
Abstract
The objective of this study was to develop a physiologically based pharmacokinetic (PBPK) model to predict the concentrations of encapsulated and free doxorubicin in plasma and tissues in mice after intravenous injection of PEGylated liposomes (Doxil®). The PBPK model used in this study contains liposomes and free doxorubicin disposition components. The free doxorubicin disposition component was used to simulate the disposition of free doxorubicin produced by mononuclear phagocyte system (MPS)-degrading liposomes. The liver, spleen, kidneys, and lungs contain an additional MPS subcompartment. These compartments are interconnected through blood and lymphatic circulation. The model was validated strictly by four doses of external observed plasma and tissue concentration–time profiles. The fold error (FE) values were almost all within threefold. The sensitivity analysis revealed that the MPS-related parameters greatly influenced the model. The predicted in vivo distribution characteristics of the doxorubicin liposomes and doxorubicin solution were consistent with the observed values. The PBPK model was established based on the physiological mechanism and parameters of practical significance that can be measured in vitro. Thus, it can be used to study the pharmacokinetic properties of liposomes. This study also provides a reference for the establishment of liposome PBPK model.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Doxil® was the first PEGylated long-circulating liposome approved by the FDA in 1995 [1]. It is primarily indicated for the treatment of AIDS-related Kaposi's sarcoma, ovarian cancer, multiple myeloma and other solid tumors [2]. Doxil® achieves high and stable accumulation of doxorubicin inside the aqueous phase of liposomes (approximately 15,000 doxorubicin molecules per vesicle) through loading via ammonium sulfate gradients [3]. The release of the encapsulated drug occurs at an exceedingly slow rate [4]. By modifying the surface of Doxil® with polyethylene glycol (PEG), it becomes difficult for the immune system to recognize, thereby prolonging its circulation time [5]. Clinical studies in humans indicated that its clearance was drastically reduced, at least 250-fold, compared to that of free doxorubicin [3]. The size of Doxil® ranged from approximately 80 nm to 90 nm. Due to its large molecular dimensions, Doxil® has difficulty crossing the dense microvascular endothelial space of normal tissue. This significantly reduces its volume of distribution, approximating the volume of blood, in comparison to the direct administration of free doxorubicin (~ 5 L/kg) [3]. This decrease in volume contributes to the mitigation of cardiotoxicity and various other associated adverse effects [6, 7]. The distribution of nanoparticles, measuring approximately 100 nm in size, is generally ascribed to two mechanisms [8]: (i) crossing the endothelial cell membrane of blood vessels to access tissues and (ii) uptake by macrophages in various tissues, such as the liver, spleen, kidney, and lungs. Lymphatic circulation also plays a role in the distribution of nanoparticles [9]. However, there have been no comprehensive studies on the mechanism of liposome distribution in various tissues. Additionally, it is challenging to distinguish the quantities of liposome-encapsulated drugs and free drugs present in each tissue using experimental techniques [10].
The physiologically based pharmacokinetic (PBPK) model has been widely used to study the pharmacokinetic mechanisms of small molecule drugs and predict in vivo pharmacokinetics and pharmacodynamics (PK/PD) under special conditions [11]. This approach offers an accurate simulation of physiological conditions and compatibility with diverse pharmacokinetic mechanisms, enabling the consideration of separate physiological parameters, drug parameters, and parameters related to drug-physiological interactions. Over the past two decades, in vivo research on the fate of nanoparticles has improved. The application of PBPK models has gradually extended to investigate the fate of nanoparticles in vivo [12,13,14,15,16,17]. At present, some PBPK models of nanoparticles include two blood compartments (arterial and venous) as well as several comprehensive tissue and organ compartments [18,19,20,21]. The distribution of nanoparticles was investigated through membrane permeability limitations. The distribution into organs such as the liver, spleen, and lungs has also taken into consideration the influence of the mononuclear phagocyte system (MPS) [22,23,24], and some studies have included the lymphatic circulation [18, 19, 23, 24] in addition to considering blood circulation. However, there are still some challenges in the establishment of nanoparticle PBPK models, such as a lack of appropriate quantitative biodistribution data, inadequate studies on multiple doses, and insufficient in vitro studies.
The relevant studies on the in vivo characteristics of liposomes based on computational pharmacokinetic models are as follows. Early in 1999, Harashima et al. [25, 26] explored the application of pharmacokinetic/pharmacodynamic (PK/PD) models to study the in vivo behavior of antitumor agents encapsulated in liposomes. They established a compartment model that included blood, tumor, and tissue. In this model, liposomes in the blood are taken up by the reticuloendothelial system (RES). At the same time, free drugs are released from liposomes in the blood and from liposomes in the interstitial fluid of the tumor at a first-order rate and are then distributed to the tissue and tumor cells. Since 2010, PK/PD models with a small number of tissue compartments have been widely used for in vivo studies of liposomes, including studies on their stability [27], intratumoral distribution [28], anti-PEG IgG-mediated clearance [29], encapsulated drug release [30] and disposition [31]. With the deepening of research on the pharmacokinetic properties of liposomes, whole-body PBPK models have been gradually established to study in vivo disposition [32], interspecies scaling [33], anti-PEG antibody-mediated clearance [34, 35], and therapeutic programs for liposomes [36]. Most of the PBPK models use a permeability-limited model, with one exception, which incorporates a deep tissue compartment beside the vascular and extravascular compartments [33]. Mager et al. [32] considered drug release from liposomes in both plasma and tissues and calculated the disposition of both liposomes and released drugs. However, these models generally do not consider the MPS or lymphatic circulation, which are important for the disposition of liposomes. Montanha et al. [37] established a whole-body PBPK model to predict the disposition of pegylated liposomal doxorubicin in rats and humans. In this model, lymphatic circulation and free drug release after uptake by the MPS were considered. However, the establishment of this model is not supported by any observed tissue concentration–time data, leading to significant uncertainties.
In this study, we developed a PBPK model for PEGylated doxorubicin liposomes (Doxil®) to simulate their distribution in various tissues in healthy mice. Compared to previously published liposomal PBPK models, this PBPK model has the following features: (i) it contains a very comprehensive distribution in plasma and 11 other tissues, especially all of which are validated by internal and external observed tissue concentration–time profiles of multiple doses; (ii) the distribution of liposomes and released doxorubicin was considered simultaneously. First, PBPK models for doxorubicin solution were established, and then they were linked to the PBPK model of liposomes to simulate the disposition of free doxorubicin produced by MPS-degrading liposomes. Notably, the predicted concentrations of liposome-encapsulated doxorubicin and free doxorubicin in the tissues were also compared with the observed data. (iii) Cell experimental data, such as the number of macrophages in each tissue and the rate of phagocytosis and degradation of nanoparticles by macrophages, were used in the model. This provides a reference for the correlation between the model parameter values and the measured data of cell experiments. Based on the PBPK model developed in this study, we explored the key parameters that influence the distribution of PEGylated liposomes.
Methods
Model structure
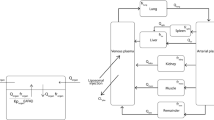
In this study, a PBPK model was established to predict the concentration–time profiles of doxorubicin in plasma and 11 other tissues following intravenous injection of PEGylated doxorubicin liposomes (Doxil®) in mice. The model incorporates two components, the disposition of liposomes and the disposition of free doxorubicin in the body, connected through the degradation of liposomes releasing doxorubicin. Therefore, it can simultaneously predict the concentrations of encapsulated and free doxorubicin in tissues, as shown in Fig. 1.

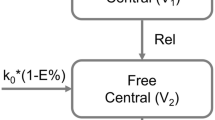
Model structure of the PBPK model in mice after intravenous injection of pegylated liposomal doxorubicin (Doxil®). A) Whole-body model. B) Tissue structure containing the MPS subcompartment of liposomes. C) Model structure of perfusion-limited distribution for free doxorubicin disposition. D) Tissue structure for liposome disposition. E) Model structure of the permeability-limited distribution of free doxorubicin
In the liposome disposition component of the model, each tissue is set according to a permeability-limited mechanism, dividing it into vascular and extravascular compartments. This is because for nanoparticle, the blood to tissue distribution is membrane-limited rather than blood flow-limited [38]. For the liver, spleen, kidneys, and lungs, an additional MPS subcompartment is included. These compartments are interconnected through blood and lymphatic circulation. The model assumes that after liposomes are phagocytized from the capillary side and enter the tissue MPS compartment, they are degraded to release free drug. The released free drugs directly enter the corresponding tissues in the free drug disposition part of the model and are processed accordingly. Doxil® has strong stability in the blood circulation and releases almost no drug [3]. Therefore, the model assumes that the degradation of liposomes in the MPS compartment is the sole pathway for drug release. The model assumes that liposomes are not directly cleared by the liver and kidneys but are instead broken down into free drugs by phagocytic cells, after which they are cleared by the liver and kidneys.
Regarding the free drug release disposition component, the lungs, heart, brain, intestine, liver, and kidneys are modeled as perfusion-limited compartments, while the remaining tissues are permeability-limited. An open-loop model was initially established in which each tissue is directly connected to the blood to determine whether to use a perfusion-limited or permeability-limited mechanism based on the fitting results. This model incorporates both hepatic clearance and renal clearance. This is because, in addition to hepatic clearance, doxorubicin is also cleared through the kidneys by 5%—12% [39, 40]. This study established and validated the PBPK model for doxorubicin solutions, which was then integrated into the PBPK model for PEGylated doxorubicin liposomes, to predict the disposition of free drugs released from liposomes.
Model equations and parameters
In vivo, the disposition of liposomes and the free drug released from liposome degradation occur concurrently. Since liposomes do not enter blood cells, the flow and volume of plasma were used in the model for the disposition of liposomes. However, for the free drug model, blood flow and volume data were used. The model includes physiological parameters and pharmacokinetic parameters for both free drug and liposome, as detailed in Tables 1, 2, and 3 [41,42,43].
Doxorubicin Solution PBPK Model
Perfusion—limited tissue.
Heart, brain and intestine:
Lungs:
Liver:
Kidneys:
where Ca_free is the concentration of doxorubicin in arterial blood; Cv_free is the concentration of doxorubicin in venous blood; Ct_free is the concentration of doxorubicin in tissue; Qt is the total blood flow entering tissue; Vt is the total volume of tissue; Kp_t is the tissue to plasma partition coefficient of doxorubicin in tissue; CLli is the intrinsic clearance rate of doxorubicin in the liver; GFR is the glomerular filtration rate of doxorubicin, which is 19 mL/h [40]; Rb is the concentration ratio of doxorubicin in whole blood to plasma reaching dynamic equilibrium, which is 1.3 [44]; and fupl is the plasma protein binding rate of doxorubicin, which has a value of 0.29 [40, 44].
Permeability—limited tissues.
Muscle, adipose, stomach, pancreas, spleen and rest of body:
where Vt_vas is the capillary volume of tissue; Vt_exv is the extravascular volume of tissue; Ct_vas_free is the concentration of doxorubicin in the vascular compartment of tissue; and Ct_exv_free is the concentration of doxorubicin in the extravascular compartment of tissue. PSt: the membrane permeability of doxorubicin in tissue.
Artery:
Vein:
where Vb is the total blood volume and QC is the cardiac output.
Doxorubicin Liposomal PBPK Model
Non-MPS tissue:
where Ct_vas is the concentration of encapsulated doxorubicin in the capillary plasma of tissue; Ct_exv is the concentration of encapsulated doxorubicin in the extravascular part of tissue; Ca is the concentration of encapsulated doxorubicin in arterial plasma; Qt_p is the plasma flow of tissue; Qt_v is the venous plasma flow from tissue; Lt is the lymphatic flow from tissue. It is 0.2% of the plasma flow [45]; Kp_n is the tissue-to-plasma partition coefficient for liposomes in each tissue (n = 1–3); CTn is the liposome membrane permeability coefficient of tissue (n = 1–4); and σ is the lymphatic reflection coefficient, which is set to 0.2 [45].
MPS-containing tissues:
MPS cells are involved in the uptake, release, and degradation of liposomes. They play a crucial role in the liposome distribution process, with the following parameters and equations for their disposition process:
where Kup_t is apparent rate constant for MPS uptake of liposomes in tissue, obtained after adjusting Kup_MPS for plasma flow and tissue weight; Kre_MPS_t is the rate constant for the release of liposomes by MPS in tissue; Kdeg_MPS_t is the rate constant for the degradation of liposomes into free drug by MPS in tissue; and Nt is the number of MPS cells per gram of tissue. In this study, the number of MPS cells was set differently for each tissue type: 10,000,000 cells/g in the liver, 30,000,000 cells/g in the lungs, 4,500,000 cells/g in the spleen, and 4,500,000 cells/g in the kidneys [22, 37]; Wt: the weight of tissue; Kup_MPS, Kre_MPS, and Kdeg_MPS are the rate constants for a single MPS cell's uptake of liposomes, release of liposomes, and degradation of liposomes into free drug, respectively.
Spleen and kidneys:
where AMPS_t is the mass of encapsulated doxorubicin in the MPS compartment of the tissue.
Liver:
Lungs:
where Cv is the concentration of encapsulated doxorubicin in venous plasma and Qlu_a is the arterial plasma flowing out from the lungs.
Artery:
Vein:
Lymph:
where Vp is the volume of the plasma compartment and Vlymph is the volume of the lymph node compartment.
Model fitting
In this study, the concentration–time profiles of doxorubicin in blood/plasma and various tissues after injecting doxorubicin solution and Doxil® were all derived from reference [46]. The experimental data were obtained by administering a 5 mg/kg dose of doxorubicin hydrochloride solution and Doxil® (calculated as doxorubicin) via intravenous injection to healthy mice. Blood, plasma, brain, adipose, heart, intestine, kidney, liver, lung, muscle, pancreas, spleen, and stomach samples were collected at 0.08, 0.17, 0.25, 0.5, 0.75, 1, 2, 4, 7, 16, 24, 48 and 72 h postadministration and analyzed using high-performance liquid chromatography tandem mass spectrometry (LC‒MS/MS) (n = 3).
Model fitting was performed using the maximum likelihood estimation method in ADAPT5 (Biomedical Simulation Resource, University of Southern California, Los Angeles, California). During the establishment of the doxorubicin liposomal PBPK model, an independent PBPK model for doxorubicin solution was first established using in vivo doxorubicin data and, upon validation, was integrated with the doxorubicin liposomal PBPK model. The parameters fitted for the doxorubicin solution model included Kp_t, PSt, and CLli. Parameters fitted for the doxorubicin liposomal PBPK model included Kp_n, CTn, Kup_MPS, Kre_MPS and Kdeg_MPS.
The variance model used in this study is as follows:
The variance for each data point i is represented as Vi, where Yi denotes the model prediction for the i-th data point. The parameters σ1 and σ2 are variance model parameters.
Internal and external validation of the model
This study undertook both internal and external validation for the developed PBPK models of doxorubicin solution and doxorubicin liposomes. During internal validation, the optimally fitted concentration–time profiles in tissues of both models were compared with the corresponding observed concentration–time profiles in tissues [46]. Quantitative assessments were conducted using the average fold error (AFE) and the absolute average fold error (AAFE) for the concentration–time profiles of each tissue. If the AFE and AAFE are both less than 2, the simulation can be considered successful.
For the external validation of the doxorubicin solution model, the established model was used to predict the concentration–time profiles in each tissue of mice after intravenous injection of doxorubicin solution at doses of 6 mg/kg, 10 mg/kg, and 12 mg/kg, and the results were compared with observed measured data from references [47, 48]. Gustafson et al. [47] administered doxorubicin solution intravenously to healthy mice at 6 mg/kg and collected tissue samples from the blood, liver, intestine, kidney, heart at 5, 10, 30, 60, 120, and 480 min for analysis using high-pressure liquid chromatography (HPLC). In addition, the concentration–time profiles in serum, liver, intestine, kidney and heart of 10 mg/kg were also obtained from this literature. Johansen, [48] administered 12 mg/kg doxorubicin solution intravenously to healthy mice and collected tissue samples from the plasma, heart, liver, kidney, and muscle at 5 min, 30 min, 2 h, 8 h, and 24 h and analyzed them using HPLC.
For the external validation of the doxorubicin liposomal PBPK model, the established model was used to predict the concentration–time profiles in various tissues of mice after intravenous injection of Doxil® at doses of 10 mg/kg, and 15 mg/kg, and the results were compared with in vivo measured data from the literature [49,50,51]. Mehrabian et al. [49] administered Caelyx® (equivalent to Doxil®, both of which are the same formulations marketed in the USA and Europe) intravenously at 10 mg/kg to healthy mice; collected plasma samples at 1, 6, 24, and 48 h; and collected samples from the spleen, lung, kidney, heart, and liver at 24 and 48 h; and analyzed them using fluorescence spectroscopy. Mashreghi et al. [50] administered Caelyx® at 10 mg/kg to mice bearing C26 colon cancer tumors via the tail vein and collected tissue samples from the plasma, liver, spleen, kidney, lung, and heart at 3, 12, 24, 48, and 72 h and analyzed them using fluorescence spectroscopy. Gheibihayat, S. M. et al. [51] injected Doxil® at 15 mg/kg into the tail vein of healthy mice and collected plasma, spleen, kidney, lung, heart and liver at 0.5, 6, 12, 24, 48, 72, 96, and 168 h, after which doxorubicin concentrations were measured via spectroscopy.
The fold error (FE) was determined by comparing the predicted and observed concentration values at each time point along the pharmacokinetic curve. A model is considered to exhibit satisfactory accuracy in predicting whether the FE ranges between 0.3 and 3 [52]. This is because external validation may bring errors by detection methods, individual variance, inter-laboratory differences, etc. The formulas are as follows:
where Cpredicted_i is the predicted concentration at time i and Cobserved_i is the observed concentration at time i.
The pharmacokinetic datasets used in the fitting and validation of PBPK models were shown in the Table 4.
Exploration of the quantities of free Doxorubicin and liposome-encapsulated doxorubicin in tissues
In the current study of drug distribution after liposome administration, the primary focus is on detecting the total amount of drug in tissues, as it is more challenging to measure the concentrations of encapsulated and free drugs in tissues separately. The doxorubicin liposomal PBPK model established in this study can simultaneously predict the pharmacokinetic curves of both encapsulated and free drugs in various tissues. Thus, the established model was used to calculate the ratio of encapsulated to free doxorubicin in tissues at each time point.
Wang et al. [10] used a two-step solid-phase extraction method to separate and measure the concentrations of free and encapsulated doxorubicin in the plasma, liver, heart, spleen, lung, and kidney. The specific dosing regimen was as follows: tumor-bearing mice received an intravenous injection of 7 mg/kg homemade PEGylated doxorubicin liposomes. After administration, plasma and tissue samples were systematically collected at intervals of 1, 12, and 36 h. This study used the doxorubicin liposomal PBPK model to predict the pharmacokinetic curves of each tissue at this dose and compared them with the measured data. Although the formulation used in the in vivo experiment was a homemade PEGylated liposome, the model in this study was based on the in vivo data of the commercial PEGylated liposome (Doxil®).
Sensitivity analysis
This research utilized a standardized sensitivity analysis methodology [8, 53] to conduct parameter sensitivity analysis on various components of the doxorubicin solution PBPK model, including Kp_t, PSt and CLli. Likewise, a similar sensitivity analysis was applied to the doxorubicin liposomal PBPK model, evaluating Kp1, Kp2, Kp3, CT1, CT2, CT3, CT4, Kup_MPS, Kdeg_MPS and Kre_MPS. The methodology included the following procedures: for the doxorubicin solution PBPK model, the impact on the area under the curve (AUC) in various tissues was calculated following a 1% increase in each parameter. For the doxorubicin liposomal PBPK model, the impact on the AUC0-4, AUC0-24, and AUC0-72 for the encapsulated drug in different tissues was assessed after a 1% parameter increase. The specific computational formulas employed are as follows:
where NSC represents the normalized sensitivity coefficient, dAUC denotes the change in the AUC after altering the parameter, and dp represents the change in the value of the parameter according to the sensitivity analysis. AUC0-4, AUC0-24, and AUC0-72 represent the early distribution, late distribution, and metabolic phases of Doxil®, respectively. A higher NSC indicates greater sensitivity of the PBPK model to changes in the corresponding parameter [8].
The impact of lymphatic circulation on the in vivo distribution of doxorubicin liposomes
This study incorporated lymphatic circulation into a doxorubicin liposomal PBPK model and investigated its impact on the in vivo distribution of liposomes. The rate of lymph flow in various organs and tissues, an essential physiological parameter, was incorporated into the model based on values cited in the literature [45]. The lymph reflection coefficient (σ) reflects the propensity of liposomes to translocate from tissues into lymphatic vessels. In this study's doxorubicin liposomal PBPK model, σ was set to 0.2 [45]. This section involved setting σ to 0, 0.2, 0.8, and 1 and then calculating the percentage exposure in each tissue.
Result
Validation of the doxorubicin solution PBPK model and doxorubicin liposomal PBPK model
The optimal fitting parameters for the doxorubicin solution PBPK model are shown in Table 2. These findings are consistent with the reported pharmacokinetic parameter characteristics of doxorubicin [3]. The best-fit concentration–time profiles for each tissue and the corresponding observed concentration–time profiles are shown in Fig. 2. They almost entirely overlap. The AFE and AAFE values for the doxorubicin solution PBPK model (Table 5) were less than 2, except for those for blood, which had an AAFE value of 2.13. This is because of the small overestimation of the last few data points of the elimination phase, as shown in Fig. 2. This study conducted an external validation of the doxorubicin solution PBPK model with three doses from two different literature sources. As shown in Fig. 3, the FE of most data points is within the range of 0.3–3. There was a slight overestimation for the last two time points of the 12 mg/kg dose concentration–time profile for blood, which may be related to individual differences in the experimental animals. In conclusion, based on internal and external validation, the doxorubicin solution PBPK model is accurate and reliable. It can be integrated into the doxorubicin liposomal PBPK model for further model development.

Doxorubicin concentration–time profiles in all tissues after intravenous injection of doxorubicin solution (5 mg/kg) in mice. The measured doxorubicin concentrations in the blood and tissues are indicated by red circles (n = 3) [46], and the black solid lines show the model fit

The optimal fitting parameters of the doxorubicin liposomal PBPK model are shown in Table 3. The best fit concentration–time profiles for each tissue and the corresponding observed profiles are depicted in Fig. 4. The AFE and AAFE values are shown in Table 5. The predicted and observed concentration–time profiles overlap significantly for most tissues, with only slight overestimations in the intestine (AAFE = 2.57) and a minor underestimation in the blood (AAFE = 2.32). This study conducted an external validation of the established doxorubicin liposomal PBPK model, drawing on data from four publications and three dosage levels. As shown in Fig. 5, the model accurately predicts the concentration–time profiles in the blood, heart, liver, and kidneys across various dosages, with only occasional discrepancies exceeding a threefold error at specific time points. However, the prediction for lung tissue was less accurate, significantly overestimating the concentration of doxorubicin in the lung tissue, with multiple time points exceeding a threefold error margin.

Doxorubicin concentration–time profiles in all tissues after intravenous injection of Doxil® at a single dose of 5 mg/kg in mice. The observed doxorubicin concentrations in plasma and tissues are indicated by blue circles (n = 3) [46], and the blue solid lines show the model fitting values. The red solid lines represent the calculated free doxorubicin concentration in tissues. The black solid lines represent the calculated encapsulated doxorubicin concentration in tissues

Exploration of the quantities of free doxorubicin and liposome-encapsulated doxorubicin in tissues
Within 72 h postadministration, the proportion of free drug in plasma fluctuated within the range of 0.01–1.64%. This is consistent with reports in the literature indicating that the proportion of free drug in circulation is less than 2% [3]. The relatively low percentage of free drug in plasma could be attributed to two factors: on the one hand, Doxil® is relatively stable in blood, showing minimal release; on the other hand, free doxorubicin has a large distribution volume [3] and high affinity for various tissues, facilitating its rapid distribution into these tissues. In other tissues, the proportion of free drug was initially low after administration but gradually increased over time, as shown in Fig. 4.
As illustrated in Fig. 6, the model developed in this study accurately predicted the concentration–time profile for both free and encapsulated drugs in the heart, lung, and kidney, with all the FEs remaining within a threefold margin of error. However, predictions for the spleen (encapsulated doxorubicin: time = 1 h, FE = 0.16, time = 36 h, FE = 0.27; free doxorubicin: time = 1 h, FE = 0.22) and liver (liposome-encapsulated doxorubicin: time = 12 h, FE = 0.18, time = 36 h, FE = 0.17; free doxorubicin: time = 12 h, FE = 0.19; time = 36 h, FE = 0.26) were less accurate, with some time points exceeding the threefold error threshold. This discrepancy may be attributed to differences in the physicochemical properties of the formulations used in the literature, which were homemade PEGylated doxorubicin liposomes, compared to the commercial PEGylated liposomal doxorubicin (Doxil®) used in this study. Additionally, the literature utilized tumor-bearing mice, whereas the model in this study was based on data from healthy mice, suggesting that physiological differences between the experimental animals may also contribute to variations in the in vivo processes of free and encapsulated drugs. However, this comparison directly confirmed that the model's ability to predict the amount of free and encapsulated drugs in each tissue was within reasonable limits.

Doxorubicin concentration–time profiles in tissues after intravenous injection of 7 mg/kg homemade PEGylated liposomes. The observed free and encapsulated doxorubicin concentrations in plasma and tissues are indicated by red and blue circles, respectively (n = 3). The red solid lines show the predicted free doxorubicin concentration. The blue solid lines show the predicted encapsulated doxorubicin concentration [10]
Sensitivity analysis
For the PBPK model of doxorubicin solution, Kp_t had a significant impact on the AUC0-72 for the corresponding tissues but had a lesser effect on other tissues. PSt had a relatively minor influence on the AUC0-72 for all tissues. CLli considerably affected the AUC0-72 for each tissue (Table 6). This finding is consistent with the characteristics of a free drug PBPK model [54].
For the doxorubicin liposomal PBPK model, Kp_n significantly influenced the AUC0-4, AUC0-48 and AUC0-72 of the encapsulated drugs in the corresponding tissues (Table 7, Table 8 and Table 9). As Kp_n increases, the drug exposure in that tissue also increases. Kp_n represents the retention of nanoparticles in the corresponding tissue. Additionally, the Kp_n of non-MPS tissues had a more pronounced effect on the AUC0-4 h of corresponding tissue (brain: NSC = 0.93, muscle: NSC = 0.89, spleen: NSC = 0.64, kidney: NSC = 0.64 in Table 7). This is because, for MPS tissues, Kp_n is not the sole factor governing liposome distribution. Liposomes can also enter tissues through uptake by MPS cells. In the model, a 1% increase in CTn had a minimal impact on the AUC0-4, AUC0-48 and AUC0-72 for all tissues. This might be because crossing the endothelial cell membrane of blood vessels is not the primary route for liposomes to enter tissues.
An increase in the concentration of the parameter Kup_MPS significantly affected the distribution of encapsulated drugs in all tissues, especially as shown in Table 9. Kup_MPS controls the rate at which nanoparticles are transported from plasma to tissues through MPS cells, and the drugs entering the MPS compartment are further degraded into free drugs. Therefore, Kup_MPS essentially acts as a clearance pathway for liposomes. As the amount of Kup_MPS increased, the AUC0-4, AUC0-48 and AUC0-72 for each tissue decreased. According to the NSC values, an increase in Kdeg_MPS had little impact on the AUC0-4, AUC0-48 and AUC0-72 for each tissue. This study further assessed the effect of Kdeg_MPS on the AUC0-72 of encapsulated drugs in MPS compartments and revealed a significant impact (liver: NSC = -0.99, spleen: NSC = -0.99, kidneys: NSC = -0.99, lungs: NSC = -0.99). This is because Kdeg_MPS is directly related to the rate of liposome degradation. The increase in the parameter Kre_MPS had almost no effect on the plasma or any other tissue, which might be due to the minimal amount of liposomes taken up by the MPS being returned to the bloodstream.
The impact of lymphatic circulation on the in vivo distribution of doxorubicin liposomes
The permeability of the lymphatic vessel walls varies for different liposomes. A σ value of 0 implies that all liposomes can enter the lymphatic circulation, whereas a σ of 1 indicates that liposomes cannot enter the lymphatic system. As shown in Fig. 7, with the change in σ, the drug exposure in the lymph center shows some variation, but the impact is relatively minor. Changes in other tissues were also not significant. This could be attributed to the fact that the volume and flow rate of lymph are much smaller than those of blood; thus, lymphatic transport is not the main pathway for the distribution of these substances.

The percentage of tissue exposed to Doxil® (% AUC0-72 h) after intravenous injection. The blue diagonal bar charts represent σ = 0, the blue dot bar charts represent σ = 0.2, the dark blue bar charts represent σ = 0.8, and the blue line bar charts represent σ = 1
Comparison of tissue distribution characteristics in mice predicted by the doxorubicin solution PBPK model and the doxorubicin liposomal PBPK model
Using the established PBPK models, the concentration–time profiles of doxorubicin in various tissues of mice were calculated for both doxorubicin solution and Doxil® administration. The area under the concentration–time profiles was calculated to compare the tissue distribution characteristics of the two formulations. As depicted in Fig. 8, PEGylated liposomes altered the tissue distribution characteristics of doxorubicin. Compared to the doxorubicin solution, PEGylated liposomes increased the plasma exposure of doxorubicin, providing it with prolonged circulation capabilities and enhancing its opportunity to enter tumor tissues. Moreover, they reduced the proportion of doxorubicin exposure in other tissues, thereby lowering toxicity.

The predicted tissue distribution of doxorubicin after intravenous injection of doxorubicin solution or Doxil® 5 mg/kg
Following Doxil® administration, doxorubicin distribution was greater in the liver (6.46%), spleen (14.76%), kidneys (9.92%), and lungs (7.26%) than in non-MPS tissues such as muscle (1.30%), stomach (1.77%), and fat (0.56%). These findings are consistent with the tissue distribution characteristics of doxorubicin solution and PEGylated liposomes reported in the literature [46].
Discussion
The doxorubicin liposomal PBPK model established in this study includes MPS compartments in the liver, spleen, kidneys, and lungs. The number of MPS cells per gram of each tissue was derived from the literature sources [22, 37, 55,56,57]. The methods for obtaining these data from the literature included separation, purification, staining, and subsequent measurement of the number of phagocytic cells in various mouse tissues via flow cytometry.
Kup_MPS, Kre_MPS and Kdeg_MPS represent the rates of uptake, release, and degradation of nanoparticles by individual MPS cells, respectively. Kup_MPS can be determined by quantitatively measuring the uptake of nanoparticles by mouse macrophage lines at different time points using colorimetric methods [22]; Kre_MPS can be obtained through model fitting [22]; and Kdeg_MPS can be calculated by fitting the release curves of nanoparticles in macrophage lines [58]. Montanha et al. [37] used the parameters obtained through the aforementioned methods as initial values in the model establishment process and performed fitting optimization. This study adopted the optimized parameter values from that publication as initial values and further optimized them during the model-building process.
The parameters of Kup_MPS, Kre_MPS and Kdeg_MPS for individual MPS cells were adjusted for tissue plasma flow and tissue weight when applied to different tissues. However, as the types and activities of phagocytic cells vary among tissues, this study introduced correction factors for Kup_MPS in different tissues: lung, 0.51; spleen, 0.01; and kidney, 0.83. These values were obtained during the model optimization process. Henrique et al. [22] reported that the activity of MPS cells fluctuates within the range of 70% ± 20%. In this study, the correction factor for the spleen was relatively low. The analysis suggested that there are two pathways for liposomes to enter the spleen: one is through transcapillary passage, and the other is uptake by the spleen's inherent phagocytic cells. However, there are no empirical data supporting the proportions of nanoparticles entering tissues through these two pathways. Therefore, there might be an overestimation of transcapillary passage and underestimation of phagocytic uptake during the model fitting process.
This study conducted rigorous internal and external validation of the accuracy of the concentration–time profile predictions for each tissue in both the doxorubicin solution model and the doxorubicin liposomal PBPK model. Additionally, the doxorubicin liposomal PBPK model was externally validated using the observed concentration–time profiles of encapsulated and free drugs measured in various tissues. The overall validation results of the model are good, but it is noteworthy that the predictive accuracy of the doxorubicin liposomal PBPK model is not as high as that of the doxorubicin solution PBPK model. For example, in external validation, the doxorubicin liposomal PBPK model tends to overestimate the doxorubicin concentration—time profile in the lung. The factors contributing to the poor prediction of lung tissue in the external validation were analyzed. On the one hand, while the concentration of liposome in the lung is significantly influenced by MPS, it is important to consider that individual variations in immune system response. It is easily influenced by health status, saturation degree and experimental environment. Consequently, this leads to discrepancies in measured data across different laboratory and dose. On the other hand, there remains a lack of comprehensive research on the PBPK modeling techniques about the uptake rates, degradation rate and release rate of nanoparticles by MPS. In this study, the parameter obtained from in vitro cell experiments and actual number of macrophages in tissues. This is a scientific approach. However, further investigation is still required to establish a reliable conversion method for translating intrinsic rates measured in vitro into their corresponding in vivo values.
The free drug release of liposome is an important in vivo process. The release mechanism was tried to add in the PBPK model. Based on the structure of the PBPK model in this study, a first-order drug release from liposome mechanism was incorporated into both the arterial and venous compartments. Then the same dataset was used for fitting. The model parameters, AFE and AAFE of these two models were compared. There was no significant difference in accuracy between the two models, and no reduction in AFE and AAFE in plasma compartments after comparison. The fitted first-order release rate constant (Kre) was 0.0002614. However, the inclusion of the release rate parameter in the plasma compartment lacks support from the measured data. This may increase the instability of the model. Particularly, if the release process is added to various tissues, it will be impossible to distinguish whether the released of free drugs is due to the degradation by MPS or the release of liposomes. This would cause a lot of uncertainty into the model. Therefore, in this study, considering the specific dosage form and model stability, we did not incorporate drug release mechanism in plasma compartment.
This study also calculated the proportion of free doxorubicin to total doxorubicin exposure in various tissues over 0–72 h, as shown in Fig. 9. In the plasma, heart, and brain, the proportion of free doxorubicin was less than 50% (plasma: 0.54%, heart: 42.88%, brain: 4.52%). This is also a theoretical basis for the reduced cardiotoxicity of doxorubicin when it is formulated into liposomes. In the pancreas and intestines, the proportion of free doxorubicin was very high (pancreas: 94.58%, intestines: 99.39%). This is attributed to the smaller CT in these organs, making it more difficult for liposomes to pass through capillary gaps into these tissues and hence decreasing the distribution of liposome-encapsulated doxorubicin. However, free doxorubicin has a greater affinity for these two tissues, and free doxorubicin generated by degradation in other tissues can more easily distribute to these organs via blood circulation. According to the doxorubicin liposomal PBPK model, the in vivo distribution of liposomes is influenced by the permeability of capillaries in various tissues and their uptake by tissue phagocytes, with the latter being the primary distribution pathway. In addition, after being taken up and degraded by phagocytes into free drug, the inherent tissue affinity of the free drug also affects its distribution.

Proportion of free doxorubicin exposure in tissues after intravenous injection of Doxil® (5 mg/kg) simulated by the doxorubicin liposomal PBPK model
A significant advantage of the PBPK model lies in its ability to facilitate species extrapolation. Specifically, the PBPK model enables prediction of drug concentration at the site of action, which is more directly correlated with drug efficacy compared to drug concentration–time profile in blood. Determining the drug concentration at the pharmacodynamic site in human bodies is often challenging using experimental methods. The application of PBPK models extrapolated from animal to humans can effectively address this issue. In general, when extrapolating from animals to humans using the PBPK model for small molecule drugs, physiological data of animals are directly substituted with human physiological data, and assuming the same Kp values between species, only the clearance needs allometric scaling. However, in the case of nanoparticles, the situation becomes more complicated. Firstly, the MPS exerts a significant impact on the disposition of nanoparticles in vivo, however, both the number and phagocytic capacity of phagocyte are different between humans and animals. This issue may be addressed by determining single human MPS cell’s uptake rate of liposomes, release rate of liposomes, and degradation rate of liposomes into free drug, respectively, then using these parameters in the human PBPK model instead corresponding parameters in the animal PBPK model. At the same time, the number of macrophages in various tissues of human should be used. Alternatively, a quantitative relationship can be established between animal phagocytosis-related parameters and human parameters to solve this problem, just like the allometric scaling of clearance. Secondly, the impact of tissue capillaries on the diffusion limitation of nanoparticles varies between humans and animals, thus, necessitating investigation into whether and how CT values change during species extrapolation. Finally, the disparities in lymphatic circulation between human and animal should be consider when extrapolation.
The model has the following limitations: i) There may be a saturation mechanism for liposome uptake by macrophages, but the model established in this study does not consider any saturation mechanisms. This is because incorporating a saturation algorithm introduces too many uncertain parameters, increasing model instability. ii) The doxorubicin solution PBPK model slightly overestimates the elimination phase in the blood compartment, possibly due to the specific affinity of doxorubicin for certain tissues [44], leading to an underestimation of the hepatic clearance rate during model fitting. The doxorubicin liposomal PBPK model slightly underestimates the elimination phase in the blood compartment at four intermediate time points, possibly related to the redistribution of doxorubicin in the body, although this mechanism is not yet clearly understood. iii) In the model establishment process, the parameters of the doxorubicin solution PBPK model were obtained using the maximum likelihood method, with almost all CV% less than 30%; the doxorubicin liposomal PBPK model was obtained through the maximum likelihood method and empirically.
Conclusion
In this study, a whole-body liposomal PBPK model in mice simulating encapsulated and free doxorubicin concentration–time profiles in plasma and tissues were established successfully and validated by four doses of observed pharmacokinetic data. In addition, the predicted in vivo distribution characteristics of the doxorubicin liposomes and solution were consistent with the observed values. Sensitivity analysis showed that Kup_MPS and Kdeg_MPS had a great influence on the disposition of liposomes. The study of the effect of the immune system on liposome phagocytosis and degradation and the addition of the corresponding parameters to the liposomal PBPK model have greatly improved the prediction accuracy of the model. Overall, the liposomal PBPK model is reliable and can be used to predict the free doxorubicin concentration–time profile in tissues, which is closely related to the pharmacodynamic effect. This model provides a reference for the establishment of a PBPK model using liposomes. It is also a useful tool for studying the pharmacokinetic properties of liposomes.
Data availability
Raw data and the model code are available from the corresponding author on request.
References
Barenholz Y. Doxil(R)–the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117–34.
Sekiya N, Imamura A. [Doxil–pegylated liposomal doxorubicin], Gan to kagaku ryoho. Cancer Chemother. 2008;35:1439–43.
Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42:419–36.
Russell LM, Hultz M, Searson PC. Leakage kinetics of the liposomal chemotherapeutic agent Doxil: The role of dissolution, protonation, and passive transport, and implications for mechanism of action. J Control Release. 2018;269:171–6.
Mohamed M, Abu Lila AS, Shimizu T, Alaaeldin E, Hussein A, Sarhan HA, Szebeni J, Ishida T. PEGylated liposomes: immunological responses. Sci Technol Adv Mater. 2019;20(1):710–24.
Safra T, Muggia F, Jeffers S, Tsao-Wei DD, Groshen S, Lyass O, Henderson R, Berry G, Gabizon A. Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2, Annals of oncology : official journal of the European Society for. Med Oncol. 2000;11:1029–33.
Li XR, Cheng XH, Zhang GN, Wang XX, Huang JM. Cardiac safety analysis of first-line chemotherapy drug pegylated liposomal doxorubicin in ovarian cancer. J Ovarian Res. 2022;15:96.
Lin Z, Monteiro-Riviere NA, Riviere JE. A physiologically based pharmacokinetic model for polyethylene glycol-coated gold nanoparticles of different sizes in adult mice. Nanotoxicology. 2016;10:162–72.
Cai S, Yang Q, Bagby TR, Forrest ML. Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv Drug Deliv Rev. 2011;63:901–8.
Wang H, Zheng M, Gao J, Wang J, Zhang Q, Fawcett JP, He Y, Gu J. Uptake and release profiles of PEGylated liposomal doxorubicin nanoparticles: A comprehensive picture based on separate determination of encapsulated and total drug concentrations in tissues of tumor-bearing mice. Talanta. 2020;208:120358.
Nestorov I. Whole body pharmacokinetic models. Clin Pharmacokinet. 2003;42:883–908.
Byun JH, Han D-G, Cho H-J, Yoon I-S, Jung IH. Recent advances in physiologically based pharmacokinetic and pharmacodynamic models for anticancer nanomedicines. Arch Pharmacal Res. 2020;43:80–99.
Cheng YH, He C, Riviere JE, Monteiro-Riviere NA, Lin Z. Meta-Analysis of Nanoparticle Delivery to Tumors Using a Physiologically Based Pharmacokinetic Modeling and Simulation Approach. ACS Nano. 2020;14:3075–95.
Li M, Zou P, Tyner K, Lee S. Physiologically Based Pharmacokinetic (PBPK) Modeling of Pharmaceutical Nanoparticles. AAPS J. 2016;19:26–42.
Yuan D, He H, Wu Y, Fan J, Cao Y. Physiologically Based Pharmacokinetic Modeling of Nanoparticles. J Pharm Sci. 2019;108:58–72.
Li M, Al-Jamal KT, Kostarelos K, Reineke J. Physiologically based pharmacokinetic modeling of nanoparticles. ACS Nano. 2010;4:6303–17.
Utembe W, Clewell H, Sanabria N, Doganis P, Gulumian M. Current Approaches and Techniques in Physiologically Based Pharmacokinetic (PBPK) Modelling of Nanomaterials. Nanomaterials. 2020;10(7):1267.
Sweeney LM, MacCalman L, Haber LT, Kuempel ED, Tran CL. Bayesian evaluation of a physiologically-based pharmacokinetic (PBPK) model of long-term kinetics of metal nanoparticles in rats. Regul Toxicol Pharmacol. 2015;73:151–63.
MacCalman L, CL T, Kuempel E. Development of a bio-mathematical model in rats to describe clearance, retention and translocation of inhaled nano particles throughout the body. J Physics Conf Ser. 2009;151(1):012028.
Liang X, Wang H, Grice JE, Li L, Liu X, Xu ZP, Roberts MS. Physiologically Based Pharmacokinetic Model for Long-Circulating Inorganic Nanoparticles. Nano Lett. 2016;16:939–45.
Chen J, Yuan M, Madison CA, Eitan S, Wang Y. Blood-brain barrier crossing using magnetic stimulated nanoparticles. J Control Release. 2022;345:557–71.
Henrique Silva A, Lima Jr E, Vasquez Mansilla M, Zysler RD, Mojica Pisciotti ML, Locatelli C, Kumar Reddy Rajoli R, Owen A, Creczynski-Pasa TB, Siccardi M. A physiologically based pharmacokinetic model to predict the superparamagnetic iron oxide nanoparticles (SPIONs) accumulation in vivo. European J Nanomed. 2017;9(2):79-90.
Aborig M, Malik PR, Nambiar S, Chelle P, Darko J, Mutsaers A, Edginton AN, Fleck A, Osei E, Wettig S. Biodistribution and Physiologically-Based Pharmacokinetic Modeling of Gold Nanoparticles in Mice with Interspecies Extrapolation. Pharmaceutics. 2019;11(4):179.
Dogra P, Butner JD, Ramírez JR, Chuang YL, Noureddine A, Brinker CJ, Cristini V, Wang Z. A mathematical model to predict nanomedicine pharmacokinetics and tumor delivery. Comput Struct Biotechnol J. 2020;18:518–31.
Harashima H, Iida S, Urakami Y, Tsuchihashi M, Kiwada H. Optimization of antitumor effect of liposomally encapsulated doxorubicin based on simulations by pharmacokinetic/pharmacodynamic modeling. J Control Release. 1999;61:93–106.
Harashima H, Tsuchihashi M, Iida S, Doi H, Kiwada H. Pharmacokinetic/pharmacodynamic modeling of antitumor agents encapsulated into liposomes. Adv Drug Deliv Rev. 1999;40:39–61.
Qin S, Seo JW, Zhang H, Qi J, Curry FR, Ferrara KW. An imaging-driven model for liposomal stability and circulation. Mol Pharm. 2010;7:12–21.
Hendriks BS, Reynolds JG, Klinz SG, Geretti E, Lee H, Leonard SC, Gaddy DF, Espelin CW, Nielsen UB, Wickham TJ. Multiscale kinetic modeling of liposomal Doxorubicin delivery quantifies the role of tumor and drug-specific parameters in local delivery to tumors. CPT Pharmacometrics Syst Pharmacol. 2012;1:e15.
McSweeney MD, Wessler T, Price LSL, Ciociola EC, Herity LB, Piscitelli JA, Zamboni WC, Forest MG, Cao Y, Lai SK. A minimal physiologically based pharmacokinetic model that predicts anti-PEG IgG-mediated clearance of PEGylated drugs in human and mouse. J Control Release. 2018;284:171–8.
Jablonka L, Ashtikar M, Gao G, Jung F, Thurn M, Preuss A, Scheglmann D, Albrecht V, Roder B, Wacker MG. Advanced in silico modeling explains pharmacokinetics and biodistribution of temoporfin nanocrystals in humans. J Control Release. 2019;308:57–70.
Matsumoto T, Masuo Y, Tanaka A, Kimura T, Ioroi T, Yamakawa T, Kitahara H, Kato Y. A physiologically based pharmacokinetic and pharmacodynamic model for disposition of FF-10832. Int J Pharm. 2022;627:122250.
Kagan L, Gershkovich P, Wasan KM, Mager DE. Dual physiologically based pharmacokinetic model of liposomal and nonliposomal amphotericin B disposition. Pharm Res. 2014;31:35–45.
Lu XF, Bi K, Chen X. Physiologically based pharmacokinetic model of docetaxel and interspecies scaling: comparison of simple injection with folate receptor-targeting amphiphilic copolymer-modified liposomes. Xenobiotica. 2016;46:1093–104.
Talkington AM, Wessler T, Lai SK, Cao Y, Forest MG. Experimental Data and PBPK Modeling Quantify Antibody Interference in PEGylated Drug Carrier Delivery. Bull Math Biol. 2021;83:123.
Talkington AM, McSweeney MD, Wessler T, Rath MK, Li Z, Zhang T, Yuan H, Frank JE, Forest MG, Cao Y, Lai SK. A PBPK model recapitulates early kinetics of anti-PEG antibody-mediated clearance of PEG-liposomes. J Control Release. 2022;343:518–27.
Howell BA, Chauhan A. A physiologically based pharmacokinetic (PBPK) model for predicting the efficacy of drug overdose treatment with liposomes in man. J Pharm Sci. 2010;99:3601–19.
Montanha MC, Howarth A, Mohamed DA, Loier E, Main L, Rosslein M, Delmaar C, Prina-Mello A, Siccardi M. A physiologically based pharmacokinetic model to predict pegylated liposomal doxorubicin disposition in rats and human, Drug Deliv. Transl Res. 2022;12:2178–86.
Li M, Panagi Z, Avgoustakis K, Reineke J. Physiologically based pharmacokinetic modeling of PLGA nanoparticles with varied mPEG content. Int J Nanomedicine. 2012;7:1345–56.
Methaneethorn J, Tengcharoen K, Leelakanok N, AlEjielat R. Population pharmacokinetics of doxorubicin: A systematic review, Asia Pac. J Clin Oncol. 2023;19:9–26.
Lee JB, Zhou S, Chiang M, Zang X, Kim TH, Kagan L. Interspecies prediction of pharmacokinetics and tissue distribution of doxorubicin by physiologically-based pharmacokinetic modeling. Biopharm Drug Dispos. 2020;41:192–205.
Stott WT, Dryzga MD, Ramsey JC. Blood-flow distribution in the mouse. J Appl Toxicol. 1983;3:310–2.
Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–5.
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13:407–84.
Dubbelboer IR, Lilienberg E, Sjogren E, Lennernas H. A Model-Based Approach To Assessing the Importance of Intracellular Binding Sites in Doxorubicin Disposition. Mol Pharm. 2017;14:686–98.
Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39:67–86.
Luo R, Li Y, He M, Zhang H, Yuan H, Johnson M, Palmisano M, Zhou S, Sun D. Distinct biodistribution of doxorubicin and the altered dispositions mediated by different liposomal formulations. Int J Pharm. 2017;519:1–10.
Gustafson DL, Rastatter JC, Colombo T, Long ME. Doxorubicin pharmacokinetics: Macromolecule binding, metabolism, and excretion in the context of a physiologic model. J Pharm Sci. 2002;91:1488–501.
Johansen PB. Doxorubicin pharmacokinetics after intravenous and intraperitoneal administration in the nude mouse. Cancer Chemother Pharmacol. 1981;5:267–70.
Mehrabian A, Dadpour S, Mashreghi M, Zarqi J, Askarizadeh A, Badiee A, Arabi L, Moosavian SA, Jaafari MR. The comparison of biodistribution of glutathione PEGylated nanoliposomal doxorubicin formulations prepared by pre-insertion and post-insertion methods for brain delivery in normal mice. IET Nanobiotechnol. 2023;17:112–24.
Mashreghi M, Zamani P, Moosavian SA, Jaafari MR. Anti-Epcam Aptamer (Syl3c)-Functionalized Liposome for Targeted Delivery Of Doxorubicin: In Vitro And In Vivo Antitumor Studies in Mice Bearing C26 Colon Carcinoma. Nanoscale Res Lett. 2020;15:1–3.
Gheibihayat SM, Jaafari MR, Hatamipour M, Sahebkar A. Improvement of the pharmacokinetic characteristics of liposomal doxorubicin using CD47 biomimickry. J Pharm Pharmacol. 2021;73:169–77.
Tan Z, Zhang Y, Wang C, Sun L. Physiologically based pharmacokinetic modeling of cefadroxil in mouse, rat, and human to predict concentration–time profile at infected tissue. Front Pharmacol. 2021;12:692741.
Deng L, Liu H, Ma Y, Miao Y, Fu X, Deng Q. Endocytosis mechanism in physiologically-based pharmacokinetic modeling of nanoparticles. Toxicol Appl Pharmacol. 2019;384:114765.
Sun L, Wang C, Zhang Y. A physiologically based pharmacokinetic model for valacyclovir established based on absolute expression quantity of hPEPT1 and its application, European journal of pharmaceutical sciences : official journal of the European Federation for. Pharm Sci. 2018;123:560–8.
Ten Hagen TL, Van Vianen W, Bakker-Woudenberg IA. Isolation and characterization of murine Kupffer cells and splenic macrophages. J Immunol Methods. 1996;193(1):81–91.
Bachler G, von Goetz N, Hungerbuhler K. A physiologically based pharmacokinetic model for ionic silver and silver nanoparticles. Int J Nanomedicine. 2013;8:3365–82.
Holt PG, Degebrodt A, Venaille T, O’leary C, Krska K, Flexman J, Farrell H, Shellam G, Young P, Penhale J, Robertson T. Preparation of interstitial lung cells by enzymatic digestion of tissue slices: preliminary characterization by morphology and performance in functional assays. Immunology. 1985;54(1):139.
Xiong XB, Huang Y, Lu WL, Zhang X, Zhang H, Nagai T, Zhang Q. Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J Control Release. 2005;107:262–75.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81603055).
The National Natural Science Foundation of China,No. 81603055,Le Sun
Author information
Authors and Affiliations
Contributions
Le Sun and Xuewei Cao conceived and designed this project. Xuewei Cao, Kejun Li, and Le Sun established the model. Jingyu Wang and Xiaoqian Xie performed the data collection and model validation. Xuewei Cao, Kejun Li and Le Sun wrote the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No ethical approval is required for this study.
Consent for publication
All authors have agreed with publication of the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cao, X., Li, K., Wang, J. et al. PBPK model of pegylated liposomal doxorubicin to simultaneously predict the concentration–time profile of encapsulated and free doxorubicin in tissues. Drug Deliv. and Transl. Res. (2024). https://doi.org/10.1007/s13346-024-01680-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s13346-024-01680-0