
Abstract
Afatinib (AT), an FDA-approved aniline-quinazoline derivative, is a first-line treatment for metastatic non-small cell lung cancer (NSCLC). Combining it with cetuximab (CX), a chimeric human-murine derivative immunoglobulin-G1 monoclonal antibody (mAb) targeting the extracellular domain of epidermal growth factor receptor (EGFR), has shown significant improvements in median progression-free survival. Previously, we developed cetuximab-conjugated immunoliposomes loaded with afatinib (AT-MLP) and demonstrated their efficacy against NSCLC cells (A549 and H1975). In this study, we aimed to explore the potential of pulmonary delivery to mitigate adverse effects associated with oral administration and intravenous injection. We formulated AT-MLP dry powders (AT-MLP-DPI) via freeze drying using tert-butanol and mannitol as cryoprotectants in the hydration medium. The physicochemical and aerodynamic properties of dry powders were well analyzed firstly. In vitro cellular uptake and cytotoxicity study revealed concentration- and time-dependent cellular uptake behavior and antitumor efficacy of AT-MLP-DPI, while Transwell assay demonstrated the superior inhibitory effects on NSCLC cell invasion and migration. Furthermore, in vivo pharmacokinetic study showed that pulmonary delivery of AT-MLP-DPI significantly increased bioavailability, prolonged blood circulation time, and exhibited higher lung concentrations compared to alternative administration routes and formulations. The in vivo antitumor efficacy study carried on tumor-bearing nude mice indicated that inhaled AT-MLP-DPI effectively suppressed lung tumor growth.
Graphical Abstract

Graphical abstract was created with BioRender software (https://biorender.com/)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Non-small cell lung cancer (NSCLC) is a prevalent malignant tumor, accounting for over 80% of lung cancer cases, and associated with high incidence and mortality rates [1]. Due to the inconspicuous nature of its early symptoms and the intricate anatomical structure of the lungs, a majority of patients are diagnosed at advanced stages. Despite the availability of comprehensive treatment approaches including radiotherapy, chemotherapy, and surgery, the overall prognosis for NSCLC patients remains unsatisfactory. In recent years, extensive research has been conducted on epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) as a promising strategy to overcome the limitations of conventional therapies by specifically targeting the epidermal growth factor receptor (EGFR) [2]. Represented by erlotinib and gefitinib, the first-generation EGFR-TKIs have demonstrated significant clinical efficacy in advanced NSCLC patients harboring EGFR-sensitive mutations, such as L858R and 19del, leading to an objective response rate exceeding 79% and prolonged patient survival [3]. Despite the initial success, clinical observations have revealed that the majority of patients experience disease progression within 7–13 months of first-line EGFR-TKI treatment, indicating the development of resistance to EGFR-TKIs [2, 4]. Research has identified the T790M mutation in EGFR as a major cause of resistance to first-generation EGFR-TKIs [5]. To address the issue of acquired resistance, researchers have been continuously exploring updated iterations of EGFR-TKIs [6]. Nevertheless, overcoming drug resistance in lung cancer patients remains a significant challenge in clinical practice.
Afatinib (AT), an orally administered covalent second-generation EGFR-TKI, effectively inhibits the phosphorylation of EGFR and HER2 kinases in the intracellular domain, leading to downstream signaling blockade. Clinical trials have demonstrated the efficacy of combining afatinib with other antitumor agents, such as cetuximab, in the treatment of NSCLC patients who are resistant to gefitinib due to the T790M mutation [7, 8]. Cetuximab (CX) is an EGFR inhibitor that has shown efficacy in various tumor types, including head and neck tumors, colorectal cancer, and NSCLC [9]. It is commercially available in injectable form. Binding specifically to the extracellular domain of EGFR on tumor cells, cetuximab inhibits receptor dimerization, tyrosine kinase phosphorylation, and downstream signal transduction, transmitting apoptotic signals to the nucleus. This mechanism leads to the inhibition of cell proliferation and promotion of apoptosis [10, 11]. The dose-related side effects of monoclonal antibodies often result in decreased quality of life and poor tolerability among patients. Consequently, the combination therapy of traditional oral formulations with injectable formulations has not been widely adopted in clinical practice. Nevertheless, due to cetuximab’s high affinity for the extracellular domain of EGFR, it has become a prominent subject of research as a specific target for EGFR. Numerous studies have successfully incorporated cetuximab into nanocarriers to enhance the targeting capabilities of the formulation [12, 13].
Immunoliposomes are actively targeted liposomes that have been modified with antibodies on their surface, enabling specific recognition and enhanced targeting of tumor cells [14, 15]. The ligands present on the surface of immunoliposomes bind to specific targets on tumor cells, forming target-ligand complexes. Upon binding stimulation, this complex is internalized via endocytosis, leading to the release of the encapsulated drug into the cytoplasm, and exerting therapeutic and cytotoxic effects. In our previous studies, we successfully conjugated cetuximab to afatinib-loaded liposomes using a thioether bond, thereby developing and evaluating a novel active targeting immunoliposomal delivery system for EGFR [16, 17]. Furthermore, injectable formulations of immunoliposomal exhibited robust drug delivery capabilities and effectively inhibited tumor growth in an NSCLC xenograft model. These findings provide evidence supporting the potential of the EGFR-targeted immunoliposomal drug delivery systems for non-small cell lung cancer treatment and pave the way for the future development of targeted formulations.
In recent years, immunoliposomes have gained increasing attention as carriers for tumor drugs, genes, vaccines, and drugs requiring penetration through the blood-brain barrier. However, several challenges still exist in the application of immunoliposomes. Firstly, the preparation process of immunoliposomes is intricate and ensuring the stability of monoclonal antibodies in novel formulations presents difficulties [14]. Secondly, antibody production costs are high and conventional dosages often necessitate large quantities [18, 19]. Thirdly, the ability of antibody-mediated liposome internalization can impact the penetration of the drug delivery system into target cells [20, 21]. Lastly, the development of formulations is currently limited to intravenous administration, thereby restricting broader clinical applicability of immunoliposomes [22].
Inhalable dosage forms offer distinct advantages in circumventing hepatic first-pass metabolism and enabling targeted drug delivery within the lungs, thereby minimizing systemic exposure [23, 24]. Dry powder inhalation (DPI) formulations for respiratory delivery present several benefits, including enhanced stability in solid-state stability and ease of handling [25, 26]. Building upon our previous research, this study aims to develop a dry powder inhalation formulation of cetuximab-modified afatinib-loaded immunoliposomes for pulmonary administration. By optimizing the administration route, we aim to reduce the drug dosage while improving its therapeutic efficacy. A series of experiments has been performed to select the optimal freeze-drying process for the immunoliposomal dry powder inhalations and investigate the relevant physicochemical properties. Additionally, analysis has been performed on aerosolization performance, as well as in vitro and in vivo efficacy and safety of the novel formulation.
Materials and methods
Materials
Afatinib (purity ≥98%) and cetuximab (purity ≥95%) were supplied by McLean Biochemical Technology (Shanghai, China). Hydrogenated soybean phospholipids (HSPC), cholesterol (CH), 1,2-distearoylsn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylenglycol)-2000] (DSPE-PEG2000), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol) 2000] (DSPE-PEG2000-Mal) were purchased from Shanghai Yuanye Biological Co. (Shanghai, China). Pentobarbital sodium and Traut’s reagent were purchased from Sigma (Barcelona, Spain). Dulbecco’s modified Eagle medium (DMEM), RPMI 1640 medium, and fetal bovine serum were obtained from Gibco (Grand Island, NY, USA). Bicinchoninic acid (BCA) kits were purchased from Solarbio (Beijing, China). 3-[4,5-dimethylthiazol-2yl]-2,5-diphenyl-tetrazolium bromide (MTT) was obtained from Dalian Meilun Biotech (Dalian, China). DAB chromogen kit was purchased from Biocare Medical (Pacheco, CA, USA). Ki-67 primary antibody was obtained from Thermo Fisher (WA, USA). All other reagents and solvents utilized in the study were of analytical grade.
Human non-small cell lung cancer cells (A549) and melanoma cells (B16) were provided from American Type Culture Collection (Manassas, VA, USA). Male Sprague–Dawley rats 200 ± 20 g and male BALB/c nude mice 25 ± 5 g were purchased from Jinan Pengyue Experimental Animal Breeding Co., Ltd (Jinan, China).
Methods
Preparation of AT-MLP
The afatinib liposome (AT-LP) was prepared using the ammonium sulfate gradient method, which was based on a previous study conducted by our research group [16, 17]. In brief, a mixture of HSPC, CH, DSPE-PEG2000, and DSPE-PEG2000-Mal was dissolved in anhydrous ethanol and then evaporated to form a lipid film under reduced pressure in a water bath at 65 °C. Subsequently, a solution of 200 mmol/L ammonium sulfate (6 mL) was added to hydrate the lipid film for 20 min. The hydrated film was sonicated in a water bath for 20 min and dialyzed in physiological saline for 6 h. Afterward, a solution of afatinib (1.5 mg/mL) was added and incubated at 60 °C for 10 min to obtain AT-LP.
To develop the afatinib-loaded immunoliposome (AT-MLP), thiolated cetuximab (pre-treated with Traut’s reagent) was incubated with AT-LPs for 20 h at 18 °C under a nitrogen atmosphere. The resulting AT-MLP was then purified using sepharose CL-4B gel filtration. The linkage of CX to AT-MLP was confirmed using the bicinchoninic acid (BCA) protein assay kit at 562 nm for mAb.
Preparation of AT-MLP-DPI
The afatinib-loaded immunoliposome dry powder inhalation (AT-MLP-DPI) was prepared by the freeze-drying method. To optimize the process, various cryoprotectants and lyophilization process parameters were carefully investigated. The selection of freeze-drying protectants was achieved by preparing various AT-MLP-DPI formulations separately with and without cryoprotectants, including sucrose, lactose, trehalose, mannitol, and tert-butanol. Four co-cryoprotectant types and three different ratios of liposomes to tert-butanol (1:0.5, 1:1, 1:2) were further considered. The lyophilization process was examined by three different pre-freezing temperatures (−20, −40, and –80 °C), two pre-freezing method (slow freezing and rapid freezing), and three varied pre-freezing times (12, 24, and 48 h), respectively.
The preparation of afatinib-loaded liposome dry powder inhalation (AT-LP-DPI) followed the same method as described above. All freeze-dried products were collected gently to minimize fragmentation. The evaluation of the lyophilized powders included assessment of appearance, particle size after redispersion, encapsulation efficacy (%), and redispersibility, which served as evaluation criteria.
Characterization of AT-MLP-DPI
Scanning electron microscopy
The surface morphology of AT-MLP-DPI was observed using scanning electron microscopy (SEM, JEOL, JEM-1400; Japan). A small amount of AT-MLP-DPI powder sample was sprinkled onto conductive tape under dry conditions. After removing excess powder, the sample was gold sprayed and examined using a scanning electron microscope operating at 5 kV and magnification ranging from 1000 to 10,000 times.
Particle size and zeta potential
The particle size and zeta potential of AT-MLP-DPI were measured using dynamic light scattering (DLS) with a laser particle size analyzer (Zeta PALS, Brookhaven Instruments Co., USA). Prior to measurement, the samples were dispersed in pure water. Each measurement was performed three times at a temperature of 25 °C.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) analysis was performed using DSC204 (NETZSCH, Germany). A 10 mg sample was placed in an aluminum pan and sealed. The samples were then heated from 30 to 250 °C at a rate of 10 °C/min under a nitrogen atmosphere.
Moisture sorption
A thermostatically dried glass weighing bottle was taken and placed in a thermostatic drying oven at 25 ± 1 °C overnight, with a saturated solution of ammonium sulfate placed at the bottom to maintain a relative humidity of 80 ± 2%. The weight (w1) of the bottle was precisely measured. An appropriate amount of the sample was then evenly spread at the bottom of the weighing bottle, and the weight (w2) was precisely measured. The bottle, along with the cap, was left open under the aforementioned thermostatic and constant humidity conditions for 24 h. After covering the weighing bottle with the cap, the weight (w3) was precisely measured.
The moisture absorption rate was calculated using the following formula:
Flowability
Tap density and bulk density: A specific quantity of AT-MLP-DPI was weighed, with three replicates per group, and placed in a graduated cylinder. The initial volume (V1) was recorded, followed by gentle vibration and observation of any volume changes in the cylinder. Once no significant change in volume was observed, the final volume (V2) of the powder in the cylinder was recorded. The difference between the weights of the cylinder before and after adding the sample represented the mass (m) of the added powder. The bulk density (ρb) and tap density (ρt) were calculated as follows: bulk density: ρb = m/V1, tap density: ρt = m/V2.
Angle of repose: The flowability of the powder was determined by measuring the angle of repose for AT-MLP-DPI using the fixed funnel method. The funnel was aligned with the center of a Petri dish, and the sample powder was poured into the dish at an inclined angle until a conical shape was formed. The height (h) and radius (r) of the cone were measured, and the tangent value of the angle of repose (tanβ = h/r) was calculated.
In vitro drug release
The in vitro release of afatinib in formulations was determined by packing 1 mg of dry powder into dialysis bags (3500 Da) containing 1 mL of release medium (PBS, pH 7.4, with 0.1% Tween80). The bags were then incubated in 30 mL of PBS (pH 7.4) and continuously agitated at 100 rpm at 37 °C. At different time points, 2 mL of sample was withdrawn from the release medium, and an equal volume of fresh release medium was added. The concentration of AT was determined using UV spectrophotometry (UV-2450; Shimadzu, Kyoto, Japan) at 343.6 nm, and the cumulative amount released in release media was calculated.
Aerodynamic particle size analysis
In order to assess the aerodynamic properties of inhalation powder, a next-generation pharmaceutical impactor (NGI) manufactured by MSP Corp. (Shoreview, MN) was employed. Approximately 6 mg of large brittle porous dry powder was manually loaded into size three HPMC capsules, and subsequently aerosolized three times using a HandiHaler® device during a single session. Aerosols were generated for a duration of 4 s at an airflow rate of 60 L/min, to achieve an inhalation volume of 4 L and a pressure drop of 4 kPa across the device. The stage cutoff size diameters were determined as follows: 8.060, 4.460, 2.820, 1.660, 0.940, 0.550, and 0.340 μm for stages 1 through 7. Subsequently, the dry powders deposited in the pre-separator, adaptor, throat, and stages 1-MOC were collected and extracted with 10 mL of a mobile phase composed of methanol and H2O in a ratio of 70:30. The obtained samples were subjected to analysis via high-performance liquid chromatography (HPLC) to quantify the AT content. Each formulation was evaluated using three replicates to ensure accuracy and reliability of the results.
For each test, the total emitted dose (TED), geometric standard deviation (GSD), mass median aerodynamic diameter (MMAD), and fine particle fraction (FPF) were calculated according to the guidelines outlined in US Pharmacopeia (USP), using the dose deposited on stages 1-MOC as the basis for calculation.
Stability
Accelerated test: To evaluate the stability of the formulation, three parallel AT-MLP-DPI dry powder samples were subjected to an accelerated test by storing them in a constant temperature and humidity chamber maintained at 40 ± 2 ℃ and 75 ± 5% relative humidity. Sampling was performed at the end of 0, 1, 2, and 3 months throughout the experimental period.
Long-term test: In addition to the accelerated test, another set of three parallel AT-MLP-DPI dry powder samples was stored under normal conditions in a standard medicine cabinet at 25 ± 2 ℃ and 60 ± 10% relative humidity. Similar to the accelerated test, samples were collected at the end of 0, 3, 6, 9, and 12 months during the experimental period. The variations of formulations in appearance, particle size, encapsulation efficiency, and redispersibility in both stability studies were evaluated.
In vitro studies
Cell culture
A549 cells and B16 cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin–streptomycin at 37 °C in an atmosphere of 5% CO2 atmosphere.
Cytotoxicity study
To evaluate the in vitro antitumor effects of inhalation powders, cytotoxicity was assessed using the MTT assay. A549 and B16 cells were seeded onto 96-well plates at a density of 1 × 104 cells per well and incubated in RPMI 1640 medium for 12 h. Subsequently, the cells were treated with various concentrations of AT solution, AT-LP-DPI, and AT-MLP-DPI, corresponding to AT concentrations of 20, 10, 5, 2.5, 1.25, and 0.625 μg/mL, for 24 and 48 h. Following removal of the culture medium and washing with PBS three times, 20 μL of MTT solution (5 mg/mL) was added to each well and incubated for 4 h. Formazan crystals were dissolved by adding 150 μL of dimethyl sulfoxide (DMSO), and the absorbance was measured at 570 nm. All experiments were repeated three times to ensure accuracy and reproducibility of the results.
Cellular uptake
To assess the cellular uptake of the formulations, coumarin 6 (C6) was utilized as a fluorescent dye for labeling purposes. Dry powders loaded with 100 ng/mL of C6 (C6-MLP-DPI and C6-LP-DPI) were prepared by using the ammonium sulfate gradient method as previously described, which was based on a previous study conducted by our research group. The C6 was encapsuled in liposomes which was armed to marking the entire liposomal delivery process. A549 cells were seeded onto 6-well plates at a density of 5 × 105 cells/well and cultured for 24 h. The culture medium was then aspirated, and the cells were rinsed with PBS three times. Subsequently, free C6, C6-MLP-DPI, and C6-LP-DPI (at concentrations of 10 μg/mL) were added and incubated for 4 h. Following incubation, the cells were washed with cold PBS (4 °C) three times, fixed with 4% paraformaldehyde for 30 min, and the cell nuclei were stained with DAPI. Finally, the cells were visualized using a Cytation 5 imaging system (BioTek, Winooski, VT, USA).
Transwell assay
A549 cells that had been starved for 12 h were seeded in the upper chamber of Transwell inserts. Various concentrations of AT, AT-LP-DPI, and AT-MLP-DPI (2.5, 5, and 10 μg/mL) in culture medium containing 1% fetal bovine serum were added to the upper chamber, while complete culture medium was added to the lower chamber. Following 24 h of incubation, the upper chamber was aspirated, and the cells were fixed with 4% paraformaldehyde for 20 min. Subsequently, the cells were stained with crystal violet for 30 min. The upper chamber was gently wiped using a cotton swab, and the cells were imaged using an inverted fluorescence microscope.
In vivo studies
Sprague-Dawley rats and BALB/c nude mice were used to explore the in vivo distribution, pharmacokinetic study, antitumor effect, and safety evaluation in this study. The rats and nude mice were housed four per cage in a 12 h light/dark cycle with food and water available ad libitum for at least 1 week. Pentobarbital sodium (3%) was used as anesthetic for rats (45 mg/kg) and nude mice (30 mg/kg). To minimize animals suffering and panic, compressed CO2 gas was employed to euthanize animals. All animal experiments were approved by Yantai University Laboratory Animal Ethics Committee, affiliated to Yantai University (approval number, YTDX 20220615).
Pharmacokinetic study
For the pharmacokinetic study, male Sprague-Dawley rats weighing 200 ± 20 g were selected for the in vivo intubation and dry powder insufflation procedure. Twenty rats were divided into five groups: AT solution (intravenous), AT solution (oral), AT solution (inhalation), AT-LP-DPI (inhalation), and AT-MLP-DPI (inhalation). To administer the drug to the lungs, we employed a handheld aerosolizer (YAN30010, Yuyan Instruments, China) and an LS-2 small animal laryngoscope (Penn-Century, Inc., Wyndmoor, PA). In each group, 4 rats were used for blood sample collection (n = 4). The rats received an AT concentration equivalent to 3 mg/kg based on their body weight. Blood samples were collected at predetermined time intervals of 5 min, 30 min, 1 h, 2 h, 4 h, 8 h, 24 h, and 48 h. To evaluate the amount of AT deposited in the lower respiratory tract, we employed a modified bronchoalveolar lavage (BAL) procedure, that 12 rats were used for BAL collection (n = 3 at 0.5 h, 8 h, and 24 h) for each group. All biological samples were stored at −20 °C until analysis. The AT concentrations in the plasma and BAL were quantified using liquid chromatography mass spectrometry (LC/MS/MS), a method previously reported by our group [16, 17].
Biodistribution study
In this study, 18 male Sprague–Dawley rats weighing 200 ± 20 g were allocated into 3 groups (n = 6) and administered AT solution, AT-LP-DPI, and AT-MLP-DPI via pulmonary administration. The rats received an equivalent AT concentration of 3 mg/kg based on their body weight. At predetermined time points (0.5 h, 8 h, and 24 h), six rats were sacrificed from each group, and the heart, liver, spleen, lung, and kidney were carefully extracted. The biological samples were homogenized with 3 mL of PBS and immediately centrifuged at 13,500 × g for 5 min at 4 °C. All biological samples were stored in a – 20 °C freezer until analysis. The AT concentrations in organs were quantified using liquid chromatography mass spectrometry (LC/MS/MS).
Antitumor efficacy
Nine NSCLC tumor-bearing mice (22 ± 2 g) were allocated randomly to 3 groups of 3 (n = 3). PBS control (intravenous), AT + CX solution (inhalation), and AT-MLP-DPI (inhalation) were administered every other day for 2 weeks. To establish the metastatic lung cancer model, B16 cells (5 × 106 cells/animal) were injected through the tail veins of male BALB/c nude mice weighing 25 ± 5 g (24 mice in total). When the body weight decreased to 17–19 g, the tumor-bearing nude mice were randomly divided into 6 groups (n = 4) according to the tumor size and administered PBS control (intravenous), AT + CX solution (intravenous), AT + CX solution (inhalation), AT-LP-DPI (inhalation), AT-MLP-DPI redispersion (intravenous), and AT-MLP-DPI (inhalation). All formulations were administrated with an equivalent AT concentration of 3 mg/kg based on body weight in both models. The body weights of the mice were recorded. Subsequently, all nude mice were sacrificed, and their organs (heart, liver, spleen, lung, and kidney) were harvested for histopathological analysis after fixation and embedding in paraffin. Lung tissues from each group of nude mice were photographed after rinsing with PBS for pulmonary metastatic node observation. The lung tissues were then blotted dry on filter paper and weighed, denoted as the wet weight (W). Subsequently, the lung tissues were placed in a 60 °C oven for 48 h until a constant weight was achieved, recorded as the dry weight (D). The W/D ratio for each group was then calculated.
Immunohistochemical analysis
The expression of Ki-67 in tumor tissues of tumor-bearing nude mice was investigated. Briefly, tumor tissue sections were placed on a high-temperature staining rack inside a pressure cooker. Citrate antigen retrieval solution was added, ensuring that the volume of the retrieval solution consistently covered the tumor tissue. An appropriate amount of peroxidase blocking agent was added to the measured area of the tumor tissue, followed by incubation with Ki-67 antibody for 1 h. Enzyme-labeled polymer was then applied, and the sections were stained using DAB chromogen kit. Subsequently, counterstaining was performed with hematoxylin, and the slides were observed under a microscope.
Statistical analysis
The data are presented as mean ± standard deviation. Statistical analysis was performed using one-way ANOVA, and a p-value of less than 0.05 was considered statistically significant in all analyses.
Results and discussion
Preparation of AT-MLP
The preparation of afatinib liposomes (AT-LP) was carried out using the ammonium sulfate gradient method, following the previous research conducted by our group. To achieve targeted tumor-cell selectivity and therapeutic activity, CX was coupled to the liposomes, resulting in the formation of “immuno-LPs.” The coupling efficiency of CX to LPs was assessed using a BCA kit, which determined the ratio between the initial and final amounts of CX conjugated to LPs. Based on our investigations, the optimal condition for AT-MLP preparation was found to be a HSPC:CX ratio of 500:1 (Mol/Mol), with a coupling efficiency of approximately 78.56%.
Preparation of AT-MLP-DPI
Screening of freeze-drying protectants
To preserve the stability and activity of CX, which was known to be sensitive to high temperature and pressure, AT-MLP was transformed into AT-MLP-DPI through the freeze-drying method. It is crucial to investigate the lyophilization process infectors, such as protective agents and freeze-drying conditions, as they significantly affect the efficacy of the freeze-dried product. In order to maintain the physicochemical stability of liposomal formulations during freeze drying, cryoprotectants were incorporated to minimize damage caused by ice crystals to the lipid vesicles. The selection and concentration of cryoprotectants can have varying effects on liposome preservation. Evaluation was conducted based on various criteria, including appearance, particle size after redispersion, encapsulation efficiency, and redispersibility.
In this study, we chose polyols or sugars as cryoprotectant, as listed in Table 1. When tert-butanol was used as the sole cryoprotectant under identical conditions, the frozen samples exhibited a porous and fragile appearance. Upon reconstitution, minimal changes in liposome size and encapsulation efficiency were observed compared to the pre-freeze-drying state, indicating good dispersibility. The next most effective cryoprotectant was mannitol, whereas the use of sucrose as a cryoprotectant led to significant alterations in particle size and encapsulation efficiency after reconstitution. Therefore, it could be concluded that employing tert-butanol as the sole cryoprotectant was advantageous in enhancing the formulation stability of liposomal freeze-dried powders. In order to obtain the best freezing-drying product, the screening of co-cryoprotectant types was conducted under a fixed total amount of cryoprotectant. When tert-butanol + mannitol was used as the co-cryoprotectant and the volume ratio of liposomes to tert-butanol was 1:0.5, the freeze-dried product exhibited increased looseness, smaller particle size, higher encapsulation efficiency, and better redispersibility.
Screening of freeze-drying process parameters
In order to preserve the activity of CX in inhalation powder, experiments were performed to optimize the freeze-drying process parameters, and results are depicted in Table 2. It has been reported in the literature that achieving optimal pre-freezing effects requires a pre-freezing temperature 10 °C below the sample’s total melting point [27, 28]. An improved state of the freeze-dried product was observed under the same freeze-drying conditions when a lower pre-freezing temperature (−80 °C) was employed. The state of the sample could be influenced by the pre-freezing time. Under the same freeze-drying conditions, a pre-freezing time of 24 h was determined to be the most optimal. Examination of pre-freezing methods revealed that, under otherwise identical conditions, dry powder inhalations prepared using slow-freezing methods demonstrated enhanced fluffiness and yield smaller particle sizes after redispersed.
Characterization of AT-MLP-DPI
Morphology
It is generally recognized that the specific surface area and morphology of API particles can significantly influence the API release characteristics, both in terms of kinetic and thermodynamic effects [29]. The variation in size, density, specific surface area, and morphology can be attributed to the particle formation process. The appearance and micro-morphology of AT-MLP-DPI were characterized using scanning electron microscopy (SEM). As shown in Fig. 1A, AT-MLP-DPI exhibited large porous particles with a homogeneous, brittle, and irregular shape. Despite their geometrically large size, the low density, high porosity, and tendency to undergo brittle fracture upon aerosolization allowed them to have an aerodynamic diameter within the respirable range.

The characterization of afatinib-loaded immunoliposomes. A SEM images of AT-MLP-DPI; B comparison of DSC results; C zeta potential and particle size of AT-MLP-DPI; D moisture sorption of AT-MLP-DPI; E in vitro release profiles
Particle size and zeta potential
After redispersion of AT-MLP-DPI in pure water, the particle size was measured as 148.2 ± 1.4 nm, and the polydispersity index (PDI) was determined to be 0.23 ± 0.02 (Fig. 1C). Compared to that of AT-MLP reported in previous work, although the particle size of AT-MLP-DPI was slightly increased, it remained conducive to effective intracellular delivery. The zeta potential of AT-MLP-DPI was −0.99 ± 0.82 mV as shown in Fig. 1C, indicating a slightly negative charge that was favorable for in vivo circulation.

In vitro deposition profile of micronized AT, AT-LP-DPI, and AT-MLP-DPI
DSC analysis
DSC analysis was performed to investigate the melting and crystallization behavior of AT in different forms, including micronized AT, AT-LP-DPI, and AT-MLP-DPI (Fig. 1B). It showed the melting endotherm peak of micronized AT was at 166.2 °C, indicating a mixture of crystalline forms, which was found to be consistent with the literature [30]. The spikes in AT-LP-DPI and AT-MLP-DPI were absent, which indicated complete drug embedding in the liposomes.
Moisture sorption
Moisture sorption is a critical factor in determining the aerosol characteristics of dry powder formulations. Literature has reported that respirable dry powders prepared with mannitol as an excipient are not significantly affected by high humidity [31]. The moisture sorption study of AT-MLP-DPI demonstrated a slow uptake of moisture as shown in Fig. 1D. Even at a relative humidity of 80%, the moisture uptake remained below 2%. It was worth noting that when tert-butanol and mannitol were used as co-lyophilization protectants, the lyophilized product exhibited poor moisture sorption, thus benefiting the storage stability of the formulation.
Flowability
Flowability is one of the key indicators for evaluating dry powder inhalers. Porous particles with large porosity exhibit good flowability and deposition performance due to their low density and porous structure. The bulk density (ρb) and tap density (ρt) of AT-MLP-DPI were measured as 0.11 ± 0.02 g/cm3 and 0.12 ± 0.04 g/cm3, and the angle of repose (tanβ) was 35.89 ± 1.17°, indicating good flowability and low density of the dry powder inhalations.
In vitro drug release
In this study, the in vitro drug release behavior of the formulation was investigated using PBS (pH 7.4) as the release medium (Fig. 1E). The results revealed a fast release of liposome DPIs at approximately 1 h, constituting a cumulative release rate of about 40%. Subsequently, a sustained-release commenced at 2 h, leading to a cumulative release rate of approximately 60%. Notably, it demonstrated that the liposomal formulations exhibited a prolonged release profile, reaching maximum release at the 12-h mark for both AT-LP-DPI and AT-MLP-DPI. Moreover, AT-MLP-DPI exhibited a slightly higher cumulative release percentage (65.8 ± 2.78%) compared to AT-LP-DPI (61.54 ± 5.68%). AT solutions displayed fast release within the initial 2 h, attaining their maximum cumulative release rate around the 8-h mark. These results collectively suggest that the lyophilization process did not significantly impact the release behavior of liposomes, thereby preserving their sustained release characteristics.
In vitro aerosol performance
Although there are many methods to determine particle size and distribution based on different geometric characteristics or physicochemical properties, aerodynamic diameter is the most relevant parameter to lung delivery and final therapeutic effect. Both gravitational settling and inertial impaction depend on the aerodynamic diameter of particles, and the size distribution directly affects the deposition and distribution of drugs in the lungs and respiratory tract. Therefore, the aerodynamic particle size distribution of the delivered API is a critical quality attribute for pulmonary inhalation products.
Figure 2 shows the in vitro deposition profiles, and Table 3 presents the TED, MMAD, FPF, and GSD values for micronized AT, AT-LP-DPI, and AT-MLP-DPI. A comparison between these values reveals interesting insights into the aerosol performance of different formulations. The FPF values for AT-LP-DPI and AT-MLP-DPI were both more than 25%, and the amount of API deposited on each stage met USP aerodynamic size distribution test. It could be attributed to factors such as the size and morphology of the liposomal particles, which may affect their ability to efficiently reach the lower respiratory tract. The MMADs and GSDs for all formulations were less than 5.0 μm, and TEDs were more than 80%, within the desired range for pulmonary delivery.
Stability
Accelerated stability study demonstrated that the particle size and encapsulation efficiency of AT-MLP-DPI remained relatively stable within a 3-month period (Table 4), suggesting that the prepared dry powder inhalation exhibited satisfactory stability over this duration. Long-term stability study revealed minimal changes in AT-MLP-DPI stored in a sealed condition at room temperature for 12 months, suggesting that the prepared dry powder inhalation could maintain stability over an extended period.
In vitro and in vivo studies
MTT assay
The cytotoxicity assay of blank-LP-DPI and blank-MLP-DPI on A549 at 24 h is shown in Fig. 3A. The viability of cells treated with blank-MLP-DPI decreased as the concentrations of CTX-modified liposomes increased, indicating the sustained effectiveness of CTX after lyophilization. This observation aligns with the findings of our previous study [16, 17]. The cytotoxicity of different preparations was evaluated on A549 and B16 cells using three different concentrations, and the cell viability results after 24 h and 48 h of exposure to the various concentrations of the formulations represented in Fig. 3B–E. The findings revealed a reduction in cell viability that was dependent on the concentration of all three formulations. Additionally, with an extended duration of culture, the cell viability decreased at the same concentration, indicating a positive correlation between the toxicity of the preparations and culture time. Significantly higher cytotoxicity was observed for AT-MLP-DPI compared to free AT and AT-LP-DPI under identical experimental conditions, indicating the advantageous properties of both AT-LP-DPI and AT-MLP-DPI. Notably, AT-MLP-DPI exhibited the most potent toxic effect by markedly reducing the viability of A549 and B16 cells.

Cytotoxicity of blank-LP-DPI and blank-MLP-DPI on A549 at 24 h (A), and different preparations on A549 at 24 h (B) and 48 h (C), and B-16 cells at 24 h (D) and 48 h (E) (*p < 0.05, **p < 0.01)
Uptake study
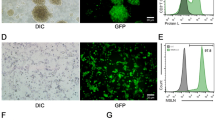
Fluorescence microscopy images of A549 cells exposed to free C6, C6-MLP-DPI, and C6-LP-DPI were acquired at 4 h (Fig. 4A). The nucleus was visualized by blue fluorescence, the formulation by green fluorescence, and the co-localization indicated intracellular uptake. At 4 h, C6-MLP-DPI exhibited a significantly higher fluorescence signal intensity compared to the other two formulations, suggesting superior cellular internalization. The results demonstrated that the modification of liposomes by CX led to an increased cell penetration ability.

CLSM images of A549 cells after 6 h of incubation with free C6 and inhalation powder (A) and representative migration images of A549 cells after treatment (B). Scale bar 20 μm
Transwell assay
The Transwell chamber was utilized to conduct A549 cell invasion experiments, wherein the primary mechanism relied on cells’ pursuit of enhanced nutrient availability, driving them to infiltrate through a matrix gel-containing membrane into the lower compartment. The experimental results are depicted in Fig. 4B, demonstrating that the control group (cells treated with PBS) exhibited robust invasive potential, characterized by a higher number of cells invading the chamber. In contrast, the invasion ability of cells treated with different formulations notably diminished, with the AT-MLP-DPI group displaying the most pronounced effects.
Tissue distribution
The results of tissue homogenate samples are presented in Fig. 5A. At 0.5 h, the AT-LP-DPIinh and AT-MLP-DPIinh groups exhibited lower concentrations in lung tissue compared to the ATinh group. At 24 h, notably elevated concentrations were detected in all organs, particularly in the lungs, within the AT-LP-DPIinh and AT-MLP-DPIinh groups. It suggested that, compared to free drug substance, the absorption of the inhalation powder in pulmonary administration was slower, resulting a longer body retention time. The results depicted in Fig. 5B illustrate the sections of major organs stained with H&E. Examination of the figures reveals that the heart, liver, spleen, lungs, and kidneys exhibited no apparent signs of necrosis or injury. Additionally, as depicted in Fig. 5C, the AT-MLP-DPIinh group exhibited a higher bronchoalveolar lavage concentration at 0.5 h, indicating that the dry powder was absorbed more slowly than the drug solution group (ATinh). This observation was consistent with the distribution of API in lung tissue.

In vivo tissue distribution and pharmacokinetic study. A In vivo tissue distribution of different preparation groups (n = 6); B tissue sections of main organs; C the AT concentration of bronchoalveolar lavage in different preparation groups (n = 3); D mean plasma concentration–time curves in rats (n = 4)
Pharmacokinetic study
Plasma concentrations were measured at different time points and analyzed using LC/MS-MS to calculate pharmacokinetic parameters. The plasma concentration results for different formulations are presented in Fig. 5D and Table 5. With intravenous administration of AT as the reference for AUC0-∞ values, the absolute bioavailability values (more than 50%) of pulmonary-administered liposomal formulations were relatively high (AT-MLP-DPIinh > AT-LP-DPIinh > ATig > ATinh). Moreover, the half-life (T1/2) and mean residence time (MRT0-∞) of pulmonary delivery groups were significantly prolonged, indicating that inhaled delivery held promise as a strategy for enhanced NSCLC treatment. When compared to the ATinh and AT-LP-DPIinh groups, the AT-MLP-DPIinh group displayed a relatively flat concentration-time curve (with lower inter-animal variability within the group), a lower Cmax value, prolonged Tmax, increased T1/2, and the highest AUC. These results suggested that, compared to other formulations, AT-MLP-DPIinh provided a slow release in the lungs, possibly due to the sustained-release effect of liposomes. Although ATinh group had a long MRT0-∞ (17.76 ± 15.56 h), the absorption of inhaled AT solution was poor, resulting in the lowest Cmax (15.83 ± 10.34 μg/L) and AUC0-∞ values (127.32 ± 35.55 μg/L*h). Therefore, the AT solution was not suitable for inhalation administration directly.
Antitumor efficacy study
To study the therapeutic effects, a tumor-growth experiment was conducted in tumor-bearing mice. Tumor growth in the control group treated with PBS was significantly increased compared with other treatment groups (Fig. 6A). These results suggested AT-MLP-DPIinh was more effective than AT + CXinh group in controlling tumor growth. The results presented in Fig. 6B demonstrate a significant reduction in tumor black nodules in each treatment group compared to the control group. The anti-metastasis potential of dry powder inhalation was assessed by counting pulmonary metastatic nodes and conducting pathological analyses of lung tissue. In accordance with the results of pharmacokinetic study, the antitumor effect of pulmonary administration was superior to that of tail vein injection for the same prescription. The lung metastasis rate was calculated by determining the ratio of the number of pulmonary nodules in each group to the number of nodules in the control group (PBS group). The lung metastasis rate of nude mice in AT-MLP-DPIiv and AT-MLP-DPIinh groups was around 62.5% and 25%, respectively. Among the treatment groups, the AT-MLP-DPIinh group exhibited the fewest black spots in the lung, indicating that inhaled AT-MLP-DPI effectively inhibited lung tumor growth and had the most prominent tumor suppression effect.

In vivo antitumor efficacy. A Macroscopic appearance of lung tumors collected from A549 tumor-bearing nude mice after 14 days of treatment (n = 3); B macroscopic appearance of lung tumors collected from B16 tumor-bearing nude mice after 14 days of treatment; C body weight changes of nude mice in each group during the experimental period; D lung tissue wet weight/dry weight (W/D) ratio in different formulation groups (*p < 0.05, **p < 0.01); E representative images of the HE-stained healthy lung tissues and tumor sections after treatment; F the expression of Ki-67 in lung tumors
Figure 6C demonstrates that, in approximately the first 8 days following the establishment of the metastatic tumor model, there were no significant fluctuations in the body weight of nude mice in each group. Around 16 days post-modeling, the body weight of nude mice in the control group and AT + CXiv group showed a decline, while in the remaining formulation groups, there was a slight increase in body weight. The results were consisted with the successful establishment of the nude mouse lung metastasis tumor model, and also indicated dry powder inhalation was safety and efficacy.
Lung cancer could induce pulmonary edema, and thus, pulmonary edema (lung wet weight/dry weight ratio, W/D) was employed to assess the extent of lung injury. As depicted in Fig. 6D, compared to the healthy group, the W/D ratio sharply increased in the control group, indicating the successful construction of the lung metastatic mouse model and the induction of pulmonary edema symptoms by metastatic tumors. Following treatments, lung tissue edema levels were significantly inhibited. The ratio in the AT-MLP-DPIinh group approached that of the normal control group, indicating that AT-MLP-DPIinh exhibited a superior therapeutic effect compared to the other groups, significantly reducing pulmonary edema conditions.
Figure 6E illustrates the lung tissue structures observed in normal mice and the control group mice. Normal mice showed intact lung tissue structures with clear alveolar structures and no infiltration of inflammatory cells. In contrast, the lung tissue of the control group mice exhibited significant lung damage, including alveolar structure destruction, alveolar septal thickening, and massive infiltration of inflammatory cells. After treatment with different formulations, the pathological symptoms improved to varying degrees. Among these groups, the AT-MLP-DPIinh group showed significant improvement in pulmonary inflammatory condition, decreased inflammatory cell infiltration, and lung tissue morphology approaching that of normal mice.
Figure 6F depicts the expression of Ki-67 in lung tumor tissues of tumor-bearing nude mice. Ki-67 staining was used to identify proliferating tumor cells, which appeared brown. The results showed that the AT-MLP-DPIinh group significantly downregulated the expression of Ki-67, indicating significant inhibition of tumor cell proliferation. Overall, these results suggested that pulmonary administration of AT-MLP-DPI provided superior tumor therapy compared to tail vein injection.
Conclusions
In this study, inhalable afatinib-loaded immune nanoparticle dry powders were successfully formulated using the freeze-drying method. We initially confirmed the physical and chemical stabilities of inhalation powders and qualitatively evaluated their intracellular uptake and efficacy in limiting tumor growth in NSCLC cancer cell lines. Furthermore, the therapeutic efficacy of the proposed formulation in treating NSCLC was supported by in vitro and in vivo studies, comparing AT-loaded polymeric nanoparticles to plain drug. The developed AT-MLP-DPI exhibited prolonged residence time and accumulated specifically in the lungs, thereby improving the pharmacokinetic profile compared to micronized AT. This study explores a novel approach for delivering afatinib through non-invasive inhalation as polymeric nanoparticles for deep lung/site-specific delivery, overcoming limitations associated with marketed oral tablets. Therefore, inhalation of AT-loaded immunoliposomes dry powders appears to be a promising strategy for enhancing NSCLC treatment.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author (Sha Liu) on reasonable request.
References
Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288–300.
Yang SR, Schultheis AM, Yu H, Mandelker D, Ladanyi M, Buttner R. Precision medicine in non-small cell lung cancer: current applications and future directions. Semin Cancer Biol. 2022;84:184–98.
Li WQ, Cui JW. Non-small cell lung cancer patients with ex19del or exon 21 L858R mutation: distinct mechanisms, different efficacies to treatments. J Cancer Res Clin Oncol. 2020;146(9):2329–38.
Song X, Cao L, Ni B, Wang J, Qin X, Sun X, Xu B, Wang X, Li J. Challenges of EGFR-TKIs in NSCLC and the potential role of herbs and active compounds: from mechanism to clinical practice. Front Pharmacol. 2023;14:1090500.
Zhong J, Bai H, Wang Z, Duan J, Zhuang W, Wang D, Wan R, Xu J, Fei K, Ma Z, et al. Treatment of advanced non-small cell lung cancer with driver mutations: current applications and future directions. Front Med. 2023;17(1):18–42.
Cooper AJ, Sequist LV, Lin JJ. Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management. Nat Rev Clin Oncol. 2022;19(8):499–514.
Cortot AB, Madroszyk A, Giroux-Leprieur E, Molinier O, Quoix E, Berard H, Otto J, Rault I, Moro-Sibilot D, Raimbourg J, et al. First-line afatinib plus cetuximab for EGFR-mutant non-small cell lung cancer: results from the randomized phase II IFCT-1503 ACE-Lung Study. Clin Cancer Res. 2021;27(15):4168–76.
Zhang G, Yan B, Guo Y, Yang H, Li X, Li J. Case report: a patient with the rare third-generation TKI-resistant mutation EGFR L718Q who responded to afatinib plus cetuximab combination therapy. Front Oncol. 2022;12:995624.
Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11(9):777–92.
Santos EDS, Nogueira KAB, Fernandes LCC, Martins JRP, Reis AVF, Neto JBV, Junior I, Pessoa C, Petrilli R, Eloy JO. EGFR targeting for cancer therapy: pharmacology and immunoconjugates with drugs and nanoparticles. Int J Pharm. 2021;592:120082.
Vacchelli E, Aranda F, Eggermont A, Galon J, Sautes-Fridman C, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: tumor-targeting monoclonal antibodies in cancer therapy. Oncoimmunology. 2014;3(1):e27048.
Petrilli R, Eloy JO, Saggioro FP, Chesca DL, de Souza MC, Dias MVS, daSilva LLP, Lee RJ, Lopez RFV. Skin cancer treatment effectiveness is improved by iontophoresis of EGFR-targeted liposomes containing 5-FU compared with subcutaneous injection. J Control Release. 2018;283:151–62.
Safaei M, Khosravian P, Kazemi Sheykhshabani S, Mardani G, Elahian F, Mirzaei SA. Enzyme-sensitive nanoparticles, smart TAT and cetuximab conjugated immunoliposomes to overcome multidrug resistance in breast cancer cells. Toxicol Appl Pharmacol. 2022;441:115989.
Merino M, Zalba S, Garrido MJ. Immunoliposomes in clinical oncology: State of the art and future perspectives. J Control Release. 2018;275:162–76.
Singh A, Myklebust NN, Furevik SMV, Haugse R, Herfindal L. Immunoliposomes in acute myeloid leukaemia therapy: an overview of possible targets and obstacles. Curr Med Chem. 2019;26(28):5278–92.
Lu X, Liu S, Han M, Yang X, Sun K, Wang H, Mu H, Du Y, Wang A, Ni L, Zhang C. Afatinib-loaded immunoliposomes functionalized with cetuximab: a novel strategy targeting the epidermal growth factor receptor for treatment of non-small-cell lung cancer. Int J Pharm. 2019;560:126–35.
Lu X, Liu S, Yang X, Han M, Sun K. Determination of tyrosine kinase inhibitor afatinib in rat plasma using LC-MS/MS and its application to in vivo pharmacokinetic studies of afatinib liposomes. J Pharm Biomed Anal. 2019;164:181–6.
Bou-Assaly W, Mukherji S. Cetuximab (erbitux). AJNR Am J Neuroradiol. 2010;31(4):626–7.
Caroline B, Sundus Y, Dawn D, Carol G, Susan M. Cost analysis of cetuximab (Erbitux) plus radiotherapy (ERT) versus concomitant cisplatin plus radiotherapy (CRT) within an NHS oncology unit (single institution): a pilot study. Br J Radiol. 2016;89(1068):20160105.
Abu Lila AS, Ishida T. Liposomal Delivery Systems: Design optimization and current applications. Biol Pharm Bull. 2017;40(1):1–10.
van Elk M, Murphy BP, Eufrasio-da-Silva T, O’Reilly DP, Vermonden T, Hennink WE, Duffy GP, Ruiz-Hernandez E. Nanomedicines for advanced cancer treatments: Transitioning towards responsive systems. Int J Pharm. 2016;515(1–2):132–64.
Eloy JO, Petrilli R, Trevizan LNF, Chorilli M. Immunoliposomes: a review on functionalization strategies and targets for drug delivery. Colloids Surf B Biointerfaces. 2017;159:454–67.
Fei Q, Bentley I, Ghadiali SN, Englert JA. Pulmonary drug delivery for acute respiratory distress syndrome. Pulm Pharmacol Ther. 2023;79:102196.
Shen AM, Minko T. Pharmacokinetics of inhaled nanotherapeutics for pulmonary delivery. J Control Release. 2020;326:222–44.
Chang RYK, Chow MYT, Khanal D, Chen D, Chan HK. Dry powder pharmaceutical biologics for inhalation therapy. Adv Drug Deliv Rev. 2021;172:64–79.
Marante T, Viegas C, Duarte I, Macedo AS, Fonte P. An Overview on Spray-Drying of Protein-Loaded Polymeric Nanoparticles for Dry Powder Inhalation. Pharmaceutics. 2020;12(11).
Carfagna M, Rosa M, Hawe A, Friess W. Lyophilization cycle design for highly concentrated protein formulations supported by micro freeze-dryer and heat flux sensor. Int J Pharm. 2023;643:123285.
Schmid B, Navalho S, Schulze PSC, Van De Walle S, Van Royen G, Schuler LM, Maia IB, Bastos CRV, Baune MC, Januschewski E, et al. Drying microalgae using an industrial solar dryer: A biomass quality assessment. Foods. 2022;11(13).
Jia W, Yawman PD, Pandya KM, Sluga K, Ng T, Kou D, Nagapudi K, Luner PE, Zhu A, Zhang S, Hou HH. Assessing the interrelationship of microstructure, properties, drug release performance, and preparation process for amorphous solid dispersions via noninvasive imaging analytics and material characterization. Pharm Res. 2022;39(12):3137–54.
Ramireddy BA, Gorantla A, Indukuri A, Beeravelly S, Sarbajna RM, Arikatla S, Jetti RR. Polymorphic Forms of Afatinib Dimaleate. 2018.
Kussendrager KD, Ellison MJ. Carrier material for dry powder inhalation. 2002.
Acknowledgements
The authors would like to thank the technical support from core facilities of Yantai University School of Pharmacy.
Funding
This study was supported by Natural Science Foundation of Shandong Province [ZR2023MH318] and Youth Innovation Team Plan of Shandong Province [2022KJ344].
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Sha Liu: conception, revision, supervision, and writing—original draft and revised draft. Daoyuan Chen: experiment, software, and writing—original draft. Xiaosu Zhu: experiment, software, analysis, and investigation. Xiaowen Wang: software and analysis. Xiao Li: methodology and analysis. Yuan Du: methodology, software, and instrument operation. Peng Zhang: reference search, analysis, and investigation. Jingwei Tian: revision, conception, and investigation. Yingjian Song: conception, revision, supervision, and final approval of draft.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All institutional and national guidelines for the care and use of laboratory animals were followed. Sprague–Dawley rats and nude mice were provided from the Jinan Pengyue Experimental Animal Breeding co., ltd (Jinan, China). The animal experiments conducted in this study adhered to the UK Animals (Scientific Procedures) Act, 1986, and associated guidelines, as well as the EU Directive 2010/63/EU for animal experiments. The ARRIVE criteria were followed in all animal experiments. During 1 week of acclimation time, the rats and nude mice were housed four per cage in a 12 h light/dark cycle with food and water available ad libitum. The dry powder formulation developed in this study was a local drug delivery preparation, and the tumor micro- environment of lung was complex in vivo. Seventy-four rats and 20 nude mice were necessarily used to explore the in vivo distribution, pharmacokinetic study, antitumor effect, and safety evaluation in this study. All animal experiments were approved by Yantai University Laboratory Animal Ethics Committee, affiliated to Yantai University (approval number, YTDX 20220615).
Consent for publication
All authors read and approved the final manuscript and agree with its publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, S., Chen, D., Zhu, X. et al. Inhaled delivery of cetuximab-conjugated immunoliposomes loaded with afatinib: A promising strategy for enhanced non-small cell lung cancer treatment. Drug Deliv. and Transl. Res. (2024). https://doi.org/10.1007/s13346-024-01536-7
Accepted:
Published:
DOI: https://doi.org/10.1007/s13346-024-01536-7