
Abstract
A major challenge in drug discovery is the identification of high affinity lead compounds that bind a particular target protein; these leads are typically identified by high throughput screens. Mass spectrometry has become a detection method of choice in drug screening assays because the target and the ligand need not be modified. Label-free assays are advantageous because they can be developed more rapidly than assays requiring labels, and they eliminate the risk of the label interfering with the binding event. However, in commonly used MS-based screening methods, detection of false positives is a major challenge. Here, we describe a detection strategy designed to eliminate false positives. In this approach, the protein and the ligands are incubated together, and the non-binders are separated for detection. Hits (protein binders) are not detectable by MS after incubation with the protein, but readily identifiable by MS when the target protein is not present in the incubation media. The assay was demonstrated using three different proteins and hundreds of non-inhibitors; no false positive hits were identified in any experiment. The assay can be tuned to select for ligands of a particular binding affinity by varying the quantity of protein used and the immobilization method. As examples, the method selectively detected inhibitors that have Ki values of 0.2 μM, 50 pM, and 700 pM. These findings demonstrate that the approach described here compares favorably with traditional MS-based screening methods.

ᅟ
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent decades, combinatorial and parallel synthesis methods have been employed to synthesize millions of library compounds that potentially could be used as new therapeutic drugs. One of the vital tasks of drug discovery is fast and effective identification of high affinity ligands from libraries containing these vast numbers of compounds. When a screening method based on fluorescent [1, 2] or chemiluminescent [3] read-out is feasible, tens of thousands of compounds can be assayed per day. Many potential drug candidates, however, cannot be probed using these standard assays because some druggable interactions, such as protein binding events, cannot be readily monitored by a change in fluorescence. In these cases, assays of lower throughput based on nuclear magnetic resonance (NMR) or surface plasmon resonance (SPR) are routinely employed [4].
Mass spectrometry (MS) is increasingly used for high throughput screening (HTS) because of its advantages of fast and sensitive detection, high specificity, and the ability to detect compounds without labels. One of the commonly used MS-based screening methods is affinity selection mass spectrometry (ASMS); this approach probes the affinity of various ligands by combining large batches of library compounds with the protein of interest and using MS to detect the binding of a small sub-set of those ligands to the protein [5, 6]. In one incarnation of the method, the direct ASMS method, any protein/ligand complex that forms is detected directly by MS. A lead is identified when the mass of the protein is shifted to include the mass of the binding ligand. The disadvantage of this approach is that the ESI process may fully or partially distort the protein–ligand complexes; determining appropriate MS parameters for each type of complexes is challenging and time consuming. In addition, the mass of the protein–ligand complex is not substantially different from that of the protein itself, and even when a binding event is detected, it can be difficult to infer the exact mass of the ligand [7].
In another embodiment of the ASMS method, often referred to as “the indirect approach,” the binding ligands are identified after the protein–ligand complex is isolated and the ligands are dissociated. This approach has shown to be highly amenable to automation, with a throughput of ~1*105 compounds per day [8, 9]. However, it has some limiting disadvantages that we seek to address herein. Namely, the ligands could bind weakly and nonspecifically to the protein, and in the ASMS assay, they would still be detected as protein binders [8–10]. In addition, the method suffers from a higher incidence of false positive hits, which can be introduced during the isolation of the ligands from the protein complex. To alleviate this problem, others have used molecular weight cut-off filters [11] or size exclusion gels [10, 12, 13] to isolate the protein complexes prior to ligand dissociation. Although these approaches are helpful, non-binding ligands may interact with these separation media as well, also leading to false positives. Additionally, these solutions do not address the problem of detecting numerous nonspecific and weak binders.
Herein we present a new MS-based high throughput screening assay that shares several advantages with the indirect ASMS method described above, but it eliminates several of the key disadvantages. In the approach described here, the protein and ligands are mixed as in the ASMS method, but the non-binding analytes are detected, instead of the binders. This detection strategy was chosen to reduce the detection of false positives—those compounds that bind weakly or nonspecifically to the protein. Only the tightest binders completely disappear from the ligand sea after protein is added, so those tightest binders are readily detected as peaks that are absent in the MS data, when the non-binding ligands are analyzed. The method described herein not only picks out the tightest binding ligands, it can also be used to detect ligands with different binding affinities, by tuning the concentration of protein used in the assay. As an additional benefit of screening in this manner, the amount of protein required and the amount of each binding ligand can be substantially reduced compared with the state-of-the-art comparator method [9]. Reduction in the amount of protein required, in particular, is a strong asset in a high throughput screening method; it can substantially reduce assay cost because the quantity of the most expensive reagent, the protein, is reduced.
Methods
Reagents
The library compounds used in this study are all FDA-approved drugs. For all the experiments conducted with 176 compounds, the drug molecules are selected from the Prestwick library, obtained from the High Throughput Screening Laboratory at the University of Kansas. The library was provided in combined batches of 176 compounds per vial, dissolved in DMSO at a concentration of 14.2 μM per compound. For all the experiments conducted with 352 compounds, the drug molecules were obtained from LOPAC1280-small scale library that was purchased from Sigma Aldrich (St. Louis, MO, USA) as individual compounds, dissolved in DMSO at a concentration of 10 mM per compound. When combining compounds for high throughput screens from the LOPAC library, any compound larger than 150 Da was considered a candidate, and no effort was made to remove compounds from the library that would likely not ionize by ESI. Pepsin, Aminolink plus coupling resin, and disposable plastic columns were acquired from Pierce Biotechnology, Inc. (Rockford, IL, USA), whereas nitrocellulose drop dialysis membranes were purchased from Fisher Scientific (Houston, TX, USA). Maltose binding protein and carbonic anhydrase were purchased from My BioSource (San Diego, CA, USA) and Sigma Aldrich (Milwaukee, WI, USA), respectively.
Binding Experiment on Agarose
Pepsin and carbonic anhydrase (CA) immobilization was carried out by adapting a procedure published elsewhere [14]. The following buffers were used for the binding experiment with pepsin: citric acid and NaHPO4 (0.1 M, pH 4.5) were used as the coupling buffer; the blocking buffer was Tris HCl (1.0 M, pH 4.5); the incubation buffer was ammonium acetate (0.02 M, pH 4.0); and the wash buffer was a solution of 1 M NaCl in coupling buffer. Similarly, for CA, the coupling buffer was phosphate buffered saline (PBS) (0.1 M, pH 7.4); the blocking buffer was Tris HCl (1.0 M, pH 7.4); the incubation buffer was ammonium acetate (0.02 M, pH 7.4); and the wash buffer was a solution of 1 M NaCl in coupling buffer. Both pepsin and CA were dissolved in 100 μL of coupling buffer and were dialyzed for 1 h with the coupling buffer using a 0.025 mm nitrocellulose drop dialysis membrane. A disposable plastic column was filled with coupling resin and washed with 2 mL of coupling buffer. Then, dissolved protein was added (in the amounts described below) followed by addition of 1 M NaCNBH4 prepared in coupling buffer, until the final concentration of NaCNBH4 is 50 mM. The column was rocked overnight and washed with 10 mL of coupling buffer followed by 5 mL of blocking buffer. Thereafter, 1 mL of blocking buffer was added, followed by 1 M NaCNBH4, until the final concentration is 50 mM, and the mixture was rocked for 2 h. Next, the column was washed with 10 mL of coupling buffer, 10 mL of wash buffer, and 15 mL of incubation buffer, respectively. Finally, the immobilized protein was transferred to Eppendorf tubes for the incubation with the library compounds.
The quantities of protein used for testing different libraries are as follows: for the limited library, 10 mg of pepsin was immobilized onto 1000 μL of resin beads. For the experiments with larger libraries, significantly less protein was used. For the 176 compound library, 450 μg of pepsin was coupled to 250 μL of resin beads. For the 352 compound library, initial experiments used 100 μg of pepsin coupled to 100 μL of resin beads. Optimization of the amount of protein used for screening was carried out using a 352 compounds library, with 100, 50, 25, 12.5, 6.25, and 3.125 μg of pepsin in 100 μL of resin beads. For the carbonic anhydrase experiments, 120, 60, 30, and 6 μg of protein was coupled to 100 μL of resin beads.
Library compounds were diluted from 14.2 μM to 150 nM in incubation buffer prior to incubation with the immobilized protein. Then, 200 μL of the library mixture was added to 100 μL of immobilized protein mixture to make the final concentration of the library compounds 100 nM. Subsequently, the mixture was rocked for 1 h at room temperature. After incubation, tubes were centrifuged at 3000 × g for 5 min; the supernatant was removed and directly used in the LC-MS analysis. All experiments using agarose were repeated and at least three times. No changes were detected in the percent of compounds that ionized in the control experiments (described later) or the identified inhibitors from the binding experiments (described above).
Alternative Binding Procedure Using Magnetic Beads
Maltose binding protein (MBP) was immobilized on N-hydroxysuccinimide (NHS)-activated magnetic beads by following the manufacturer’s protocol (Pierce NHS-Activated Magnetic Beads, Thermo Scientific, Rockford (Illinois), USA). Briefly, after washing the magnetic beads per the manufacturer’s instructions, 100 μg of maltose binding protein in coupling buffer (PBS, 0.1 M; pH 7.4) was added to 300 μL of magnetic beads and slowly rocked overnight at room temperature. The supernatant was removed and the immobilized protein was washed with 1 mL of coupling buffer. After that, 300 μL of quenching buffer (Tris.HCl, 1.0 M, pH 7.4) was added and slowly rocked for 2 h. The supernatant was removed and the immobilized protein was washed with 300 μL of washing buffer (1M NaCl in coupling buffer) followed by 2 mL of ammonium acetate (0.02 M, pH 8.0) incubation buffer. The supernatant was removed again and 50 μL of 352 compounds library mixture and a known inhibitor of maltose binding protein, maltotriose, was added. The final concentration of the library compounds was 100 nM. The mixture was then rocked for 1 h at room temperature. After incubation, the supernatant was removed and directly used in the LC-MS analysis. It was assayed three times to assure that the results were reproducible. No changes were detected in the percent of compounds that ionized in the control experiment (described next) or the inhibitors identified in the binding experiment (described above).
Control Experiments
Coupling resin (agarose or magnetic beads) without protein was used for the control experiments. It was treated in the same way as described for its corresponding binding experiment, except no protein was added. The same sets of library compounds, at the same concentrations as described above, were combined with the resin. For each control experiment, all conditions were identical to the matched binding experiment, and the LC-MS analysis, described below, was also conducted identically, for both the control experiments and the binding experiments.
Capture and Release Experiment, an Alternative Testing Method
The 352 compound library and a known strong inhibitor, ethoxzolamide, was incubated with 60 μg of immobilized carbonic anhydrase. The concentrations of the library compounds and the incubation conditions were replicated from the binding experiment. After incubation, the supernatant was removed, and the immobilized protein was washed five times with incubation buffer. In each washing step, 1 mL of incubation buffer was added and vortexed for 10 s, followed by centrifugation at 3000 × g for 5 min. After washing was complete, 200 μL of acetonitrile was added to denature the protein, and the sample was kept at 70 °C for 10 min. The sample was centrifuged at 3000 × g for 5 min and the supernatant was analyzed using LC/MS.
LC/MS Analysis
Liquid chromatography/mass spectrometry analysis was done using an Acquity UPLC system (Waters Corporation, Milford, MA, USA) coupled to an Orbitrap Velos Pro mass spectrometer. Mobile phase A was 99.9% water with 0.1% formic acid, and mobile phase B was 99.9% MeOH with 0.1% formic acid. For each run, 5 μL of the supernatant was injected onto a C18 Hypersil Gold column (particle size: 5 μm; 1 mm i.d × 100 mm, 175 Å, Thermo Electron Corporation, Thermo Fisher Scientific, Pittsburgh, Pam YSA) at a flow rate of 50 μL/min. The following multi-step gradient was used for the limited library: the column was equilibrated at 98% solvent A for 5 min. Solvent B was linearly increased to 30% in 3 min, followed by a linear increase of solvent B to 60% in 10 min, and a final linear increase to 98% in 2 min, where the solvent composition was maintained for an additional 3 min. The multi-step gradient used for the large libraries (176 compounds, and 352 compounds) was as follows: 100% solvent A for 5 min, then a linear increase of B to 10% in 5 min, followed by the linear increase of B to 60% in the next 30 min, followed by another linear increase to 85% in an additional 10 min, followed by a linear increase to 95% in the final 7 min, where the column was maintained for another 3 min. A short wash and a blank run were carried out to ensure there was no sample carryover between runs. The eluent was diverted to waste for 5 min at the beginning of each run except for the experiments conducted with maltose binding protein, where the time was reduced to 1 min, to prevent salts and DMSO from entering the MS source. The mass spectrometer was operated in the positive ion mode with a 3 kV potential on the ESI needle, and the capillary temperature was set at 250 °C. Full scan MS data were acquired at a mass range of m/z 150–1000 using the Orbitrap mass analyzer at a resolution of 30,000 for m/z 400.
All LC-MS data were interrogated using the MS analysis software, Apex (Sierra Analytics, Modesto, CA, USA), which identifies compounds based on both mass match and a match of the isotopic distribution. The molecular formulas were input, and the software extracted chromatograms of each compound from the.raw data files. All analyses were conducted using a 5 ppm mass accuracy threshold and an MS similarity score of 0.05. The spectral data of the compounds that were not identified by the software were manually identified. A mass accuracy of 5 ppm and the correct isotopic cluster pattern, especially the presence of C13 isotopic peak, were required for a positive identification during manual analysis. Blank runs for both the binding and control experiments were analyzed in order to eliminate any false identification of compounds that could arise due to background contaminants.
Results and Discussion
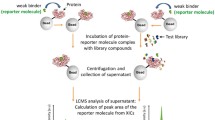
Figure 1 shows a workflow of the screening method described in this manuscript. Coupling resin is used for both the binding assay and the control experiment. In the binding assay, the protein is immobilized on the resin. The resin, without protein, is used for the control experiment. After immobilization, the remaining active sites on the resin, for both the binding and control experiments, are blocked using Tris. The resins (agarose, or magnetic beads) are then incubated with the library compounds, and the unbound compounds are detected by analyzing the supernatants using LC-MS. The compounds that are absent in the spectrum acquired from the binding experiment compared with that of the control are considered bound to the protein. It should be noted here that the control experiment is used to determine which compounds are detectable in the assay. Any compound not detectable in the control experiment could be assayed by a different ionization method, such as APCI. However, these compounds are not false positives or false negatives. They are simply “unassayable in this experiment.”

Workflow used herein. The inhibitors/binders were incubated with the protein (and, separately, with blocked beads), removed, and analyzed by LC-MS. Strong binders were identified by comparing the spectral data of the control and binding experiments. The binders are present in the data for the control experiment but absent in the data for the binding experiment
The effectiveness of the approach was demonstrated in several experiments. First, the feasibility of the method was assessed using immobilized pepsin and a limited (seven compound) protease inhibitor library, so the results could be compared directly with a previous method upon which this one was loosely based [14]. Next, larger libraries of drug-like candidates were incubated with the protein target, to demonstrate that the method was amenable to larger sets of compounds. Subsequently, the amount of protein was reduced, to demonstrate the method was scalable to very large screens where thousands of compounds could be tested. We demonstrated that the assay could be tuned to only identify inhibitors with the strongest binding constants by optimizing the amount of protein used. Finally, we demonstrated that the assay was transferrable to other protein/inhibitor analyses and to detecting inhibitors with Ki’s approaching the micromolar range.
Feasibility Study
A well-studied, limited protease inhibitor library [14] was used for proof of concept. Pepsin, immobilized on agarose, was incubated with a small number of protease inhibitors, including one strong pepsin inhibitor, pepstatin A, and the raw data from this experiment is shown in Figure 2. Overlaid selected ion chromatograms (SICs) of the seven compound inhibitory library incubated with blocked resin beads (from the control experiment) and after incubation with immobilized pepsin (from the binding experiment) are shown in Figure 2a and b, respectively.

Selected ion chromatograms of known protease inhibitors, from the (a) control and (b) binding experiments. Pepstatin A (blue trace) was not detected in the binding experiment (b) because it is a strong binder compared with other compounds. Seven known protease inhibitors were used in both experiments. They are (in order of increasing retention time): antipain (black); leupeptin (red); bestatin (green); (+)-diisopropyl L tartarate (olive); N-acetyl-3,5-diiodo-Tyr (orange); peptstatin A (blue); cholic acid (purple)
Analysis of data from both the control experiment (Figure 2a) and binding experiment (Figure 2b) shows only one compound, pepstatin A, is absent in the binding experiment, indicating it is the single strong-binding inhibitor. This result is significant because pepstatin A is the strongest binder to pepsin in the group of compounds. In earlier work by Cancilla et al. [14], several compounds were identified as potential inhibitors, including pepstatin A. In the previous experiment, any compound whose MS signal was reduced, compared to the signal obtained prior to incubation with the protein, was identified as a potential inhibitor. Therefore, weak and/or nonspecific binders were detected as possible hits. In contrast to the protocol by Cancilla et al., the strongest binders can be exclusively identified if the detection strategy requires that the binder is completely absent after incubation with the protein. This change in detection strategy was intended to eliminate false positives and weak binders that are commonly detected in MS-based screening assays. The method was further adapted for high throughput screens by reducing the protein requirements and increasing the number of compounds to be analyzed in one batch, as described next.
Testing Large Libraries
The number of compounds that were incubated with immobilized pepsin was increased ~20-fold in order to investigate the method’s capacity for screening large numbers of compounds. Figure 3 illustrates the results from screening 176 compounds in one analysis while simultaneously scaling back the protein consumption by more than a factor of 20, from 10 mg to 450 μg. Figure 3a shows a representative TIC from this experiment, demonstrating that the library compounds elute throughout the course of the experiment. Each compound in the library was searched individually in the dataset using Apex, as described in the Experimental section. Comparison of data from the control experiment and binding experiment shows only one compound, the known inhibitor, is present in the control and absent in the binding experiment. Data in Figure 3b and c show the selected ion chromatograms for pepstatin A, demonstrating that it is identifiable as a pepsin binder. These data clearly demonstrate the method’s potential for high specificity, high throughput screens.

(a) Selected ion chromatogram (SIC) of pepstatin A in (a) overlaid on TIC. (b) SIC of pepstatin A in the control experiment, and (c) in the presence of 450 μg of pepsin in the binding experiment. Pepstatin A was not detected in the binding experiment because it is a strong binder. A large library of 176 compounds and the known inhibitor (each with 100 nM concentration) were used in all experiments
In an effort to further test the method, the number of library compounds was doubled again (from 176 to 352), and the amount of pepsin used for the binding experiment was further reduced from 450 μg to 100 μg. Again, only the known inhibitor is absent comparing the data from the control experiment and the binding experiment; see Supplemental Figure 1. The number of detectable compounds in the control experiment was 313, or ~88% of the total number of compounds assayed. The compounds that were not detectable in the control experiment (~12%) would need to be assayed by another method. They are not false positives or false negatives. Overall, the results from the 176 compound library and the 352 compound library demonstrate that large numbers of compounds can be analyzed in batches and that the protein necessary for these types of experiments is substantially lower than originally reported in Reference [14].
Optimization of the Protein Amount
One important measure of a good of high throughput screening method is the ability to test large numbers of compounds on small quantities of protein. Therefore, we asked the question: what is the minimum quantity of protein needed to detect pepstatin A, while still screening >350 compounds at a time? Figure 4 illustrates the MS data collected in a number of experiments where the amount of pepsin was systematically reduced in order to determine the minimum amount of the protein needed for assay to remain successful. (A successful assay is one in which the known, strong inhibitor, peptstatin A, does not appear in the selected ion chromatogram when analyzing the supernatant from the binding experiment.) Several quantities of pepsin were tested, including 50, 25, 12.5, and 6.25 μg. In each case, the pepstatin A was not detected in the supernatant from the binding experiments. However, when the amount of pepsin was reduced to 3.125 μg, the inhibitor finally appeared in the chromatogram (Figure 4b). These data demonstrate that the high throughput screening campaign could be conducted on a scale that consumed as low as 6 μg (171 pmol) of protein to screen batches of >350 compounds. We expect that this protein quantity is at the low end of what one would want to use in this kind of assay, particularly if weaker-binding ligands are targeted. This inhibitor has a very low Ki, 45 pM, so it is likely to be a very strongly binding inhibitor. Additional experiments, below, indicate that the assay can be done on less than 100 μg of protein per well, even when lower-affinity binders are to be detected. In each of the experiments described above, all the other compounds that are observed in the control experiment, except the inhibitor, were also observed in the binding experiments, demonstrating that zero false positives were identified, which is a key strength of this screening method.

Selected ion chromatogram of pepstatin A in (a) control experiment overlaid on TIC, and in the presence of (b) 0 μg (control), 50, 25, 12.5, 6, and 3 μg of pepsin in binding experiments. A library of 352 compounds and the known inhibitor (each with 100 nM concentration) were used in all experiments
Application to a New Protein, Carbonic Anhydrase (CA)
A second protein target, carbonic anhydrase (CA) immobilized on agarose beads, along with the 352 compound library described above, was used to further demonstrate the feasibility of this screening method. The library was spiked with two known CA inhibitor standards; ethoxzolamide with Ki of 700 pM, and sulpiride with Ki of 63 μM. The data in Figure 5a show the TIC for the control experiment, and Figure 5b contains the data from the binding experiment. In each case, the SICs of two known inhibitors, ethoxzolamide (red) and sulpiride (blue) are overlaid in the Figures. Inserts show the mass spectral data for each inhibitor. Approximately 88% (312) of the compounds were detected from the 352 compounds in the control experiment (Figure 5a). All 312 compounds that were observed in the control experiment were also observed in the binding experiment, except ethoxzolamide, the strong inhibitor (Figure 5b). Even though sulpiride is known to be a CA inhibitor, it was not identified as such in this experiment since the Ki is rather high (63 μM). These data demonstrate that only the strongest binders are identified using this approach.

TIC of the (a) control and (b) binding experiments (with 120 μg of CA), including selected ion chromatograms of two known CA inhibitors, ethoxzolamide (red), and sulpiride (blue), overlaid. Ethoxzolamide was not detected in the binding experiment because it is a strong binder. Every other compound detected in the control experiment (a) was also detected in the binding experiment (b), including the weak binder. This experiment shows that zero false positives were detected and that the assay is selective for the strongest binder, when more than one binder is present
We optimized the assay to determine the minimum amount of protein required to detect ethoxzolamide. These data are presented in Supplemental Figure 2. When 60 μg of CA was used, ethoxzolamide was not detected. When the amount of CA is reduced in half, significant spectral data were observed for the inhibitor, and even more intense spectral data were observed with the use of 6 μg of CA. These results indicate that approximately 60 μg of the protein is needed to identify this inhibitor, which has a Ki of 700 pM. We demonstrated in Figure 4 that the amount of protein required for this assay is substantially less, if stronger binding inhibitors are to be identified. Specifically, just 6 μg of protein was necessary to test 352 compounds and selectively identify an inhibitor with a Ki of 45 pM. In comparing the data herein for carbonic anhydrase (6 pmol protein per compound was required to identify a compound with a Ki of 700 pM) and the previous assay (0.5 pmol of protein per compound was required to identify a compound with a Ki of 45 pM), one can see that the amount of protein required for the assay is roughly proportional to the Ki for the compound to be detected. The strongest binding inhibitors can be detected with very low quantities of protein.
Detecting Lower Affinity Ligands
To screen for the inhibitors in Ki ~ μM range using the immobilization conditions described above, a large amount of protein would be required. To successfully conduct the HAMS assay with lower affinity ligands, the protein can be immobilized on magnetic beads instead of agarose. This change in immobilization conditions is necessary because the beads can then be removed from the supernatant without centrifugation, so weak protein–ligand complexes are not disturbed while separating the proteins from the ligands.
To demonstrate that the HAMS method can identify inhibitors with Ki’s approaching the μM range, a screening was conducted using maltose binding protein (MBP), immobilized on magnetic beads, and a 352 compound library, which was spiked with a known MBP inhibitor, maltotriose, with a Ki of ~ 0.2 μM. The data in Supplemental Figure 3a show the SIC of maltotriose in the control experiment, where the peak is clearly detectable. The maltotriose peak completely disappears in the binding experiment, as shown in Supplemental Figure 3b. The data clearly illustrate that maltotriose binds to the protein and can readily be detected as a hit in the HAMS assay. Similar to the previous assays, not one false positive hit was detected. Every compound that appeared in the control experiment was also detected in the binding experiment, except the known inhibitor. These data show that it is fully feasible to screen for inhibitors with Ki approaching = 0.2 μM using the HAMS assay. In this case, less than 7 pmol of protein was needed per compound screened.
Comparison to Alternative Testing Methods
One major benefit of the described method is its consistent ability to identify only the strongest inhibitors, substantially eliminating false positive interactions. Other MS-based screening methods have not yet been capable of identifying only the strongest inhibitor present. With this in mind, we asked the question: Did we just get lucky and test a bunch of compounds that would not show up as false positives in other assays either? To answer the question, the library of 352 compounds was used to determine how many false positives would have been identified as carbonic anhydrase binders in the ASMS method, a state-of-the-art screening approach.
To screen the compounds using the ASMS method, the 352 potential binders (used in Figure 5) were incubated with CA; the non-binders were removed, and the protein was washed using stringent conditions, as described in the Experimental section. The protein was deactivated, and released compounds were analyzed using LC-MS, following the procedure described in Reference [9]. Using this approach, one could still detect the presence of the strong inhibitor. However, in addition to this compound, 30 other non-inhibiting compounds were also detected. (See Supplemental Table 1.) This experiment emphasizes the advantage of the newly described protocol over existing methods. Even though the protein was washed thoroughly prior to releasing the bound compounds, almost 10% of the ligands were detected as potential binders. Thus, the traditional high throughput screening approach may leave the investigators with numerous compounds for further investigation, when in fact only one of them would be desirable to detect in this case, the known inhibitor with Ki in the pM range.
Even though the ASMS method suffers from a large number of false positives, it is perhaps best described as complementary to the HAMS method, instead of inferior to it, when all metrics are considered. The ASMS method’s two key advantages are its rapid throughput and simple data analysis. The throughput of the HAMS method is about 5000 compounds per day, whereas the ASMS method can process more than twice as many compounds in the same timeframe. Additionally, the ASMS method, which relies on detecting inhibitors based on the presence of their MS signal, has a simpler data analysis workflow compared to HAMS, which relies on disappearance of the analyte’s signal. Therefore, these two methods are quite complementary. A method that could deliver the speed and simplicity of ASMS, along with the complete absence of false positives, like HAMS, would be a remarkable advancement in the field.
Investigation of the Undetected Compounds
In all the large screening assays we conducted, ~88% of compounds were observed for both the control and binding experiments. In other words, ~12% of the compounds were not detected in the control experiment and, therefore, not assayable. They were not false positives or false negatives. They were simply not testable compounds. If these compounds were not detectable due to the assay conditions or due to matrix effects, that would represent a potential weakness that would need to be considered prior to selecting this assay for a large screen. Therefore, we examined the reasons behind their absence in the data sets.
The undetectable compounds from the carbonic anhydrase screen were tested individually, to determine if they were undetected due to poor ionization efficiency, or due to the assay, or due to matrix effects. Each compound was prepared individually and subjected to direct infusion experiments, but none of the compounds were detected in the positive ion mode during these experiments, indicating that all the compounds that possibly can be observed using positive mode ESI were successfully detected in the carbonic anhydrase screen. The undetected compounds also were individually tested in negative ion mode, and only 17 were detected. These compounds could potentially be detected if the LC-MS analyses were to be repeated in negative ion mode; however, the value of that experiment would have to be balanced against the analysis time required to perform it. In sum, 7% of the compounds did not ionize at all by ESI. Those compounds were, therefore, not detected in the screen. Of the remaining 93% that were ionizable by ESI, 312 out of 329 were detected in one chromatographic run, using only positive ion mode, with no gradient optimization performed. Overall, these experiments demonstrate that the number of detectable compounds is not limited by the assay conditions or matrix effects; rather, it is directly related to the ability of the compounds to ionize by ESI-MS. Every MS-based assay suffers from the fact that it cannot detect compounds that do not ionize. The HAMS assay is unique in that it can identify these compounds as “untestable,” and they are not mistakenly characterized as false positives or false negatives.
Conclusion
Inhibitors were quickly identified for three different proteins by comparing the mass spectra of the library molecules before and after incubation with immobilized protein. In every experiment conducted, all the compounds that were observed in the control experiment were also observed in the binding experiments, except the known inhibitors. This observation indicates that the method described herein is unprecedented for MS-based assays in that the false positive detection rates are exceedingly low, approaching zero. This method was used to selectively detect inhibitors that have Ki values of 50 pM (with pepsin), 700 pM (with carbonic anhydrase), and the minimum amount of protein needed for these assays, 0.5 and 6 pmol per compound, respectively, was used for screening. These results indicate that one can control the affinity of the ligands detected by controlling the amount of protein used in the assay, a feature that has not been demonstrated by other MS-based screening assays. By using magnetic beads for protein immobilization, inhibitors with Ki’s in the micromolar range could also be assayed, using just 7 pmol of protein per compound screened. Finally, the method compares very favorably with the state-of-the-art MS-based assay (ASMS) [9], where the false-positive identification rate was nearly 10% for the ligands assayed herein.
References
Johnston, P.A., Foster, C.A., Shun, T.Y., Skoko, J.J., Shinde, S., Wipf, P., Lazo, J.S.: Development and implementation of a 384-well homogeneous fluorescence intensity high-throughput screening assay to identify mitogen-activated protein kinase phosphatase-1 dual-specificity protein phosphatase inhibitors. Assay Drug Dev. Technol. 5, 319–332 (2007)
Lim, M.J., Liu, Z.Y., Braunschweiger, K.I., Awad, A., Rothschild, K.J.: Correlated matrix-assisted laser desorption/ionization mass spectrometry and fluorescent imaging of photocleavable peptide-coded random bead-arrays. Rapid Commun. Mass Spectrom. 28, 49–62 (2014)
Brabek, J., Constancio, S.S., Shin, N.Y., Pozzi, A., Weaver, A.M., Hanks, S.K.: CAS promotes invasiveness of Src-transformed cells. Oncogene 23, 7406–7415 (2004)
Makley, L.N., Gestwicki, J.E.: Expanding the number of ‘druggable’ targets: non-enzymes and protein–protein interactions. Chem. Biol. Drug Des. 81, 22–32 (2013)
Deng, G.J., Sanyal, G.: Applications of mass spectrometry in early stages of target based drug discovery. J. Pharm. Biomed. 40, 528–538 (2006)
Jonker, N., Kool, J., Irth, H., Niessen, W.M.A.: Recent developments in protein-ligand affinity mass spectrometry. Anal. Bioanal. Chem. 399, 2669–2681 (2011)
Gross, M.L., Chen, G., Pramanik, B.N.: Protein and Peptide Mass Spectrometry in Drug Discovery, p. 464. Wiley, Hoboken (2012)
Muckenschnabel, I., Falchetto, R., Mayr, L.M., Filipuzzi, I.: SpeedScreen: label-free liquid chromatography-mass spectrometry-based high-throughput screening for the discovery of orphan protein ligands. Anal. Biochem. 324, 241–249 (2004)
O'Connell, T.N., Ramsay, J., Rieth, S.F., Shapiro, M.J., Stroh, J.G.: Solution-based indirect affinity selection mass spectrometry-a general tool for high-throughput screening of pharmaceutical compound libraries. Anal. Chem. 86, 7413–7420 (2014)
Annis, D.A., Nazef, N., Chuang, C.C., Scott, M.P., Nash, H.M.: A general technique to rank protein-ligand binding affinities and determine allosteric versus direct binding site competition in compound mixtures. J. Am. Chem. Soc. 126, 15495–15503 (2004)
Johnson, B.M., Nikolic, D., van Breemen, R.B.: Applications of pulsed ultrafiltration-mass spectrometry. Mass Spectrom. Rev. 21, 76–86 (2002)
Zehender, H., Mayr, L.M.: Application of high-throughput affinity-selection mass spectrometry for screening of chemical compound libraries in lead discovery. Expert Opin. Drug Discovery 2, 285–294 (2007)
Brown, N., Zehender, H., Azzaoui, K., Schuffenhauer, A., Mayr, L.M., Jacoby, E.: A chemoinformatics analysis of hit lists obtained from high-throughput affinity-selection screening. J. Biomol. Screen. 11, 123–130 (2006)
Cancilla, M.T., Leavell, M.D., Chow, J., Leary, J.A.: Mass spectrometry and immobilized enzymes for the screening of inhibitor libraries. Proc. Natl. Acad. Sci. U. S. A. 97, 12008–12013 (2000)
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental figure 1
Selected Ion Chromatogram of Pepstatin A in (A) Control experiment overlaid on TIC, (B) in the control experiment, and in the presence of (C) 100 μg of pepsin in the binding experiment. 352 library compounds and known inhibitor (each with 100 nM concentration) were used in all experiments. (DOCX 91 kb)
Supplemental figure 2
(A) SIC of Ethoxzolamide in (A) the control experiment overlaid on TIC and in the presence of (B) 0 μg (Control), 120 μg, 60 μg, 30 μg, and 6 μg of CA in binding experiments. 352 library compounds and two known CA inhibitors (each with 100 nM concentration) were used in all experiments. (DOCX 192 kb)
Supplemental figure 3
Selected ion chromatograms of known maltose binding protein (MBP) inhibitor: maltotriose, in (A) control and in the presence of (B) 100 μg of MBP in the binding experiments. 352 library compounds and known inhibitor (each with 100 nM concentration) were used in all experiments. (DOCX 52 kb)
Supplemental Table 1
The list of compounds that were detected as false positives in the capture and release experiment. (DOCX 584 kb)
Rights and permissions
About this article

Cite this article
Imaduwage, K.P., Go, E.P., Zhu, Z. et al. HAMS: High-Affinity Mass Spectrometry Screening. A High-Throughput Screening Method for Identifying the Tightest-Binding Lead Compounds for Target Proteins with No False Positive Identifications. J. Am. Soc. Mass Spectrom. 27, 1870–1877 (2016). https://doi.org/10.1007/s13361-016-1472-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-016-1472-3