantly intermediate and poor prognosis, Garzon et al [34] found that miRNA expression was closely correlated with specific cytogenetic and molecular abnormalities, such as t(11q23), isolated trisomy 8, and FLT3-ITD mutations. Moreover, high expression levels of miR-191 and miR-199a were found to be unfavorably associated with overall and event-free survival unfavorably. Significant correlation of miR-191 and miR-199a with survival highlights the prognostic value of this small subset of miRNAs.
NPM1 mutations are primary events that precede acquisition of FLT3-ITD or other mutations and occur in association with FLT3-ITD mutations in approximately 35 % of AML patients [24, 40]. The analysis of these two mutations is most commonly used for outcome prediction of CN-AML patients. Uncovering the role of miRNAs in patients carrying NPM1 and FLT3-ITD revealed a signature of up-regulation of miR-10a, miR-10b as well as several members of the let-7 and miR-29 families, distinguishing NPM1-mutated from NPM1-unmutated cases [41]. Several studies on the clinical impact in AML subgroups revealed that the subset of AML carrying NPM1 mutations without concomitant FLT3-ITD is a prognostically favorable subgroup and can be classified in the molecular low-risk CN-AML group [24, 42].
Statistical analysis of miRNA microarray data revealed a prominent miRNA signature in AML patients with NPM1 mutations. This miRNA expression signature includes the up-regulation of miR-10a, miR-10b, miR-196a, and miR-196b, all of which reside in a genomic cluster of HOX genes [24]. Because miR-196a directly targets HOXB8 transcript [43], an aberrant regulatory circuit including NPM1, HOX genes, and miRNAs might be considered in the development of NPM1-mutated AML [24]. Also, Garzon et al [41] were able to find down-regulation of miR-204 and miR-128a as an additional miRNA expression signature in NPM1-mutated AML patients. They showed that miR-204 repressed expression of HOXA10 and myeloid ecotropic viral integration site 1 (MEIS1), two members of the HOX gene cluster. This would suggest that HOX up-regulation observed in NPM1-mutated AML is attributed, at least in part, to the loss of HOX regulator miRNAs.
It has been found that miR-155 is overexpressed in a subset of AML patients (particularly, monocytic FAB types M4 and M5), and that enforced overexpression of miR-155 in normal mouse hematopoietic stem cells causes a myeloproliferative disorder [44]. Two groups reported independently that miR-155 was up-regulated in AML patients harboring FLT3-ITD mutation, suggesting a role of this miRNA in the highly proliferative phenotype of this subset of AML [24, 34]. However, blocking FLT3 signaling using a potent FLT3 inhibitor or overexpressing FLT3-ITD in mouse myeloid precursor cells had no significant effect on miR-155 expression. Thus, up-regulation of miR-155 in AML samples seems to be independent from FLT3 signaling [41]. Taken together, these data suggest that targeting miR-155 with agents such as miR-155-specific antagomirs in combination with FLT3-ITD inhibitors may provide a beneficial treatment option for this subset of AML patients [22].
2.2 MicroRNA expression in acute lymphoblastic leukemia
Acute lymphoblastic leukemia (ALL), one of the most common malignancies observed in pediatric leukemia patients, originates from the clonal proliferation of lymphoid progenitor cells in BM and is characterized by recurring genetic abnormalities including chromosomal aneuploidies and a number of chromosomal rearrangements [45].
In a large-scale genome-wide analysis of 17 ALL and 52 AML cases, Mi et al [46] observed miRNA expression signatures which accurately discriminate ALL from AML. Of the 27 differentially expressed miRNAs (6 up-regulated and 21 down-regulated in ALL compared with AML) (see Table 2), four (let-7b, miR-128a, miR-128b, and miR-223) were most discriminatory with a diagnostic accuracy of 97–99 %. Among these miRNAs, miR-128a and miR-128b were significantly up-regulated, whereas let-7b and miR-223 were significantly down-regulated in ALL compared with AML. A genome-wide miRNA expression profiling of pediatric acute leukemia conducted by Zhang et al [47] showed that the most highly expressed miRNAs in pediatric ALL were miR-34a, miR-128a, miR-128b, and miR-146a, while highly expressed miRNAs in pediatric AML were miR-100, miR-125b, miR-335, miR-146a, and miR-99a, which were significantly different from those observed in adult AML cases [34, 41, 46]. This pediatric-specific miRNA pattern indicates that the regulatory networks between pediatric and adult acute leukemias may be significantly different. Also, miRNA profiles revealed an expression pattern characterized by high expression levels of miR-7, miR-198, and miR-663 and low expression levels of miR-126, miR-222, miR-551a, and miR-345 in pediatric ALL with central nervous system (CNS) relapse compared to non-CNS relapsed ALL. This miRNA cascade may serve as a clinically useful biomarker for predicting pediatric ALL associated with CNS relapse [47].
Schotte et al [48] identified an aberrant expression of 19 selected miRNAs in ALL patients. Quantification of miRNAs in ALL samples compared to normal CD34+ cells revealed that 14 miRNAs (miR-128a, miR-142-3p, miR-142-5p, miR-150, miR-151-5p, miR-181a, miR-181b, miR-181c, miR-193a, miR-30e-5p, miR-34b, miR-365, miR-582, and miR-708) were up-regulated, whereas five miRNAs (miR-100, miR-125b, miR-99a, miR-196b, and let-7e) were down-regulated. Among eight miRNAs differentially expressed between MLL and non-MLL precursor B-ALL samples, miR-708 was from 250-fold up to 6,500-fold more highly expressed in 57 TEL-AML1, BCR-ABL1, E2A-PBX1, hyperdiploid and other B-ALL samples compared with 20 MLL-rearranged and 15 T-ALL samples. On the contrary, the expression of miR-196b showed a 500-fold increase in MLL-rearranged and an 800-fold increase in 5 of 15 T-ALL samples compared to other B-ALL samples.
MiRNA expression profiles of ALL samples compared with control CD19+ B-cells revealed that the five most highly expressed miRNAs were miR-128b, miR-204, miR-218, miR-331, and miR-181b-1. The most represented miRNA in ALL samples was miR-128b, which was 436.5-fold higher compared with normal CD19+ B-cells. The four miRNAs with the lowest expression levels in ALL samples were miR-135b, miR-132, miR-199 s, miR-139, and miR-150 [49] (Table 2). The miR-17-92 cluster was also found to be up-regulated in ALL samples [49], as previously reported for a range of hematopoietic malignancies, particularly some types of lymphomas [50]. Because the miR-17-92 cluster targets many genes involved in apoptotic pathways and favors the survival of B-cell progenitors [51], the aberrant expression of miRNA members of the cluster might be considered as a possible mechanism in the development of some types of leukemias and lymphomas.
Enforced expression of the miR-17-92 cluster in the hematopoietic system accelerated disease onset and progression in a transgenic mouse model of B-cell lymphoma [52]. This suggests that dysregulation of the miR-17-92 cluster contributes to lymphomagenesis by repressing tumor suppressor gene(s) [53]. The proapoptotic protein BIM is the most likely target of the miR-17-92 cluster in lymphomagenesis [54]. By establishing a strain of miR-17-92 knockout mice, Ventura et al [51] demonstrated that the loss of miR-17-92 led to elevated BIM protein levels and, subsequently, inhibition of B-cell development. This suggests that down-regulation of BIM mRNA and protein levels induced by miR-17-92 overexpression might be considered as a possible anti-apoptotic mechanism in lymphomagenesis [55]. PTEN is also a known target of the miR-17-92 cluster, and its dysregulation also might contribute to lymphomagenesis [56]. In murine lymphoma and leukemia models, the oncogenic activity of the miR-17-92 cluster was found to be exerted by miR-19-mediated down-regulation of PTEN and suppression of apoptosis [57, 58]. By generating transgenic mice with elevated miR-17-92 expression in lymphocytes, Xiao et al [56] demonstrated that miR-17-92 encoded miRNAs suppressed expression of two tumor suppressors, PTEN and BIM, as functionally important targets. It seems that down-regulation of PTEN and BIM protein expression by miR-17-92 miRNAs contributes to the lymphoproliferative and autoimmunity phenotype observed in miR-17-92 transgenic mice, and to lymphoma development in patients carrying amplifications of the miR-17-92 cluster.
Inomata et al [53] revealed that silencing of two miR-17-92 encoded miRNAs (miR-17 and miR-20a) in Jeko-1 cells derived from mantle cell lymphoma led to up-regulation of CDKN1A/p21, resulting in G1-S arrest and decreased cell growth. On the contrary, upon transfection of miR-17-19b-1 (a miR-17-92 variant), expression of p21, but not BIM, was suppressed in SUDHL4 cells, which were derived from diffuse large B-cell lymphoma (DLBCL) with aberrant BCL-2 overexpression. Also, suppression of both BIM and p21 expression levels was observed in transfected Raji cells, which were derived from a Burkitt lymphoma with aberrant c-MYC overexpression. These results revealed that CDKN1A/p21 is likely an essential target of miR-17-92 during B-cell lymphomagenesis, and that the miR-17-92 cluster down-regulates expression of distinct targets in different B-cell lymphoma subtypes.
2.3 MicroRNA expression in chronic myeloid leukemia
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder arising from neoplastic transformation of a single hematopoietic stem cell. The hallmark of the disease, in >95 % of the patients, is the presence of a Philadelphia (Ph) chromosome which arises from a reciprocal translocation t(9; 22) (q34; q11) that, in turn, creates a BCR-ABL fusion oncogene [59, 60]. The BCR-ABL oncogene produces a constitutively active tyrosine kinase recruiting and activating several molecular pathways that ultimately lead to abnormal cellular adhesion, enhanced proliferation, and inhibition of apoptosis [60, 61].
CML is associated with up-regulation of the miR-17-92 cluster, which is transcriptionally regulated by c-MYC [62]. The miR-17-92 cluster can also be induced by members of the E2F family of transcription factors [63], while miR-17-5p and miR-20a, two miRNAs in the cluster, directly target E2F1 in a negative feedback loop of transcriptional regulation [64]. E2F1 is induced by c-MYC and creates a reciprocal positive feedback loop by inducing c-MYC expression [64]. Owing to c-MYC-induced expression of the miR-17-92 cluster, it seems that this autoregulatory feedback loop may provide fine tuning to control the opposing proliferative and apoptotic functions of E2F and that interaction between the cluster and c-MYC modulates E2F1 expression [63–65].
Venturini et al [66] found that the miR-17-92 cluster is overexpressed in CML CD34+ cells from patients in chronic phase, but not in blast crisis, compared with normal CD34+ cells. They also showed that treatment of human myeloid cell line K562 with imatinib, anti-BCR-ABL RNA interference or anti-c-MYC RNA interference resulted in the down-regulation of miR-17-92. These data demonstrated that the expression of miR-17-92 miRNAs in CML cell lines depend on BCR-ABL tyrosine kinase activity and add miRNAs to the signaling network affected by the BCR-ABL oncoprotein. Additionally, miR-203, which is genetically and epigenetically silenced in CML, controls BCR-ABL oncogene expression. Re-expression of miR-203 reduces ABL1 and BCR-ABL fusion protein levels and dramatically inhibits the proliferation of tumor cells in an ABL1-dependent manner. Thus, miR-203 is able to modulate the expression of tumor-specific translocation proteins [67].
Although the BCR-ABL oncoprotein up-regulates the expression of oncogenic miRNAs and down-regulates tumor suppressor miRNAs favoring leukemic transformation, there is also convincing evidence that points to abnormal expression of miRNAs independent of BCR-ABL activity. Agirre et al [68] identified an abnormal miRNA expression profile in CD34+ and mononuclear cells from patients with CML compared with healthy controls. Expression analysis of 157 miRNAs revealed that miR-10a, miR-150, and miR-151 were down-regulated, whereas miR-96 was up-regulated in both CD34+ and BM mononuclear cells of CML patients at diagnosis compared to healthy controls. Interestingly, this study showed that down-regulation of miR-10a was not dependent on BCR-ABL activity and resulted in increased upstream stimulator factor 2 transcription factor (USF2)-mediated cell growth of CML cells, supporting the potential role of a miRNA in the abnormal behavior of CML.
The BCR-ABL tyrosine kinase inhibitor imatinib is the first-line therapy for newly diagnosed CML. To evaluate whether imatinib treatment of CML patients can normalize characteristic miRNA expression profiles. Flamant et al [69] determined the repertoire of miRNAs expressed in leukemic cells from newly diagnosed patients with CML, prior to and within the first 2 weeks during imatinib therapy. They observed a significantly increased expression of miR-150 and miR-146a and a decreased expression of miR-142-3p and miR-199b-5p in peripheral blood mononuclear cells of CML patient after 2 weeks of imatinib therapy. The aberrant expression levels of these miRNAs were tending towards normal levels after 2 weeks of imatinib therapy. Despite the clinical success obtained with the use of imatinib, approximately 20–25 % of patients do not respond to the therapy, owing to intolerance or drug resistance. San José-Enériz et al [70] reported a distinct signature consisting of 19 miRNAs which were differentially expressed between imatinib-resistant and -responder CML patients: 18 of them were down-regulated (miR-7, miR-23a, miR-26a, miR-29a, miR-29c, miR-30b, miR-30c, miR-100, miR-126, miR-134, miR-141, miR-183, miR-196b, miR-199a, miR-224, miR-326, miR-422b, and miR-520a) while only one was up-regulated (miR-191) in imatinib-resistant CML patients. Among the predicted targets of these miRNAs are several membrane transporters that belong to the ATP binding cassette superfamily of transmembrane transporters, which have been implicated in resistance to chemotherapy. Taken together, these findings put more emphasis on the significance of miRNA profiling to predict clinical resistance to imatinib in patients with newly diagnosed CML (Table 3).
2.4 MicroRNA expression in chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL), the most common type of leukemia among adults, arises from a malignant clone of B-cells and is characterized by multiple and recurrent chromosomal abnormalities, of which deletions in chromosome 13q (del13q14) occur in more than 50 % of all CLL cases. Other chromosomal alterations include 11q deletions (11q-; 18 %), trisomy of chromosome 12 (12 %), and 17p deletions (17p-; 7 %) [71].
The first evidence for the involvement of miRNAs in hematologic malignancies was described in CLL. The miR-15a/miR-16-1 cluster resides at chromosome 13q14.3, a genomic region frequently lost or down-regulated in B-cell CLLs [72]. A 10 kb minimally-deleted region (MDR) which contains the deleted in leukemia (DLEU) 2 gene as well as the miR-15a/miR-16-1 cluster has been identified in 13q14. Klein et al [73] generated transgenic mice that either lacked the MDR (containing both DLEU2 and miR-15a/miR-16-1) or two miRNA genes only. The deletion of the MDR caused B-cell lymphoproliferative disorders, recapitulating the spectrum of CLL-associated phenotypes, including monoclonal B-cell lymphocytosis (MBL), the presumed low-penetrance premalignant stage, classic CLL/small cell lymphocytic leukemia (SLL), and the low-penetrance aggressive progression stage of diffuse large B-cell lymphoma (DLBCL). The significantly more aggressive phenotype displayed by the MDR-deleted mice compared to mice with the miR-15a/miR-16-1 deletion raises the possibility that 13q14 deletions of different size (including or excluding the DLEU5 locus), may affect the clinical behavior of CLL in humans. These findings are in line with the previous observation that miR-15a/miR-16-1 deletion was associated with the indolent form of disease [72]. Calin et al [71] identified germ-line and somatic mutations in the primary precursors of miR-15a and miR-16-1 in approximately 10 % of CLL patients. A similar point mutation adjacent to the miR-16-1 region in the New Zealand Black (NZB) mouse model of human CLL has been associated with a low expression level of miR-16-1, and mice harboring this mutation developed a B-cell lymphoproliferative disease highly reminiscent of CLL [74]. MiR-16-1 expression was decreased in subpopulations of NZB B-cells, and overexpression of miR-16 in an NZB-derived malignant B-1 cell line led to cell cycle alterations and increased apoptosis. This suggests that altered expression of the miR-15a/miR-16-1 cluster is an important molecular lesion in CLL and that these miRNAs may be targets for therapeutic efficacy in this disease [74, 75].
Among direct targets of the miR-15a/miR-16-1 cluster, a number of mRNAs encoding gene products critically involved in regulating cell proliferation and apoptosis have been identified. Most remarkably, the target genes include several proliferation-associated genes such as cyclins (CCND1 and CCND3) and cyclin-dependent kinases (CHK6) as well as genes involved in apoptosis (BCL-2) [76]. Mir-15a and miR-16-1 are severely down-regulated in 70 % of patients with CLL and overexpression of this pair of miRNAs induces apoptosis by targeting the anti-apoptotic gene BCL-2 [77]. Calin et al [78] performed a high-throughput profiling of genes modulated by the miR-15a/16-1 cluster in a leukemic cell line model (MEG-01) and in primary CLL samples and identified a signature of common genes whose silencing characterizes the miR-15a/miR-16-1-induced phenotype in CLL. They proved that the miR-15a/miR-16-1 cluster targets several cancer-related genes (such as MCL-1, BCL-2, JUN, MSH-2, or WT-1) that directly or indirectly affect apoptosis and cell cycle regulation. Also, it has been shown that miR-16-1 negatively regulated cellular growth and cell cycle progression of human mantle cell lymphomas by targeting CCND1, a cell cycle regulator which promotes G1 to S-phase progression [79]. Linsley et al [80] showed that miR-16-down-regulated transcripts were enriched with cell cycle regulatory genes (CDK6, CDC27, CARD10, and C10orf46) whose silencing caused an accumulation of cells in G0/G1. Simultaneous siRNA-mediated silencing of these genes was more effective in blocking cell cycle progression than silencing of any of the genes individually. Thus, miR-16 coordinately regulates cell cycle regulators that may function synergistically to control cell cycle progression.
MiR-155 maps within, and is processed from, an exon of the non-coding RNA known as B-cell integration cluster (BIC), and has been shown to be up-regulated in different human B-cell neoplasms, suggesting a role for miR-155 in B-cell lymphomagenesis [81, 82]. MiRNA expression profiling of 56 CLL patients revealed that miR-155 was dramatically overexpressed in almost every patient analyzed, suggesting that miR-155 overexpression is a general characteristic of CLL [82]. Several studies showed that vast overexpression of miR-155 under the Eμ-myc promoter resulted in the development of B-cell malignancies in mice [83–87]. Costinean et al [83] showed that the transgenic mice model of CLL carrying a miR-155 transgene whose expression is targeted to B-cells (Eμ-mmu-miR-155) exhibited initially a polyclonal preleukemic pre-B-cell proliferation evident in the spleen and BM, followed by a frank B-cell malignancy. This model demonstrates a role for miR-155 in the development of B-cell malignancies, favoring the capture of secondary genetic changes for full transformation, and suggests that miR-155 is directly involved in the induction of polyclonal B-cell expansion.
The miR-34 family and particularly miR-34a as direct, conserved p53 target genes have been shown to mediate some of the p53-dependent effects including apoptosis, cell cycle arrest, and senescence [88–91]. Previous publications showed that miR-34a depends on the intactness of the p53 pathway to exert its functional effects through direct targeting CDK4, CDK6, CCND1, CCNE2, MET, BCL2, and E2F transcription factor 3 [92–95]. Therefore, despite being transcriptionally induced by p53, miR-34a creates a positive feedback loop by affecting factors upstream of p53 that in turn lead to p53 activation [89, 95, 96]. It has been shown that CLLs, with TP53 mutations and 17p deletions are resistant to chemotherapy. One study showed that 17p13/P53 deletion was associated with down-regulation of miR-34a expression in patients with B-CLL [97]. This finding would suggest that miR-34a is a part of the resistance network in CLL. A large cohort study has revealed that low expression of miR-34a in CLL is associated with p53 inactivation, chemotherapy-refractory disease, impaired DNA damage response, and apoptosis resistance irrespective of 17p deletion/TP53 mutation, suggesting that miR-34a may serve as a marker for poor prognosis for CLL [98].
Pekarsky et al [99] found that miR-29b and miR-181b act as tumor suppressors in aggressive CLL by targeting T-cell leukemia/lymphoma 1 (TCL-1), an oncogene that is overexpressed in CLL cases with 11q-. The expression levels of miR-29b and miR-181b were inversely correlated with TCL-1 expression levels in CLL patients, suggesting that TCL-1 is a target of both miRNAs [99]. High expression levels of TCL-1 are associated with high expression of a 70-kDa zeta-associated protein (ZAP-70) and an unmutated immunoglobulin heavy-chain variable-region (IgVH), factors that indicate a more aggressive CLL [100]. Supporting these findings, TCL-1 transgenic mouse models displayed a disease state closely resembling aggressive, treatment-resistant human CLL [101, 102].
To investigate whether the expression of miRNA genes predicts the clinical course of CLL, Calin et al [71] performed genome-wide expression profiling with a miRNA microchip in a large number of CLL samples from patients with available clinical data. The miRNA expression profiles of 94 samples of CLL cells revealed that there is a unique miRNA expression signature, composed of 13 miRNA genes (out of 190 analyzed), which was associated with disease progression. Interestingly, the expression levels of the members of the signature were found to be correlated with CLL prognostic factors such as ZAP-70 expression and IgVH mutated/unmutated status (see Table 4). Although two other independent studies found non-overlapping miRNA signatures, each of these expression signatures was found to be correlated with the mutational status of IgVH, a prognostic factor for aggressive CLL [82, 103]. Stamatopoulos et al [104] also revealed that down-regulation of miR-29c and miR-223, associated with progression of disease from Binet stage A to C and poor prognostic factors for CLL, can significantly predict treatment-free survival (TFS) and overall survival (OS). Additionally, a quantitative real-time polymerase chain reaction (qRT-PCR) score combining miR-29c and miR-223 with well-established prognostic factors ZAP-70 and lipoprotein lipase (LPL) was developed to stratify treatment and death risk in CLL patients (from 0 to 4 poor prognostic markers) [104, 105] (Table 4). MiR-21 and miR-181b, two unfavorable prognostic factors independent of other clinical pathologic factors, have been found to be differentially expressed in CLL patients. High miR-21 expression in patients with a poor prognosis predicted OS and low miR-181b expression in therapy-refractory cases significantly predicted TFS [106]. Rossi et al [106] developed a “21FK” score (miR-21 qRT-PCR, fluorescence in situ hybridization, karyotype) to stratify OS in CLL patients with chromosome 17p deletions and found that the survival of patients with a low score was significantly higher than those with high scores. Comparing the relative power of the 21FK score with the currently used survival and prognostic factors (such as ZAP-70 and IgVH status) highlighted the efficacy of the 21FK score for distinguishing between good and poor prognosis CLL patients.
3 MicroRNA expression profiling as a novel clinical tool
A growing body of literature, summarized above and in Table 5, describes the role of miRNAs in hematopoietic malignancies including the four leukemias discussed. Comparing the expression profiles between distinct sets of healthy and leukemic cells has led to the successful identification of miRNA signatures of specific leukemia types. However, based on the large number of miRNAs in the human genome and the broad variety of mRNAs regulated by these molecules, some miRNA members of the identified signatures in different types of leukemia are surprisingly common.
Different leukemia types and subtypes have specific miRNA signatures that may be useful as diagnostic and prognostic markers. Additionally, there is a growing body of evidence that points to the potential roles of miRNAs as promising novel biomarkers for leukemia detection [107, 108]. A miRNA expression profiling conducted by Tanaka et al [109] showed that down-regulation of miR-92 in human plasma could be considered as a novel biomarker for acute leukemia patients. Furthermore, miR-683 was found to be equally expressed in all the samples (regardless of age, sex or leukemia classification) so the ratio of miR-92a/miR-638 in plasma may be able to distinguish healthy individuals from acute leukemia patients.
Also, miRNA expression profiles have been shown to change during treatment and the modulation of some miRNAs increases the sensitivity of tumor cells to chemotherapeutic agents [15]. The ability of miRNA profiling to predict therapeutic outcome in well-established anti-cancer/leukemia therapies could be considered as the most promising application of miRNAs, but needs to be further investigated. For example, alterations in miRNA expression profiles could provide information about sensitivity or resistance of certain tumor types to different treatments before starting any therapy. Moreover, changes in miRNA expression during a therapy might offer a tool for the control of success of treatment [110]. It was shown that the expression levels of certain miRNAs dysregulated in CML were significantly restored after 2 weeks of imatinib therapy [69]. A group of 19 miRNAs was identified as possible predictors of clinical resistance to imatinib in patients with newly diagnosed CML [70]. Moussay et al [111] identified a set of genes and miRNAs that could predict the clinical outcome of CLL patients and refine the prognosis before fludarabine therapy. Differential expression of the sulfatase SULF2 and of miR-29a, miR-181a, and miR-221 was observed between resistant and sensitive patients before treatment with fludarabine. These results could be of interest for clinical trials and may help to predict the clinical outcome of CLL patients before or early during their treatment.
Restoring the expression levels of tumor suppressive miRNAs using viral or liposomal delivery of mimic miRNAs [15, 112], or sequence-specific knockdown of oncogenic miRNAs by chemically engineered oligonucleotides termed “antagomirs” or locked nucleic acid (LNA)-anti-miR oligonucleotides, represents a potential novel therapeutic approach for the treatment of cancer and leukemia [113, 114]. In leukemia cells, a miR-19-specific antagomir caused an increase in the expression levels of silenced tumor suppressors including PTEN and produced anti-proliferative effects, thereby opposing miR-19-driven leukemogenesis [115]. Targeting MLL-AF9 transformed/miR-196b overexpressing BM progenitor cells with an antagomir against miR-196-b abrogated the proliferative capacity of these cells in vitro [35]. This suggests that the approach based on antagonizing the activity of even a single miRNA may have therapeutic potential against leukemia [116]. Interestingly, preliminary data validate the effectiveness of antagomirs as specific silencers of endogenous miRNAs in mice [113, 117]. Krützfeldt et al [113] showed that intravenous administration of antagomirs against miR-16, miR-122, miR-192 and miR-194 led to specific and long-lasting reduction of corresponding miRNA levels in mice. The advantage of the high metabolic stability of LNA-anti-miRs, due in part to enhanced selectivity and nuclease resistance, their small size and lack of acute toxicity, implies that LNA-anti-miRs may be well-suited for the development of novel therapeutic approaches targeting cancer-related miRNAs [118]. Elmén et al [119] demonstrated that the simple systemic delivery of an unconjugated LNA-anti-miR efficiently antagonized the liver-expressed miR-122 in non-human primates. Moreover, LNA-anti-miR-122 (SPC3649) is the first miRNA-targeted drug to enter human clinical trials and is currently undergoing phase 1 clinical studies to test the safety of its intravenous administration in healthy volunteers (www.santaris.com).
The above data indicate that modulation of miRNA activity may represent a promising therapeutic approach for leukemia patients, either alone or in combination with currently used therapies. However, there are several obstacles ahead to be resolved prior to consideration of conducting miRNA-based clinical therapy including dosage, stability, specificity, efficacy, safety, and problems of delivery to the target [38, 105, 120, 121]. Moreover, some miRNAs may present activities as both oncogene and tumor suppressor in different contexts. For example, the miR-17-92 cluster, one of the first and well-studied oncogenic miRNAs, seems to be frequently overexpressed in a range of human malignancies [122, 123], including some types of leukemias and lymphomas [20, 24, 50, 124]. However, there is a body of evidence highlighting tumor suppressive activity of the miR-17-92 cluster, which contrasts with the hypothesized oncogenic role seen in other cancers [125]. This implies that the cellular context of miRNA expression decisively controls the function of a miRNA, emphasizing the need for a better insight into expression patterns and potential roles of candidate miRNA(s) in other tissues to avoid undesirable side effects [38].
4 Conclusion and future direction
The published observations discussed in this review demonstrate that miRNAs are involved in the pathogenesis of leukemias and that miRNA profiling could be used for the classification of leukemias, establishing specific diagnoses and offering prognostic values in the near future. The association of miRNA dysregulation with leukemogenesis and the functional analysis of specific miRNAs demonstrate the feasibility of manipulating miRNA expression levels as a potential strategy for developing efficient “personalized” therapies against leukemias. Obviously, the potential involvement of candidate miRNAs in the pathogenesis of leukemias needs to be further investigated as the field moves toward clinical applications.
Abbreviations
- MiRNAs:
-
MicroRNAs
- AML:
-
Acute myeloid leukemia
- CN-AML:
-
Cytogenetically normal acute myeloid leukemia
- ALL:
-
Acute lymphoblastic leukemia
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- Ph:
-
Philadelphia chromosome
- BM:
-
Bone marrow
- USF2:
-
Upstream transcription factor 2
- BCL-2:
-
B-cell leukemia/lymphoma-2
- E2F-1:
-
E2 transcription factor family-1
- TCL-1:
-
T-cell leukemia/lymphoma1
- MCL-1:
-
Myeloid cell leukemia 1
- Itch:
-
Itchy E3 ubiquitin protein ligase homolog
- C/EBPα:
-
CCAAT/enhancer binding protein alpha
- PLK2:
-
Polo-like kinase 2
- AE:
-
AML1/ETO
- FLT3-ITD:
-
Internal tandem duplication of FMS-like tyrosine kinase 3
- HOX:
-
Homeobox
- MEIS1:
-
Myeloid ecotropic viral integration site 1
- c-Kit:
-
v-Kit hardy-zuckerman 4 feline sarcoma viral oncogene homolog
- CNS:
-
Central nervous system
- MDR:
-
Minimally-deleted region
- DLEU:
-
Deleted in leukemia
- DLBCL:
-
Diffuse large B-cell lymphoma
- MBL:
-
Monoclonal B-cell lymphocytosis
- IgVH :
-
Immunoglobulin heavy-chain variable-region
- ZAP-70:
-
70-kDa zeta-associated protein
- LPL:
-
Lipoprotein lipase
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- OS:
-
Overall survival
- TFS:
-
Treatment-free survival
- OG:
-
Oncogene
- TS:
-
Tumor suppressor
References
X. Liu, K. Fortin, Z. Mourelatos, MicroRNAs: biogenesis and molecular functions. Brain Pathol 18, 113–121 (2008)
S. Babashah, M. Soleimani, The oncogenic and tumour suppressive roles of microRNAs in cancer and apoptosis. Eur. J. Cancer 47, 1127–1137 (2011)
R.C. Lee, R.L. Feinbaum, V. Ambros, The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854 (1993)
B.J. Reinhart, F.J. Slack, M. Basson, A.E. Pasquinelli, J.C. Bettinger, A.E. Rougvie, H.R. Horvitz, G. Ruvkun, The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906 (2000)
M.J. Bueno, I. Perez de Castro, M. Malumbres, Control of cell proliferation pathways by microRNAs. Cell Cycle 7, 3143–3148 (2008)
Y. Lee, M. Kim, J. Han, K.H. Yeom, S. Lee, S.H. Baek, V.N. Kim, MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060 (2004)
G.M. Borchert, W. Lanier, B.L. Davidson, RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 13, 1097–1101 (2006)
Y. Lee, K. Jeon, J.T. Lee, S. Kim, V.N. Kim, MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 21, 4663–4670 (2002)
G. Hutvagner, P.D. Zamore, A microRNA in a multiple-turnover RNAi enzyme complex. Science 297, 2056–2060 (2002)
R. Garzon, G.A. Calin, C.M. Croce, MicroRNAs in Cancer. Annu Rev Med 60, 167–179 (2009)
M. Garofalo, C.M. Croce, microRNAs: Master regulators as potential therapeutics in cancer. Annu. Rev. Pharmacol. Toxicol. 51, 25–43 (2011)
O.A. Kent, J.T. Mendell, A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene 25, 6188–6196 (2006)
L. Ma, R.A. Weinberg, MicroRNAs in malignant progression. Cell Cycle 7, 570–572 (2008)
A. Esquela-Kerscher, F.J. Slack, Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer 6, 259–269 (2006)
G.A. Calin, C.M. Croce, MicroRNA signatures in human cancers. Nat. Rev. Cancer 6, 857–866 (2006)
M. Fabbri, R. Garzon, M. Andreeff, H.M. Kantarjian, G. Garcia-Manero, G.A. Calin, MicroRNAs and noncoding RNAs in hematological malignancies: molecular, clinical and therapeutic implications. Leukemia 22, 1095–1105 (2008)
S. Yendamuri, G.A. Calin, The role of microRNA in human leukemia: a review. Leukemia 23, 1257–1263 (2009)
J. Kluiver, B.J. Kroesen, S. Poppema, A. van den Berg, The role of microRNAs in normal hematopoiesis and hematopoietic malignancies. Leukemia 20, 1931–1936 (2006)
R. Garzon, C.M. Croce, MicroRNAs in normal and malignant hematopoiesis. Curr. Opin. Hematol. 15, 352–358 (2008)
Z. Li, J. Lu, M. Sun, S. Mi, H. Zhang, R.T. Luo, P. Chen, Y. Wang, M. Yan, Z. Qian, M.B. Neilly, J. Jin, Y. Zhang, S.K. Bohlander, D.E. Zhang, R.A. Larson, M.M. Le Beau, M.J. Thirman, T.R. Golub, J.D. Rowley, J. Chen, Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. U. S. A. 105, 15535–15540 (2008)
H. Seca, G.M. Almeida, J.E. Guimaraes, M.H. Vasconcelos, miR signatures and the role of miRs in acute myeloid leukaemia. Eur. J. Cancer 46, 1520–1527 (2010)
H. Zhao, D. Wang, W. Du, D. Gu, R. Yang, MicroRNA and leukemia: tiny molecule, great function. Crit Rev Oncol Hematol 74, 149–155 (2010)
A. Dixon-McIver, P. East, C.A. Mein, J.B. Cazier, G. Molloy, T. Chaplin, T. Andrew Lister, B.D. Young, S. Debernardi, Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS One 3, e2141 (2008)
M. Jongen-Lavrencic, S.M. Sun, M.K. Dijkstra, P.J. Valk, B. Lowenberg, MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood 111, 5078–5085 (2008)
C. Nervi, F. Fazi, F. Grignani, Oncoproteins, heterochromatin silencing and microRNAs: a new link for leukemogenesis. Epigenetics 3, 1–4 (2008)
F. Fazi, A. Rosa, A. Fatica, V. Gelmetti, M.L. De Marchis, C. Nervi, I. Bozzoni, A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 123, 819–831 (2005)
F. Fazi, S. Racanicchi, G. Zardo, L.M. Starnes, M. Mancini, L. Travaglini, D. Diverio, E. Ammatuna, G. Cimino, F. Lo-Coco, F. Grignani, C. Nervi, Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 12, 457–466 (2007)
J.B. Johnnidis, M.H. Harris, R.T. Wheeler, S. Stehling-Sun, M.H. Lam, O. Kirak, T.R. Brummelkamp, M.D. Fleming, F.D. Camargo, Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451, 1125–1129 (2008)
F. Rosenbauer, S. Koschmieder, U. Steidl, D.G. Tenen, Effect of transcription-factor concentrations on leukemic stem cells. Blood 106, 1519–1524 (2005)
M. Bousquet, C. Quelen, R. Rosati, V. Mansat-De Mas, R. La Starza, C. Bastard, E. Lippert, P. Talmant, M. Lafage-Pochitaloff, D. Leroux, C. Gervais, F. Viguie, J.L. Lai, C. Terre, B. Beverlo, C. Sambani, A. Hagemeijer, P. Marynen, G. Delsol, N. Dastugue, C. Mecucci, P. Brousset, Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J Exp Med 205, 2499–2506 (2008)
L. Fontana, E. Pelosi, P. Greco, S. Racanicchi, U. Testa, F. Liuzzi, C.M. Croce, E. Brunetti, F. Grignani, C. Peschle, MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol 9, 775–787 (2007)
S. Mi, Z. Li, P. Chen, C. He, D. Cao, A. Elkahloun, J. Lu, L.A. Pelloso, M. Wunderlich, H. Huang, R.T. Luo, M. Sun, M. He, M.B. Neilly, N.J. Zeleznik-Le, M.J. Thirman, J.C. Mulloy, P.P. Liu, J.D. Rowley, J. Chen, Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc. Natl. Acad. Sci. U. S. A. 107, 3710–3715 (2010)
P. Wong, M. Iwasaki, T.C. Somervaille, F. Ficara, C. Carico, C. Arnold, C.Z. Chen, M.L. Cleary, The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res. 70, 3833–3842 (2010)
R. Garzon, S. Volinia, C.G. Liu, C. Fernandez-Cymering, T. Palumbo, F. Pichiorri, M. Fabbri, K. Coombes, H. Alder, T. Nakamura, N. Flomenberg, G. Marcucci, G.A. Calin, S.M. Kornblau, H. Kantarjian, C.D. Bloomfield, M. Andreeff, C.M. Croce, MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood 111, 3183–3189 (2008)
R. Popovic, L.E. Riesbeck, C.S. Velu, A. Chaubey, J. Zhang, N.J. Achille, F.E. Erfurth, K. Eaton, J. Lu, H.L. Grimes, J. Chen, J.D. Rowley, N.J. Zeleznik-Le, Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood 113, 3314–3322 (2009)
G. Marcucci, K. Maharry, M.D. Radmacher, K. Mrozek, T. Vukosavljevic, P. Paschka, S.P. Whitman, C. Langer, C.D. Baldus, C.G. Liu, A.S. Ruppert, B.L. Powell, A.J. Carroll, M.A. Caligiuri, J.E. Kolitz, R.A. Larson, C.D. Bloomfield, Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J. Clin. Oncol. 26, 5078–5087 (2008)
M.L. Choong, H.H. Yang, I. McNiece, MicroRNA expression profiling during human cord blood-derived CD34 cell erythropoiesis. Exp. Hematol. 35, 551–564 (2007)
J. Chen, O. Odenike, J.D. Rowley, Leukaemogenesis: more than mutant genes. Nat. Rev. Cancer 10, 23–36 (2010)
G. Marcucci, M.D. Radmacher, K. Maharry, K. Mrozek, A.S. Ruppert, P. Paschka, T. Vukosavljevic, S.P. Whitman, C.D. Baldus, C. Langer, C.G. Liu, A.J. Carroll, B.L. Powell, R. Garzon, C.M. Croce, J.E. Kolitz, M.A. Caligiuri, R.A. Larson, C.D. Bloomfield, MicroRNA expression in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 358, 1919–1928 (2008)
C. Thiede, S. Koch, E. Creutzig, C. Steudel, T. Illmer, M. Schaich, G. Ehninger, Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood 107, 4011–4020 (2006)
R. Garzon, M. Garofalo, M.P. Martelli, R. Briesewitz, L. Wang, C. Fernandez-Cymering, S. Volinia, C.G. Liu, S. Schnittger, T. Haferlach, A. Liso, D. Diverio, M. Mancini, G. Meloni, R. Foa, M.F. Martelli, C. Mecucci, C.M. Croce, B. Falini, Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc. Natl. Acad. Sci. U. S. A. 105, 3945–3950 (2008)
B. Falini, I. Nicoletti, M.F. Martelli, C. Mecucci, Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc + AML): biologic and clinical features. Blood 109, 874–885 (2007)
S. Yekta, I.H. Shih, D.P. Bartel, MicroRNA-directed cleavage of HOXB8 mRNA. Science 304, 594–596 (2004)
R.M. O'Connell, D.S. Rao, A.A. Chaudhuri, M.P. Boldin, K.D. Taganov, J. Nicoll, R.L. Paquette, D. Baltimore, Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med 205, 585–594 (2008)
R. Crazzolara, L. Bendall, Emerging treatments in acute lymphoblastic leukemia. Curr Cancer Drug Targets 9, 19–31 (2009)
S. Mi, J. Lu, M. Sun, Z. Li, H. Zhang, M.B. Neilly, Y. Wang, Z. Qian, J. Jin, Y. Zhang, S.K. Bohlander, M.M. Le Beau, R.A. Larson, T.R. Golub, J.D. Rowley, J. Chen, MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc. Natl. Acad. Sci. U. S. A. 104, 19971–19976 (2007)
H. Zhang, X.Q. Luo, P. Zhang, L.B. Huang, Y.S. Zheng, J. Wu, H. Zhou, L.H. Qu, L. Xu, Y.Q. Chen, MicroRNA patterns associated with clinical prognostic parameters and CNS relapse prediction in pediatric acute leukemia. PLoS One 4, e7826 (2009)
D. Schotte, J.C. Chau, G. Sylvester, G. Liu, C. Chen, V.H. van der Velden, M.J. Broekhuis, T.C. Peters, R. Pieters, M.L. den Boer, Identification of new microRNA genes and aberrant microRNA profiles in childhood acute lymphoblastic leukemia. Leukemia 23, 313–322 (2009)
D.L. Zanette, F. Rivadavia, G.A. Molfetta, F.G. Barbuzano, R. Proto-Siqueira, W.A. Silva-Jr, R.P. Falcao, M.A. Zago, miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. Braz J Med Biol Res 40, 1435–1440 (2007)
A. Ota, H. Tagawa, S. Karnan, S. Tsuzuki, A. Karpas, S. Kira, Y. Yoshida, M. Seto, Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 64, 3087–3095 (2004)
A. Ventura, A.G. Young, M.M. Winslow, L. Lintault, A. Meissner, S.J. Erkeland, J. Newman, R.T. Bronson, D. Crowley, J.R. Stone, R. Jaenisch, P.A. Sharp, T. Jacks, Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132, 875–886 (2008)
L. He, J.M. Thomson, M.T. Hemann, E. Hernando-Monge, D. Mu, S. Goodson, S. Powers, C. Cordon-Cardo, S.W. Lowe, G.J. Hannon, S.M. Hammond, A microRNA polycistron as a potential human oncogene. Nature 435, 828–833 (2005)
M. Inomata, H. Tagawa, Y.M. Guo, Y. Kameoka, N. Takahashi, K. Sawada, MicroRNA-17-92 down-regulates expression of distinct targets in different B-cell lymphoma subtypes. Blood 113, 396–402 (2009)
L. O'Connor, A. Strasser, L.A. O'Reilly, G. Hausmann, J.M. Adams, S. Cory, D.C. Huang, Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17, 384–395 (1998)
C. Dong, M. Ji, C. Ji, microRNAs and their potential target genes in leukemia pathogenesis. Cancer Biol Ther 8, 200–205 (2009)
C. Xiao, L. Srinivasan, D.P. Calado, H.C. Patterson, B. Zhang, J. Wang, J.M. Henderson, J.L. Kutok, K. Rajewsky, Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol. 9, 405–414 (2008)
P. Mu, Y.C. Han, D. Betel, E. Yao, M. Squatrito, P. Ogrodowski, E. de Stanchina, A. D'Andrea, C. Sander, A. Ventura, Genetic dissection of the miR-17 92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 23, 2806–2811 (2009)
V. Olive, M.J. Bennett, J.C. Walker, C. Ma, I. Jiang, C. Cordon-Cardo, Q.J. Li, S.W. Lowe, G.J. Hannon, L. He, miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 23, 2839–2849 (2009)
C.L. Sawyers, Chronic myeloid leukemia. N. Engl. J. Med. 340, 1330–1340 (1999)
M.W. Deininger, J.M. Goldman, J.V. Melo, The molecular biology of chronic myeloid leukemia. Blood 96, 3343–3356 (2000)
T.G. Lugo, A.M. Pendergast, A.J. Muller, O.N. Witte, Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 247, 1079–1082 (1990)
Y.A. Mian, N.J. Zeleznik-Le, MicroRNAs in leukemias: emerging diagnostic tools and therapeutic targets. Curr Drug Targets 11, 801–811 (2010)
K. Woods, J.M. Thomson, S.M. Hammond, Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J. Biol. Chem. 282, 2130–2134 (2007)
K.A. O'Donnell, E.A. Wentzel, K.I. Zeller, C.V. Dang, J.T. Mendell, c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839–843 (2005)
Y. Sylvestre, V. De Guire, E. Querido, U.K. Mukhopadhyay, V. Bourdeau, F. Major, G. Ferbeyre, P. Chartrand, An E2F/miR-20a autoregulatory feedback loop. J. Biol. Chem. 282, 2135–2143 (2007)
L. Venturini, K. Battmer, M. Castoldi, B. Schultheis, A. Hochhaus, M.U. Muckenthaler, A. Ganser, M. Eder, M. Scherr, Expression of the miR-17-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood 109, 4399–4405 (2007)
M.J. Bueno, I. Perez de Castro, M. Gomez de Cedron, J. Santos, G.A. Calin, J.C. Cigudosa, C.M. Croce, J. Fernandez-Piqueras, M. Malumbres, Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell 13, 496–506 (2008)
X. Agirre, A. Jimenez-Velasco, E. San Jose-Eneriz, L. Garate, E. Bandres, L. Cordeu, O. Aparicio, B. Saez, G. Navarro, A. Vilas-Zornoza, I. Perez-Roger, J. Garcia-Foncillas, A. Torres, A. Heiniger, M.J. Calasanz, P. Fortes, J. Roman-Gomez, F. Prosper, Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol Cancer Res 6, 1830–1840 (2008)
S. Flamant, W. Ritchie, J. Guilhot, J. Holst, M.L. Bonnet, J.C. Chomel, F. Guilhot, A.G. Turhan, J.E. Rasko, Micro-RNA response to imatinib mesylate in patients with chronic myeloid leukemia. Haematologica 95, 1325–1333 (2010)
E. San Jose-Eneriz, J. Roman-Gomez, A. Jimenez-Velasco, L. Garate, V. Martin, L. Cordeu, A. Vilas-Zornoza, P. Rodriguez-Otero, M.J. Calasanz, F. Prosper, X. Agirre, MicroRNA expression profiling in Imatinib-resistant Chronic Myeloid Leukemia patients without clinically significant ABL1-mutations. Mol Cancer 8, 69 (2009)
G.A. Calin, M. Ferracin, A. Cimmino, G. Di Leva, M. Shimizu, S.E. Wojcik, M.V. Iorio, R. Visone, N.I. Sever, M. Fabbri, R. Iuliano, T. Palumbo, F. Pichiorri, C. Roldo, R. Garzon, C. Sevignani, L. Rassenti, H. Alder, S. Volinia, C.G. Liu, T.J. Kipps, M. Negrini, C.M. Croce, A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 353, 1793–1801 (2005)
G.A. Calin, C.D. Dumitru, M. Shimizu, R. Bichi, S. Zupo, E. Noch, H. Aldler, S. Rattan, M. Keating, K. Rai, L. Rassenti, T. Kipps, M. Negrini, F. Bullrich, C.M. Croce, Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 99, 15524–15529 (2002)
U. Klein, M. Lia, M. Crespo, R. Siegel, Q. Shen, T. Mo, A. Ambesi-Impiombato, A. Califano, A. Migliazza, G. Bhagat, R. Dalla-Favera, The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17, 28–40 (2010)
E.S. Raveche, E. Salerno, B.J. Scaglione, V. Manohar, F. Abbasi, Y.C. Lin, T. Fredrickson, P. Landgraf, S. Ramachandra, K. Huppi, J.R. Toro, V.E. Zenger, R.A. Metcalf, G.E. Marti, Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood 109, 5079–5086 (2007)
E. Salerno, B.J. Scaglione, F.D. Coffman, B.D. Brown, A. Baccarini, H. Fernandes, G. Marti, E.S. Raveche, Correcting miR-15a/16 genetic defect in New Zealand Black mouse model of CLL enhances drug sensitivity. Mol. Cancer Ther. 8, 2684–2692 (2009)
U. Klein, R. Dalla-Favera, New insights into the pathogenesis of chronic lymphocytic leukemia. Semin Cancer Biol 20, 377–383 (2010)
A. Cimmino, G.A. Calin, M. Fabbri, M.V. Iorio, M. Ferracin, M. Shimizu, S.E. Wojcik, R.I. Aqeilan, S. Zupo, M. Dono, L. Rassenti, H. Alder, S. Volinia, C.G. Liu, T.J. Kipps, M. Negrini, C.M. Croce, miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. U. S. A. 102, 13944–13949 (2005)
G.A. Calin, A. Cimmino, M. Fabbri, M. Ferracin, S.E. Wojcik, M. Shimizu, C. Taccioli, N. Zanesi, R. Garzon, R.I. Aqeilan, H. Alder, S. Volinia, L. Rassenti, X. Liu, C.G. Liu, T.J. Kipps, M. Negrini, C.M. Croce, MiR-15a and miR-16-1 cluster functions in human leukemia. Proc. Natl. Acad. Sci. U. S. A. 105, 5166–5171 (2008)
R.W. Chen, L.T. Bemis, C.M. Amato, H. Myint, H. Tran, D.K. Birks, S.G. Eckhardt, W.A. Robinson, Truncation in CCND1 mRNA alters miR-16-1 regulation in mantle cell lymphoma. Blood 112, 822–829 (2008)
P.S. Linsley, J. Schelter, J. Burchard, M. Kibukawa, M.M. Martin, S.R. Bartz, J.M. Johnson, J.M. Cummins, C.K. Raymond, H. Dai, N. Chau, M. Cleary, A.L. Jackson, M. Carleton, L. Lim, Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol. Cell. Biol. 27, 2240–2252 (2007)
P.S. Eis, W. Tam, L. Sun, A. Chadburn, Z. Li, M.F. Gomez, E. Lund, J.E. Dahlberg, Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. U. S. A. 102, 3627–3632 (2005)
V. Fulci, S. Chiaretti, M. Goldoni, G. Azzalin, N. Carucci, S. Tavolaro, L. Castellano, A. Magrelli, F. Citarella, M. Messina, R. Maggio, N. Peragine, S. Santangelo, F.R. Mauro, P. Landgraf, T. Tuschl, D.B. Weir, M. Chien, J.J. Russo, J. Ju, R. Sheridan, C. Sander, M. Zavolan, A. Guarini, R. Foa, G. Macino, Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood 109, 4944–4951 (2007)
S. Costinean, N. Zanesi, Y. Pekarsky, E. Tili, S. Volinia, N. Heerema, C.M. Croce, Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 103, 7024–7029 (2006)
C.H. Lawrie, S. Soneji, T. Marafioti, C.D. Cooper, S. Palazzo, J.C. Paterson, H. Cattan, T. Enver, R. Mager, J. Boultwood, J.S. Wainscoat, C.S. Hatton, MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int. J. Cancer 121, 1156–1161 (2007)
T.H. Thai, D.P. Calado, S. Casola, K.M. Ansel, C. Xiao, Y. Xue, A. Murphy, D. Frendewey, D. Valenzuela, J.L. Kutok, M. Schmidt-Supprian, N. Rajewsky, G. Yancopoulos, A. Rao, K. Rajewsky, Regulation of the germinal center response by microRNA-155. Science 316, 604–608 (2007)
B.J. Scaglione, E. Salerno, M. Balan, F. Coffman, P. Landgraf, F. Abbasi, S. Kotenko, G.E. Marti, E.S. Raveche, Murine models of chronic lymphocytic leukaemia: role of microRNA-16 in the New Zealand Black mouse model. Br. J. Haematol. 139, 645–657 (2007)
A. Rodriguez, E. Vigorito, S. Clare, M.V. Warren, P. Couttet, D.R. Soond, S. van Dongen, R.J. Grocock, P.P. Das, E.A. Miska, D. Vetrie, K. Okkenhaug, A.J. Enright, G. Dougan, M. Turner, A. Bradley, Requirement of bic/microRNA-155 for normal immune function. Science 316, 608–611 (2007)
G.T. Bommer, I. Gerin, Y. Feng, A.J. Kaczorowski, R. Kuick, R.E. Love, Y. Zhai, T.J. Giordano, Z.S. Qin, B.B. Moore, O.A. MacDougald, K.R. Cho, E.R. Fearon, p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 17, 1298–1307 (2007)
T.C. Chang, E.A. Wentzel, O.A. Kent, K. Ramachandran, M. Mullendore, K.H. Lee, G. Feldmann, M. Yamakuchi, M. Ferlito, C.J. Lowenstein, D.E. Arking, M.A. Beer, A. Maitra, J.T. Mendell, Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26, 745–752 (2007)
L. He, X. He, L.P. Lim, E. de Stanchina, Z. Xuan, Y. Liang, W. Xue, L. Zender, J. Magnus, D. Ridzon, A.L. Jackson, P.S. Linsley, C. Chen, S.W. Lowe, M.A. Cleary, G.J. Hannon, A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134 (2007)
X. He, L. He, G.J. Hannon, The guardian's little helper: microRNAs in the p53 tumor suppressor network. Cancer Res. 67, 11099–11101 (2007)
L. He, X. He, S.W. Lowe, G.J. Hannon, microRNAs join the p53 network–another piece in the tumour-suppression puzzle. Nat. Rev. Cancer 7, 819–822 (2007)
J.S. Wei, Y.K. Song, S. Durinck, Q.R. Chen, A.T. Cheuk, P. Tsang, Q. Zhang, C.J. Thiele, A. Slack, J. Shohet, J. Khan, The MYCN oncogene is a direct target of miR-34a. Oncogene 27, 5204–5213 (2008)
C. Welch, Y. Chen, R.L. Stallings, MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 26, 5017–5022 (2007)
D. Asslaber, J.D. Pinon, I. Seyfried, P. Desch, M. Stocher, I. Tinhofer, A. Egle, O. Merkel, R. Greil, microRNA-34a expression correlates with MDM2 SNP309 polymorphism and treatment-free survival in chronic lymphocytic leukemia. Blood 115, 4191–4197 (2010)
M. Yamakuchi, M. Ferlito, C.J. Lowenstein, miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. U. S. A. 105, 13421–13426 (2008)
M.K. Dijkstra, K. van Lom, D. Tielemans, F. Elstrodt, A.W. Langerak, M.B. van 't Veer, M. Jongen-Lavrencic, 17p13/TP53 deletion in B-CLL patients is associated with microRNA-34a downregulation. Leukemia 23, 625–627 (2009)
T. Zenz, J. Mohr, E. Eldering, A.P. Kater, A. Buhler, D. Kienle, D. Winkler, J. Durig, M.H. van Oers, D. Mertens, H. Dohner, S. Stilgenbauer, miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood 113, 3801–3808 (2009)
Y. Pekarsky, U. Santanam, A. Cimmino, A. Palamarchuk, A. Efanov, V. Maximov, S. Volinia, H. Alder, C.G. Liu, L. Rassenti, G.A. Calin, J.P. Hagan, T. Kipps, C.M. Croce, Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 66, 11590–11593 (2006)
M.S. Nicoloso, T.J. Kipps, C.M. Croce, G.A. Calin, MicroRNAs in the pathogeny of chronic lymphocytic leukaemia. Br. J. Haematol. 139, 709–716 (2007)
R. Bichi, S.A. Shinton, E.S. Martin, A. Koval, G.A. Calin, R. Cesari, G. Russo, R.R. Hardy, C.M. Croce, Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc. Natl. Acad. Sci. U. S. A. 99, 6955–6960 (2002)
X.J. Yan, E. Albesiano, N. Zanesi, S. Yancopoulos, A. Sawyer, E. Romano, A. Petlickovski, D.G. Efremov, C.M. Croce, N. Chiorazzi, B cell receptors in TCL1 transgenic mice resemble those of aggressive, treatment-resistant human chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 103, 11713–11718 (2006)
S. Marton, M.R. Garcia, C. Robello, H. Persson, F. Trajtenberg, O. Pritsch, C. Rovira, H. Naya, G. Dighiero, A. Cayota, Small RNAs analysis in CLL reveals a deregulation of miRNA expression and novel miRNA candidates of putative relevance in CLL pathogenesis. Leukemia 22, 330–338 (2008)
B. Stamatopoulos, N. Meuleman, B. Haibe-Kains, P. Saussoy, E. Van Den Neste, L. Michaux, P. Heimann, P. Martiat, D. Bron, L. Lagneaux, microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood 113, 5237–5245 (2009)
W.C. Cho, MicroRNAs: potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int. J. Biochem. Cell Biol. 42, 1273–1281 (2010)
S. Rossi, M. Shimizu, E. Barbarotto, M.S. Nicoloso, F. Dimitri, D. Sampath, M. Fabbri, S. Lerner, L.L. Barron, L.Z. Rassenti, L. Jiang, L. Xiao, J. Hu, P. Secchiero, G. Zauli, S. Volinia, M. Negrini, W. Wierda, T.J. Kipps, W. Plunkett, K.R. Coombes, L.V. Abruzzo, M.J. Keating, G.A. Calin, microRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood 116, 945–952 (2010)
S. Gilad, E. Meiri, Y. Yogev, S. Benjamin, D. Lebanony, N. Yerushalmi, H. Benjamin, M. Kushnir, H. Cholakh, N. Melamed, Z. Bentwich, M. Hod, Y. Goren, A. Chajut, Serum microRNAs are promising novel biomarkers. PLoS One 3, e3148 (2008)
P.S. Mitchell, R.K. Parkin, E.M. Kroh, B.R. Fritz, S.K. Wyman, E.L. Pogosova-Agadjanyan, A. Peterson, J. Noteboom, K.C. O'Briant, A. Allen, D.W. Lin, N. Urban, C.W. Drescher, B.S. Knudsen, D.L. Stirewalt, R. Gentleman, R.L. Vessella, P.S. Nelson, D.B. Martin, M. Tewari, Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. U. S. A. 105, 10513–10518 (2008)
M. Tanaka, K. Oikawa, M. Takanashi, M. Kudo, J. Ohyashiki, K. Ohyashiki, M. Kuroda, Down-regulation of miR-92 in human plasma is a novel marker for acute leukemia patients. PLoS One 4, e5532 (2009)
R. Hummel, D.J. Hussey, J. Haier, MicroRNAs: predictors and modifiers of chemo- and radiotherapy in different tumour types. Eur. J. Cancer 46, 298–311 (2010)
E. Moussay, V. Palissot, L. Vallar, H.A. Poirel, T. Wenner, V. El Khoury, N. Aouali, K. Van Moer, B. Leners, F. Bernardin, A. Muller, P. Cornillet-Lefebvre, A. Delmer, C. Duhem, F. Ries, E. van Dyck, G. Berchem, Determination of genes and microRNAs involved in the resistance to fludarabine in vivo in chronic lymphocytic leukemia. Mol Cancer 9, 115 (2010)
F. Meng, R. Henson, M. Lang, H. Wehbe, S. Maheshwari, J.T. Mendell, J. Jiang, T.D. Schmittgen, T. Patel, Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology 130, 2113–2129 (2006)
J. Krutzfeldt, N. Rajewsky, R. Braich, K.G. Rajeev, T. Tuschl, M. Manoharan, M. Stoffel, Silencing of microRNAs in vivo with 'antagomirs'. Nature 438, 685–689 (2005)
U.A. Orom, S. Kauppinen, A.H. Lund, LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene 372, 137–141 (2006)
K.J. Mavrakis, A.L. Wolfe, E. Oricchio, T. Palomero, K. de Keersmaecker, K. McJunkin, J. Zuber, T. James, A.A. Khan, C.S. Leslie, J.S. Parker, P.J. Paddison, W. Tam, A. Ferrando, H.G. Wendel, Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol 12, 372–379 (2010)
D. Schotte, R. Pieters, M.L. Den Boer, MicroRNAs in acute leukemia: from biological players to clinical contributors. Leukemia 26, 1–12 (2012)
J. Krutzfeldt, S. Kuwajima, R. Braich, K.G. Rajeev, J. Pena, T. Tuschl, M. Manoharan, M. Stoffel, Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 35, 2885–2892 (2007)
J. Elmen, M. Lindow, A. Silahtaroglu, M. Bak, M. Christensen, A. Lind-Thomsen, M. Hedtjarn, J.B. Hansen, H.F. Hansen, E.M. Straarup, K. McCullagh, P. Kearney, S. Kauppinen, Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 36, 1153–1162 (2008)
J. Elmen, M. Lindow, S. Schutz, M. Lawrence, A. Petri, S. Obad, M. Lindholm, M. Hedtjarn, H.F. Hansen, U. Berger, S. Gullans, P. Kearney, P. Sarnow, E.M. Straarup, S. Kauppinen, LNA-mediated microRNA silencing in non-human primates. Nature 452, 896–899 (2008)
A.W. Tong, J. Nemunaitis, Modulation of miRNA activity in human cancer: a new paradigm for cancer gene therapy? Cancer Gene Ther 15, 341–355 (2008)
W. Wu, M. Sun, G.M. Zou, J. Chen, MicroRNA and cancer: Current status and prospective. Int. J. Cancer 120, 953–960 (2007)
Y. Hayashita, H. Osada, Y. Tatematsu, H. Yamada, K. Yanagisawa, S. Tomida, Y. Yatabe, K. Kawahara, Y. Sekido, T. Takahashi, A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 65, 9628–9632 (2005)
S. Volinia, G.A. Calin, C.G. Liu, S. Ambs, A. Cimmino, F. Petrocca, R. Visone, M. Iorio, C. Roldo, M. Ferracin, R.L. Prueitt, N. Yanaihara, G. Lanza, A. Scarpa, A. Vecchione, M. Negrini, C.C. Harris, C.M. Croce, A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U. S. A. 103, 2257–2261 (2006)
G.A. Calin, C.G. Liu, C. Sevignani, M. Ferracin, N. Felli, C.D. Dumitru, M. Shimizu, A. Cimmino, S. Zupo, M. Dono, M.L. Dell'Aquila, H. Alder, L. Rassenti, T.J. Kipps, F. Bullrich, M. Negrini, C.M. Croce, MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. U. S. A. 101, 11755–11760 (2004)
Z. Yu, C. Wang, M. Wang, Z. Li, M.C. Casimiro, M. Liu, K. Wu, J. Whittle, X. Ju, T. Hyslop, P. McCue, R.G. Pestell, A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol 182, 509–517 (2008)
H. Becker, G. Marcucci, K. Maharry, M.D. Radmacher, K. Mrozek, D. Margeson, S.P. Whitman, Y.Z. Wu, S. Schwind, P. Paschka, B.L. Powell, T.H. Carter, J.E. Kolitz, M. Wetzler, A.J. Carroll, M.R. Baer, M.A. Caligiuri, R.A. Larson, C.D. Bloomfield, Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J. Clin. Oncol. 28, 596–604 (2010)
Y. Pekarsky, A. Koval, C. Hallas, R. Bichi, M. Tresini, S. Malstrom, G. Russo, P. Tsichlis, C.M. Croce, Tcl1 enhances Akt kinase activity and mediates its nuclear translocation. Proc. Natl. Acad. Sci. U. S. A. 97, 3028–3033 (2000)
R. Garzon, F. Pichiorri, T. Palumbo, M. Visentini, R. Aqeilan, A. Cimmino, H. Wang, H. Sun, S. Volinia, H. Alder, G.A. Calin, C.G. Liu, M. Andreeff, C.M. Croce, MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene 26, 4148–4157 (2007)
D. Sampath, G.A. Calin, V.K. Puduvalli, G. Gopisetty, C. Taccioli, C.G. Liu, B. Ewald, C. Liu, M.J. Keating, W. Plunkett, Specific activation of microRNA106b enables the p73 apoptotic response in chronic lymphocytic leukemia by targeting the ubiquitin ligase Itch for degradation. Blood 113, 3744–3753 (2009)
F. Isken, B. Steffen, S. Merk, M. Dugas, B. Markus, N. Tidow, M. Zuhlsdorf, T. Illmer, C. Thiede, W.E. Berdel, H. Serve, C. Muller-Tidow, Identification of acute myeloid leukaemia associated microRNA expression patterns. Br. J. Haematol. 140, 153–161 (2008)
C. Langer, M.D. Radmacher, A.S. Ruppert, S.P. Whitman, P. Paschka, K. Mrozek, C.D. Baldus, T. Vukosavljevic, C.G. Liu, M.E. Ross, B.L. Powell, A. de la Chapelle, J.E. Kolitz, R.A. Larson, G. Marcucci, C.D. Bloomfield, High BAALC expression associates with other molecular prognostic markers, poor outcome, and a distinct gene-expression signature in cytogenetically normal patients younger than 60 years with acute myeloid leukemia: a Cancer and Leukemia Group B (CALGB) study. Blood 111, 5371–5379 (2008)
C. Xiao, D.P. Calado, G. Galler, T.H. Thai, H.C. Patterson, J. Wang, N. Rajewsky, T.P. Bender, K. Rajewsky, MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 131, 146–159 (2007)
K. Machova Polakova, T. Lopotova, H. Klamova, P. Burda, M. Trneny, T. Stopka, J. Moravcova, Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer 10, 41 (2011)
R. Garzon, F. Pichiorri, T. Palumbo, R. Iuliano, A. Cimmino, R. Aqeilan, S. Volinia, D. Bhatt, H. Alder, G. Marcucci, G.A. Calin, C.G. Liu, C.D. Bloomfield, M. Andreeff, C.M. Croce, MicroRNA fingerprints during human megakaryocytopoiesis. Proc. Natl. Acad. Sci. U. S. A. 103, 5078–5083 (2006)
H. Bruchova, D. Yoon, A.M. Agarwal, J. Mendell, J.T. Prchal, Regulated expression of microRNAs in normal and polycythemia vera erythropoiesis. Exp. Hematol. 35, 1657–1667 (2007)
R.W. Georgantas 3rd, R. Hildreth, S. Morisot, J. Alder, C.G. Liu, S. Heimfeld, G.A. Calin, C.M. Croce, C.I. Civin, CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc. Natl. Acad. Sci. U. S. A. 104, 2750–2755 (2007)
G. Marcucci, K. Mrozek, M.D. Radmacher, R. Garzon, C.D. Bloomfield, The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood 117, 1121–1129 (2011)
S. Debernardi, S. Skoulakis, G. Molloy, T. Chaplin, A. Dixon-McIver, B.D. Young, MicroRNA miR-181a correlates with morphological sub-class of acute myeloid leukaemia and the expression of its target genes in global genome-wide analysis. Leukemia 21, 912–916 (2007)
U. Thorsteinsdottir, A. Mamo, E. Kroon, L. Jerome, J. Bijl, H.J. Lawrence, K. Humphries, G. Sauvageau, Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood 99, 121–129 (2002)
N. Felli, L. Fontana, E. Pelosi, R. Botta, D. Bonci, F. Facchiano, F. Liuzzi, V. Lulli, O. Morsilli, S. Santoro, M. Valtieri, G.A. Calin, C.G. Liu, A. Sorrentino, C.M. Croce, C. Peschle, MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. U. S. A. 102, 18081–18086 (2005)
S.M. Tanner, J.L. Austin, G. Leone, L.J. Rush, C. Plass, K. Heinonen, K. Mrozek, H. Sill, S. Knuutila, J.E. Kolitz, K.J. Archer, M.A. Caligiuri, C.D. Bloomfield, A. de La Chapelle, BAALC, the human member of a novel mammalian neuroectoderm gene lineage, is implicated in hematopoiesis and acute leukemia. Proc. Natl. Acad. Sci. U. S. A. 98, 13901–13906 (2001)
Acknowledgements
The authors wish to thank David Leif Anderson for valuable comments on the manuscript. This work was supported by a grant from Tarbiat Modares University.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Babashah, S., Sadeghizadeh, M., Tavirani, M.R. et al. Aberrant microRNA expression and its implications in the pathogenesis of leukemias. Cell Oncol. 35, 317–334 (2012). https://doi.org/10.1007/s13402-012-0095-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-012-0095-3