
Abstract
The cardioprotective effect of microRNAs (miRNAs) on myocardial ischemic-reperfusion (I/R) injury has been documented. Here, we aim to decipher the mechanism of miR-24 delivered by human umbilical cord mesenchymal stem cell-derived extracellular vesicles (hUC-MSC-EVs) in myocardial I/R injury after dexmedetomidine (DEX) preconditioning. We collected and identified hUC-MSCs and extracted EVs, which were co-cultured with DEX-preconditioned hypoxia/reoxygenation (H/R) cardiomyocyte models or injected into I/R mouse models. The cardiomyocytes and myocardial injury were evaluated by molecular biology experiments. miR-24 was highly expressed in hUC-MSC-EVs. hUC-MSC-EVs could transfer miR-24 into cardiomyocytes where miR-24 augmented cell viability and inhibited cell apoptosis after DEX preconditioning. In the co-culture system of RAW264.7 macrophages with hUC-MSC-EVs, miR-24 promoted M2-type polarization of macrophages and reduced M1-type macrophage polarization. Mechanistically, miR-24 targeted KEAP1 and inhibited its expression, resulting in disruption of the Nrf2/HO-1 signaling. In vivo data confirmed that miR-24 delivered by hUC-MSC-EVs enhanced the suppressing effect of DEX preconditioning on inflammation and apoptosis in rats following myocardial I/R injury. Overall, miR-24 delivered by hUC-MSC-EVs can promote M2 polarization of macrophages and enhance the protective effect of DEX preconditioning on myocardial I/R injury by down-regulating the KEAP1/Nrf2/HO-1 signaling axis.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute myocardial infarction (AMI) is one of the leading causes of death and disability in the world. Although reperfusion therapy is a reasonable method to prevent further myocardial necrosis and protect surviving cells, the myocardial reperfusion process will cause myocardial cell damage, which is “myocardial reperfusion injury,” and contribute to the final myocardial infarction [1, 2]. Ischemia–reperfusion (I/R) injury is a complex procedure correlated with multiple factors, including oxidative stress and inflammatory response, which can cause myocardial dysfunction [3]. Dexmedetomidine (DEX) preconditioning has been reported to alleviate myocardial I/R injury [4].
Cardiomyocytes are the working cells of the heart and are also the most susceptible to I/R injury. Acute myocardial I/R leads to apoptosis, necrosis, and inflammatory response determining the size of the final myocardial infarction after acute myocardial ischemia to different degrees, so regulating cell death and inflammatory response becomes an important therapeutic target for cardiac protection [1]. Macrophages that infiltrate after infarction show a high degree of phenotypic diversity. Among them, M1 macrophages promote the progress of inflammatory response, M2 macrophages exert a certain anti-inflammatory effect, and they jointly regulate the immune microenvironment around the infarction by changing their functional characteristics [5]. Macrophage subpopulations can mediate cell protection and repair of infarcted heart, and can also play an active role by activating the anti-apoptotic program in cardiomyocytes [6].
Extracellular vesicles (EVs) are membrane vesicles that are released by a heterogeneous group of cells and encompass exosomes and microvesicles, originating from the endosomal system or the plasma membrane [7]. EVs can interact with recipient cells to transfer their contents of proteins, lipids, and RNAs [8]. microRNAs (miRNAs) shuttled by EVs have been highlighted to be tumor biomarkers and potential therapeutic vehicles due to their roles in regulating the pathological processes in disease conditions, including cardiovascular diseases [9, 10]. Mesenchymal stem cell-derived EVs (MSC-EVs) can change the polarization state of macrophages through its content miR-182, and inhibit myocardial damage by promoting the transformation of M1 macrophages (pro-inflammatory) into M2 macrophages (anti-inflammatory) [11]. miR-24 was reported to prevent against myocardial I/R injury by regulating the NF-κB/TNF-α pathway [12]. miR-24-3p is able to inhibit RIPK1 and plays a cardioprotective role in myocardial I/R injury [13]. It should be noted that our bioinformatics prediction found kelch like ECH associated protein 1 (KEAP1) as the target gene of miR-24 in myocardial I/R injury. Notably, the inactivation of KEAP1/Keap-nuclear factor-erythroid 2-related factor 2 (Nrf-2) signaling pathway in the presence of Yiqi Huoxue recipe repressed cardiomyocyte apoptosis triggered by heart failure [14]. Nrf-2 can induce inflammation in early myocardial I/R through regulation of macrophages [15]. The critical role of Nrf2/heme oxygenase-1 (HO-1) pathway in myocardial ischemia has also been documented [16]. Elevated expression of HO-1 was observed in the heart of mice post myocardial infarction [17]. In light of the aforementioned evidence, we tried to explore the underlying mechanism concerning MSC-EV miR-24 in myocardial I/R injury and to discuss the possible role of macrophage polarization during this process, with the involvement of the KEAP1/Nrf-2/HO-1 axis.
Materials and methods
Ethics statement
This study was implemented in the light of the Declaration of Helsinki and the guidelines issued by National Institutes of Health (Bethesda, MA) and ratified by the clinical ethics committee of The First Affiliated Hospital, Hengyang Medical School, University of South China. Human umbilical cords were obtained from infants with parental consent. Animal experiments were implemented in the light of the institutional animal care and use committee of The First Affiliated Hospital, Hengyang Medical School, University of South China.
Bioinformatics analysis
Microarray datasets GSE53211 and GSE159814 were downloaded from the Gene Expression Omnibus (GEO) database [18]. GSE53211 includes 9 myocardial infarction samples and 4 healthy control samples. GSE159814 contains 3 hUC-MSC samples. Differential analysis was performed on the GSE53211 dataset using the R language “limma” package with a threshold of p < 0.05 [19]. TargetScan (|cumulative weighted context + + score|> 0.1) [20] and miRmap (miRmap score > 90) [21] databases were used to predict miRNA target gene. The GeneCards database was used to obtain genes related to myocardial I/R injury [22].
Characterization of hUC-MSCs
Fresh umbilical cords were gained from infants and washed twice with PBS to remove blood at 4 °C. Then, the umbilical cords were separated into 10 mm3 samples which were detached for 4 h in a solution containing 0.1% hyaluronidase, collagenase type I, and 3 mM CaCl2. Afterwards, the samples were incubated in DMEM (Thermo Fisher Scientific Inc., Waltham, MA) appended to 10% FBS (Pan Biotech, Aidenbach, Germany) in a 5% CO2 cell incubator at 37 °C. The medium was renewed every 4 days, and non-adherent cells were removed.
When the hUC-MSCs grew to approximately 80% confluence, they were passaged at 1:3. Flow cytometry was implemented for testing the expression of surface markers such as CD73 (V B-CD73.3, BD Biosciences, Franklin Lakes, NJ), CD90 (AB_395969), CD105 (AB_11154045), CD34 (V_MA22), and CD45 (AB_647424) for the identification of the multipotency of hUC-MSCs. In differentiated hUC-MSCs, CD73, CD90, and CD105 were highly expressed, while CD34 and CD45 were poorly expressed. Alizarin red staining (ScienCell, Carlsbad, CA) was implemented to test the osteogenic differentiation of hUC-MSCs. Adipogenic differentiation and cartilage differentiation were confirmed by oil red O staining (Shanghai Yuanye Biotechnology Co., Ltd., Shanghai, China) and Alcian Blue staining (Shanghai Haling Biological Technology Co., Ltd., Shanghai, China), respectively. The cells at passage 3–6 were chosen for the subsequent experiment.
Isolation and identification of hUC-MSC-EVs
EVs were isolated from hUC-MSCs. Briefly, the hUC-MSCs at passage 3–5 were cultured in serum-free medium at a density of 2 × 106 cells/100 mm for 24 h. Next, the cell culture supernatant was centrifuged at 2000 g and 4 °C for 20 min to get rid of cell debris, and ultra-centrifuged at 100,000 g and 4 °C for 1 h using an ultracentrifuge machine (Beckman Coulter Optima L-100 XP, Miami, FL), with the supernatant discarded. Thereafter, the pellet was resuspended in 10 mL PBS and centrifuged at 110,000 g and 4 °C for 90 min. The pellet was harvested, resuspended in 100 µL PBS, and stored at − 80 °C until use.
To characterize the isolated EVs, the EVs were submitted to fixation in 1% glutaraldehyde, staining with 1% phosphotungstic acid and observation under a JEM-2100 transmission electron microscope (TEM, JEOL, Tokyo, Japan). In addition, a NanoSight NS300 system (Malvern Instruments, Malvern, UK) was used for nanoparticle tracking analysis (NTA). The Brownian motion of EVs was tracked and the size and concentration were calculated using the Stokes–Einstein equation. Western blot analysis was employed for testing the expression of EV specific surface markers CD63 (1: 2000, Abcam Inc., Cambridge, UK, ab216130, rabbit antibody), tumor susceptibility gene 101 (TSG101; 1: 5000, Abcam, ab125011, rabbit antibody), ALG-2 interacting protein X (ALIX; 1: 1000, Abcam, ab275387, rabbit antibody), and calnexin (1: 5000, Abcam, ab133615, rabbit antibody).
In the in vitro experiment, EVs resuspended in PBS were supplemented to the cultured cells at a concentration of 0.5 × 1011–1 × 1011 particles/0.5 × 105 cells.
Myocardial I/R injury mouse model construction after DEX preconditioning
Seventy-two male C57BL/6 mice (aged 8–10 weeks old, Model Animal Research Center of Nanjing University, Nanjing, China) were housed in a specific-pathogen free environment. The mice were intraperitoneally injected with DEX (T2524, 20 µg/kg; purchased from Target Mol, Boston, MA) for 1 h [23, 24] and then randomly sham-operated, subjected to I/R modeling, or subjected to I/R modeling, further treated with EVs-negative control (NC) mimic, EVs-miR-24-mimic, EVs-NC inhibitor or EVs-miR-24-inhibitor, 12 mice per group. All mice were anesthetized with an isoflurane vaporizer and ventilated with an HX-300S animal ventilator. The left chest cavity of the mice was opened, and the left anterior descending (LAD) coronary artery was sutured utilizing 8–0 Prolene suture to produce occlusion. Thirty minutes after local ischemia induction, the ligature was released and the mice were reperfused for 60 min. Finally, the chest was sutured, and the catheter was pulled out to allow the mice to resume spontaneous breathing. hUC-MSC-EVs (2 × 107, 30 µL PBS) or PBS alone was injected at 5 sites around the border area of the infarcted heart using a No. 31 Hamilton syringe. In addition, sham-operated mice received the same operations without the ligation of the LAD coronary artery. After the experiment, the mice were anesthetized with isoflurane and then euthanized, with the heart collected.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted with TRIzol reagent (16,096,020, Thermo Fisher Scientific) and then reversely transcribed and quantified using Takara PrimeScript RT master mix kits (RR037B, Takara, Dalian, China, for mRNA detection) or TaqMan MicroRNA Reverse Transcription Kit (4,366,596, Thermo Fisher Scientific, for miRNA detection). RT-qPCR was implemented utilizing the SYBR Premix Ex Taq II kit (Takara, Cat No. 2) with an equal amount of cDNA in a 20 µL reaction mixture. All the mRNA primers are shown in Supplementary Table 1. TaqMan probes were purchased from Thermo Fisher Scientific (miR-24, Thermo Fisher Scientific, Cat No. 1). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was utilized as the internal reference gene for mRNA abundance. The relative content of the target gene was expressed as the ratio of the target gene to the internal reference gene, followed by plotting.
Next, the loading of miR-24 in EVs was measured. With the specific miR-24 primer provided by TaqMan® MicroRNA Detection Kit (Applied Biosystems Inc., Carlsbad, CA), different concentrations of the synthetic miR-24-cDNA standards (10−2–10−9 µM) were used to perform RT-qPCR to obtain an absolute quantitative standard curve. The negative logarithm of the miR-24-cDNA concentration (X axis) was constructed against the Ct value (Y axis). miR-24 in 2 µg EVs was gained, reversely transcribed, and subjected to RT-qPCR detection. The absolute content of miR-24 was then determined referring to the generated standard curve. Using the internal reference gene, U6, as a reference, the relative content of miR-24 was calculated, followed by plotting.
Cell transfection and H/R model construction
RAW264.7 cells (CL-0190, Procell, Wuhan, China) and HL-1 cells (Cat# SCC065, Millipore, Billerica, MA) were cultured in a 6-well plate cell culture dish at a density of 2 × 105 cells/mL. Upon reaching 80% confluence, hUC-MSCs, HL-1 cells, and RAW264.7 cells were transfected utilizing lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA). After transfection, RAW264.7 cells were reacted with 1 µg/mL LPS (Sigma, St Louis, MO) for 24 h to induce polarization [25]. hUC-MSCs were manipulated with miR-24-inhibitor, inhibitor-NC, miR-24-mimic, or NC-mimic. HL-1 cells were manipulated with miR-24-mimic, NC-mimic, short hairpin RNA (sh)-KEAP1, sh-NC, or sh-Nrf2. The above transfection materials were synthesized by GenePharma (Shanghai, China). The transfected cells were cultured at 37 °C with 5% CO2 for 48 h for subsequent experiments.
Cells were pretreated with 1 nM DEX for 60 min [26] before H/R modeling. The H/R cell model was constructed by hypoxic pretreatment of HL-1 cells in a three-gas incubator (Thermo Fisher Scientific) with 94% N2, 1% O2, and 5% CO2. Hypoxia was conducted for 1, 3, 6, and 12 h, and reoxygenation for 1, 6, 12, and 24 h, respectively. Finally, 6 h of hypoxia and 12 h of reoxygenation were selected as the best nodes for subsequent experiments.
Internalization of EVs by cardiomyocytes and macrophages
PKH67 Green Fluorescent Cell Linker Kit (Sigma) was applied to label EVs. EVs were resuspended in 0.5 mL diluent C provided by the kit, incubated with 4 µL PKH67 for 4 min and then supplemented with 10 mL DMEM/F12 medium appended to 10% FBS to stop staining. Centrifugation was conducted at 100,000 g and 4 °C g to obtain EVs. The obtained PKH67-labeled EVs were added to HL-1 cell or macrophage culture medium and incubated for 8 h. Next, the cells were submitted to fixation utilizing 4% paraformaldehyde for 10 min, and staining with phalloidin (cytoskeleton staining, 40734ES75, Shanghai Yeasen Biotechnology Co., Ltd., Shanghai, China) for 30 min and 4′,6-diamidino-2-phenylindole (Sigma) for 5 min. Finally, a laser scanning confocal microscope (FV1000, Olympus Optical Co., Ltd, Tokyo, Japan) was employed to collect fluorescence images.
Luciferase assay
The targeting between miR-24 and KEAP1 was verified by the dual luciferase reporter assay. Wild-type (WT) and mutant type (MUT) miR-24 and KEAP1 3′untranslated region (3′UTR) were artificially synthesized, which were then cloned to the pGL3 plasmid containing an ARE-Luc reporter (Yeasen) by T4 DNA ligase after double restriction enzyme digestion (NheI and BglII). After Sanger sequencing verification, the HL-1 cells were transfected with pGL3-KEAP1-WT, pGL3-KEAP1-MUT, or pGL3-control plasmid, respectively, together with miR-24-mimic or NC-mimic for 12 h. Luciferase activity was checked in the Dual Luciferase Reporter Assay System (E1910, Promega Corporation, Madison, WI) using a GloMAX20/20 luminometer (Promega).
Determination of superoxide dismutases (SOD) and glutathione peroxidase (GSH-Px)
HL-1 cells were homogenized in ice-cold saline. The activity of SOD was determined using a commercially available detection kit (A001-3–2, Nanjing Jiancheng Institute of Bioengineering, Nanjing, China). GSH-Px activity was tested with GSH-Px Activity Assay Kit (A005-1–2, Jiancheng). The samples were finally analyzed using a spectrophotometer.
Cell counting kit-8 (CCK-8) assay
The cell viability was detected by CCK-8 (Dojindo, Kyushu Island, Japan), and the absorbance of the samples was measured at 450 nm utilizing a micro-plate reader (Molecular Devices, LLC, Sunnyvale, CA).
Enzyme-linked immunosorbent assay (ELISA)
ELISA kit was used to detect the level of inflammation-related factors IL-6 (ab222503, Abcam) and TNF-α (ab208348, Abcam) in the myocardial tissues of the I/R mice.
Flow cytometry
Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kits (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) were adopted to test cell apoptosis. Cells following 48 h of culture were harvested, treated with 0.25% ethylenediamine tetraacetic acid-free trypsin, and resuspended in 500 µL of binding buffer. The cell suspension was stained using 5 µL annexin V-FITC-labeled specific antibody and propidium iodide for 15–20 min in the dark, after which the cell sorting was performed by flow cytometry and data were analyzed with FASCDiva 4.1 software (BD Biosciences).
Histology assay
Cell apoptosis detection was implemented utilizing terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) Apoptosis Detection Kit (C1088, Beyotime, Shanghai, China, green fluorescence). The cell suspension was spread on a coverslip, fixed with 4% paraformaldehyde for 1 h, treated with 0.1% Triton X-100 (Beyotime) at 4 °C for 3 min, and treated with 50 µL of TUNEL solution at 37 °C for 1 h in a dark room. After the slide was sealed with anti-fluorescence quenching mounting solution, the fluorescence intensity was checked under a fluorescence microscope, and the number of TUNEL-positive cells was computed in five randomly chosen high-power fields.
To detect the pathological changes of myocardial tissues following modeling, histology studies were performed, including hematoxylin–eosin (HE) staining and Masson’s trichrome staining. The tissue block was removed from the myocardial tissue and fixed with 10% formalin and Bouin’s fixative solution (HE staining, which was performed utilizing HE staining kits (C0105S, Beyotime)) or BD-Pharmingen (Masson’s trichrome staining), and rinsed with running water to remove the fixative solution from the tissue for 30 min. After gradient dehydration with alcohol, paraffin embedding, sectioning, and staining were performed following the standard procedure. Images were taken by Aperio (Leica Biosystems, Buffalo Grove, IL) under a confocal microscope (Nikon, Melville, NY) and analyzed by Image J software.
5-Triphenyl tetrazolium chloride (TTC) staining
TTC assay was performed to detect the infarct size of the mouse heart. In short, the mice were euthanized utilizing intraperitoneal injection of overdose sodium pentobarbital (100 mg/kg) after reperfusion, and the heart was taken out. The residual blood was washed with pre-cooled PBS solution, and the LAD was ligated again. The heart was slowly injected with 0.3 mL of 1% Evans blue, frozen in a − 80 °C freezer for 2 h, and cut into 2-mm-thick sections vertically along the longitudinal axis of the heart. The sections were then stained in 1% TTC solution in a 37 °C water bath for 30 min (dark condition) and fixed in 10% paraformaldehyde solution for 24 h. Next, partial sections were dried with filter paper, arranged in a row from the apex to the bottom of the heart, and photographed using a Sony camera. ImageJ software was employed to assess the area of myocardial infarction. The non-infarct size is the blue-stained area; the infarct size is the gray-white myocardium; and the red is the area at risk.
Transthoracic echocardiography
Evaluation of mouse heart function was conducted utilizing Acuson Sequoia 512 color Doppler ultrasound by professionals with the assistance of 62c-S probe (frequency: 8.5 mHz) at the scanning speed of 100 mm/s [27]. The left ventricular end-diastolic diameter (LVEDD, mm), left ventricular end-systolic diameter (LVESD, mm), left ventricular end-diastolic volume (LVEDV, µL), and left ventricular end-systolic volume (LVESV, µL) were measured for 3 consecutive cardiac cycles, with the average value obtained.
Western blot
To determine related protein expression, the total protein from cells was extracted using RIPA lysis buffer (R0010, Beijing Solarbio Science & Technology Co., Ltd., Beijing, China), and the protein concentration was tested with the help of a bicinchoninic acid protein assay kit (20201ES76, Yeasen). After separation by polyacrylamide gel electrophoresis, the protein was transferred onto a polyvinylidene fluoride membrane by the wet transfer method which was then blocked with 5% bovine serum albumin for 1 h at room temperature and probed with the diluted rabbit primary antibodies against KEAP1 [1:1000, 2524, Cell Signaling Technologies (CST), Beverly, MA], Nrf2 (1:1000, 2171, CST), heme oxygenase-1 (HO-1; 1:500, 18,583, CST), B-cell lymphoma 2 (Bcl-2; 1:1000, 16540S, CST), iNOS (1:1000, 13,120), Arg1(1:1000, 93,668), caspase-3, cleaved caspase-3 (1:1000, 19,677–1-AP, Proteintech, Wuhan, China), BAX (1:1000, 50,599–2-Ig, Proteintech, China), and GAPDH (1:10,000, ab181602, Abcam) at 4 °C overnight. The next day, the membrane was re-probed with the horseradish peroxidase-labeled goat anti-rabbit IgG (ab205718, 1:20,000, Abcam) for 1 h. Finally, chemiluminescence reagents were used to develop images. ImageJ 1.48 software (National Institutes of Health, Bethesda, Maryland) was chosen for gray value analysis. The density of target protein band was normalized to GAPDH, followed by plotting.
Statistical analysis
All data were analyzed utilizing SPSS 22.0 software (IBM Corp. Armonk, NY) with the help of unpaired t-test (two groups) and one-way analysis of variance (multiple groups). To ensure the accuracy of experimental results, all in vitro experiments were repeated at least 3 times. Measurement data were presented as mean ± standard deviation. p < 0.05 was concluded as statistically significant.
Results
Identification of the hUC-MSCs and hUC-MSC-EVs
hUC-MSCs were isolated from clinical tissue samples, and their surface markers were tested by flow cytometry. The results showed that the isolated hUC-MSCs were positive for CD73, CD90, and CD105 but negative for CD34 and CD45 (Fig. 1A). The results of oil red O staining, Alizarin red staining, and Alcian blue staining assays showed that the isolated hUC-MSCs had the characterization of adipogenic, osteogenic, and chondrogenic differentiation (Fig. 1B). These results pinpointed the successful isolation of hUC-MSCs.

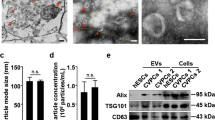
Characterization of hUC-MSCs and hUC-MSC-EVs. A. The identification of the hUC-MSC surface markers by flow cytometry. B The identification of differentiation ability of hUC-MSCs, (i) analysis of osteogenic differentiation using Alizarin red staining; (ii) analysis of adipogenic differentiation by oil red O staining; (iii) analysis of chondrogenic differentiation using Alcian blue staining. C EV morphology under a TEM. The EVs are shown by the red arrows. (D) The size distribution of hUC-MSC-EVs measured by NTA. E Expression analyses of EV surface markers using Western blot. * p < 0.05 vs. hUC-MSCs group. Data were expressed as mean ± standard deviation. Comparison between two groups was performed by unpaired t-test. The experiment was repeated three times
EVs were then isolated from hUC-MSCs. Under a TEM, the isolated EVs presented with obvious heterogeneity and were round or elliptical membranous vesicles, and the membranous structure could be seen on the periphery of the vesicles. Low electron density components occurred in the center (Fig. 1C). At the same time, NTA results showed that EVs had irregular Brownian motion with a diameter of 30–150 nm (Fig. 1D). Western blot results clarified that hUC-MSC-EVs were positive for CD63, TSG101, and ALIX and negative for the endoplasmic reticulum marker Calnexin [28] (Fig. 1E).
hUC-MSC-EVs deliver miR-24 to suppress M1 polarization of macrophages and apoptosis of cardiomyocytes after DEX preconditioning
EVs play a critical role in cellular communication by transferring miRNAs [29, 30]. We screened 16 differentially expressed miRNAs related to myocardial infarction from the GSE53211 dataset which includes 9 myocardial infarction samples and 4 healthy control samples (Fig. 2A), and obtained the top 10 miRNAs that were significantly enriched in three hUC-MSC-EV samples from the GSE159814 dataset (Fig. 2B), where miR-24 (miR-24-3p) belongs to the intersection of the two datasets (Fig. 2C).

hUC-MSC-EVs deliver miR-24 to inhibit cardiomyocyte apoptosis and M1 polarization of macrophages while polarizing macrophages toward M2 phenotype after DEX preconditioning. A Heatmap of differentially expressed miRNAs between healthy control samples (n = 4) and myocardial infarction samples (n = 9) in the GSE53211 dataset. B Heatmap of the top 10 hUC-MSCs-EV-miRNAs from the GSE159814 dataset. C Venn diagrams from the two miRNA datasets. D The uptake of EVs by HL-1 cells under a confocal microscopy. E Relative expression of miR-24 in hUC-MSCs and hUC-MSC-EVs detected by RT-qPCR. F Relative expression of miR-24 in HL-1 cells treated with hUC-MSC-EVs alone or combined with miR-24-mimic detected by RT-qPCR. A cardiomyocyte model of DEX-preconditioned H/R was established in HL-1 cells. G Viability of HL-1 cells treated with hUC-MSC-EVs alone or combined with miR-24-mimic measured using CCK-8. H Apoptosis detection of HL-1 cells treated with hUC-MSC-EVs alone or combined with miR-24-mimic by flow cytometry. I The expression of apoptosis-related proteins in the HL-1 cells treated with hUC-MSC-EVs alone or combined with miR-24-mimic detected by Western blot. J The phagocytosis of RAW264.7 cells to EVs observed under a laser confocal microscope. K RT-qPCR detection of miR-24 expression in RAW264.7 cells treated with hUC-MSC-EVs alone or combined with miR-24 mimic. L Western blot was used to detect the expression changes of key proteins of M1 and M2 macrophage polarization in RAW264.7 cells in response to hUC-MSC-EVs alone or combined with miR-24 mimic. M The positive rates of M1 and M2 polarized cell surface markers in RAW264.7 cells in response to hUC-MSC-EVs alone or combined with miR-24 mimic detected by flow cytometry. In the graph, for Western blot, data are normalized to GAPDH expression; for RT-qPCR, data of miR-24 relative expression are normalized to U6 expression while the data of the remaining genes are normalized to GAPDH expression and relative to the PBS group or the DEX + PBS group. * p < 0.05. Data were expressed as mean ± standard deviation. Comparison between two groups was performed by unpaired t-test, and multi-group data comparison was performed by one-way ANOVA. Cell experiments were repeated three times
For validation, we first co-cultured PKH67-labeled EVs with HL-1 cells for 48 h. Confocal microscope observation revealed that HL-1 cells internalized PKH67-labeled EVs (Fig. 2D). In addition, an increase in the expression of miR-24 was found in hUC-MSCs and hUC-MSC-EVs treated with miR-24-mimic, and a decline in the expression of miR-24 was seen in hUC-MSCs and hUC-MSC-EVs treated with miR-24-inhibitor. The above results showed significantly changed expression of miR-24 in hUC-MSCs and hUC-MSC-EVs following transfection with miR-24-mimic or miR-24-inhibitor (Supplementary Fig. 1A). After verifying the transfection efficiency, we constructed a co-culture system of hUC-MSC-EVs with HL-1 cells to investigate the effect of hUC-MSC-EVs on the expression of miR-24 in HL-1 cells. An increase in the expression of miR-24 was noted in the HL-1 cells co-cultured with hUC-MSC-EVs. The expression of miR-24 was significantly elevated in response to hUC-MSC-EVs miR-24-mimic; in the presence of hUC-MSC-EVs miR-24-inhibitor, the expression of miR-24 was decreased (Fig. 2E; Supplementary Fig. 1B). These results suggest that hUC-MSC-EVs carrying miR-24 can be internalized by cardiomyocytes.
As shown in Fig. 2F and Supplementary Fig. 1C, after DEX preconditioning, lower expression of miR-24 was found in H/R-induced HL-1 cells than that in PBS-treated cells. Upon co-culture with hUC-MSC-EVs, the HL-1 cells exhibited increased miR-24 expression to a comparable level to that of PBS treatment. However, further treatment with miR-24-mimic resulted in increased miR-24 expression, but further treatment with miR-24-inhibitor led to a decline in the miR-24 expression.
Furthermore, we assessed the effect of miR-24 on the viability and apoptosis of cardiomyocytes. After DEX preconditioning, the viability of H/R-induced cardiomyocytes was decreased and the apoptosis was increased, which could be recovered upon co-culture with hUC-MSC-EVs to a comparable level to that of PBS treatment. In addition, further miR-24 overexpression promoted cell viability and inhibited cell apoptosis in H/R-induced cardiomyocytes upon co-culture with hUC-MSC-EVs, while additional miR-24 inhibition resulted in opposite results (Fig. 2G, H; Supplementary Fig. 1D, E).
Additionally, after DEX preconditioning, the protein expression of Bcl-2-associated X protein (Bax) and the ratio of cleaved caspase-3/caspase-3 were increased, and the protein expression of Bcl-2 was reduced in the H/R cells. On the contrary, co-culture with hUC-MSC-EVs led to opposite results. Opposite to additional miR-24 overexpression, additional miR-24 inhibition caused an enhancement in the protein expression of Bax and ratio of cleaved caspase-3/caspase-3 yet a decrease in the protein expression of Bcl-2 (Fig. 2I; Supplementary Fig. 1F).
Cardiomyocyte injury is closely related to the continuous chronic inflammatory infiltration in the microenvironment [31]. Thus, we further investigated the effect of miR-24 in hUC-MSC-EVs on macrophages involved in inflammatory infiltration in the microenvironment. To clarify the regulatory effect of hUC-MSC-EVs delivering miR-24 on macrophage polarization, we co-cultured macrophages RAW264.7 with hUC-MSC-EVs. Under a confocal microscope, RAW264.7 cells were observed to internalize PKH67-labeled EVs (Fig. 2J). Furthermore, expression of miR-24 was increased in RAW264.7 cells co-incubated with hUC-MSC-EVs, the effect of which could be augmented by further treatment with miR-24-mimic but reversed by additional treatment with miR-24-inhibitor (Fig. 2K; Supplementary Fig. 1G). These results indicated that hUC-MSC-EVs could be internalized by RAW264.7 cells and alter the expression of miR-24 in RAW264.7 cells.
Furthermore, we detected the expression of iNOS (M1 polarization-related protein) and Arg-1 (M2 polarization-related protein) in LPS-induced RAW264.7 cells. Western blot results identified an elevation of iNOS expression but a decline of Arg-1 expression in RAW264.7 cells following LPS induction, indicating that LPS treatment induced M1 polarization of macrophages and suppressed M2 polarization. Additionally, co-culture with hUC-MSC-EVs decreased iNOS expression yet enhanced Arg-1 expression in RAW264.7 cells, which proved that hUC-MSC-EVs promoted M2 polarization while arresting M1 polarization. In addition, the expression of iNOS protein was decreased, while that of Arg-1 was elevated in RAW264.7 cells co-cultured with hUC-MSC-EVs + miR-24-mimic, indicating that macrophages polarized to M2 phenotype and M1 macrophage polarization was repressed. Conversely, upon co-culture with hUC-MSC-EVs + miR-24-inhibitor, the expression of iNOS protein was increased, while that of Arg-1 was suppressed in RAW264.7 cells, implying that macrophages polarized to M1 phenotype and M2 macrophage polarization was repressed (Fig. 2L; Supplementary Fig. 1H).
For further validation, we then detected the expression of M1 polarized cell surface marker CD86 and M2 polarized cell surface marker CD206 using flow cytometry. The results showed that the proportion of CD86+CD206− was increased while that of CD86−CD206+ was decreased in the presence of LPS. Conversely, hUC-MSC-EVs led to contrary results. In response to LPS + EVs-miR-24-mimic, the proportion of CD86+CD206− was reduced while that of CD86−CD206+ was increased; opposite results were observed in the presence of LPS + EVs-miR-24-inhibitor (Fig. 2M; Supplementary Fig. 1I). Collectively, these data demonstrated that miR-24 delivered by hUC-MSC-EVs inhibited H/R-induced cardiomyocyte apoptosis after DEX preconditioning, polarized macrophages toward M2 phenotype, and inhibited macrophages polarized to M1 phenotype.
hUC-MSC-EVs deliver miR-24 to alleviate I/R injury in mice after DEX preconditioning
Next, we pinpointed the in vivo effect of miR-24 delivered by hUC-MSC-EVs in I/R injury in mice after DEX preconditioning. RT-qPCR results showed lower miR-24 expression in the myocardial tissue of the I/R mice after DEX preconditioning, which could be reversed by additional treatment with EVs-NC-mimic or EVs-NC-inhibitor. EVs-miR-24-mimic further increased while EVs-miR-24-inhibitor diminished the expression of miR-24 in DEX-preconditioned I/R mice (Fig. 3A; Supplementary Fig. 2A).

hUC-MSC-EVs deliver miR-24 to attenuate myocardial I/R injury in mice after DEX preconditioning. A mouse model of DEX-preconditioned I/R was constructed. A miR-24 expression in the myocardial tissue of mice treated with hUC-MSC-EVs alone or combined with miR-24 mimic detected by RT-qPCR. B Detection of myocardial infarct size of mice in response to hUC-MSC-EVs alone or combined with miR-24 mimic by TTC staining. The infarct size is white and the live myocardium is red. Image J software was used to evaluate the infarct size (n = 5). C HE and Masson’s trichrome staining analysis of the degree of myocardial damage and fibrosis of mice in response to hUC-MSC-EVs alone or combined with miR-24 mimic. In Masson’s trichrome staining, myocardial collagen fibers are stained blue and myocardial fibers are stained red. D TUNEL-positive cells in myocardial tissues of mice in response to hUC-MSC-EVs alone or combined with miR-24 mimic. E The detection of SOD activity in myocardial tissues of mice in response to hUC-MSC-EVs alone or combined with miR-24 mimic by ELISA. F The detection of GSH-Px activity in myocardial tissues of mice in response to hUC-MSC-EVs alone or combined with miR-24 mimic by ELISA. G The expression of IL-6 and TNF-α detected by ELISA in myocardial tissues of mice in response to hUC-MSC-EVs alone or combined with miR-24 mimic. In the graph, for RT-qPCR, data of miR-24 relative expression are normalized to U6 expression and relative to the DEX + Sham group. * p < 0.05 vs. sham-operated mice. # p < 0.05 vs. I/R mice and p < 0.05 vs. I/R mice treated with EVs-NC-mimic. Data were expressed as mean ± standard deviation. Comparison between two groups was performed by unpaired t-test, and multi-group data comparison was performed by one-way ANOVA. n = 8 mice upon each treatment
In addition, as depicted in Supplementary Table 2, LVEDV, LVEDD, LVESV, and LVESD were increased, but left ventricular fractional shortening (LVEEF) and left ventricular ejection fractions (LVEFS) were reduced in DEX-preconditioned I/R mice. Additionally, opposite to additional miR-24 overexpression, further miR-24 inhibition induced an upward inclination in the LVEDV, LVEDD, LVESV, and LVESD yet a downward inclination in LVEEF and LVEFS in DEX-preconditioned I/R mice in response to hUC-MSC-EVs treatment. Furthermore, TCC staining analysis suggested enlarged infarct size of myocardial tissues in DEX-preconditioned I/R mice, which could be reversed in the presence of hUC-MSC-EVs. EV-miR-24-mimic treatment reduced the infarct size of myocardial tissues in DEX-preconditioned I/R mice, while a contrary finding was found in the absence of miR-24 (Fig. 3B; Supplementary Fig. 2B).
The results of HE staining and Masson’s trichrome staining revealed that myocardial tissue damage and myocardial fiber collagen deposition in DEX-preconditioned I/R mice were alleviated by hUC-MSC-EVs, the effect of which was enhanced by miR-24 overexpression but reversed by miR-24 inhibition (Fig. 3C; Supplementary Fig. 2C). In addition, treatment with hUC-MSC-EVs impeded the cell apoptosis in the myocardial tissue of DEX-preconditioned I/R mice; additional miR-24 overexpression suppressed the cell apoptosis, while further miR-24 inhibition triggered it (Fig. 3D; Supplementary Fig. 2D).
Meanwhile, we found through ELISA enhanced SOD and GSH-Px activities yet inhibited expression of IL-6 and TNF-α in the myocardial tissue of I/R mice treated with hUC-MSC-EVs after DEX preconditioning. Overexpression of miR-24 further enhanced the above trends, while contrasting results were noted upon knockdown of miR-24 (Fig. 3E–G; Supplementary Fig. 2E-G). In summary, after DEX preconditioning, miR-24 delivered by hUC-MSC-EVs could inhibit I/R-induced inflammation and cardiomyocyte apoptosis, alleviate the cardiomyocyte damage, and elevate the activity of SOD and GSH-Px, thus improving myocardial I/R injury after DEX preconditioning.
miR-24 targets KEAP1 and suppresses its expression
Subsequently, we sought to dissect out the downstream mechanism of miR-24 in myocardial I/R injury with DEX preconditioning. We predicted the target genes of miR-24 through TargetScan and miRmap databases and obtained the intersection with the top 500 genes related to myocardial I/R injury obtained from the GeneCards database, and a total of 12 candidate genes were obtained (Fig. 4A). Recent study has shown that miR-24 targets the KEAP1 gene to participate in the process of myocardial I/R injury [32]. We discovered an increase in the expression of KEAP1 in DEX-preconditioned H/R-induced cells (Fig. 4B, C). The TargetScan database predicted the presence of miR-24 binding sites in the 3′UTR of KEAP1 (Fig. 4D). Luciferase assay results further showed that the luciferase activity of KEAP1-3′UTR-WT was reduced in the presence of miR-24 overexpression while there was no changes in the luciferase activity of KEAP1-3′UTR-MUT (Fig. 4E). In HL-1 cells manipulated with miR-24-mimic, KEAP1 expression was downregulated (Fig. 4F, G). These results suggested that miR-24 could target KEAP1 and inhibit KEAP1 expression.

KEAP1 is a direct target of miR-24. A The Venn diagram of the target genes of miR-24 predicted by TargetScan and miRmap databases and the top 500 myocardial I/R injury-related genes obtained from the GeneCards database. B KEAP1 expression in H/R-induced cells after DEX preconditioning detected by RT-qPCR. C KEAP1 protein expression in H/R-induced cells after DEX preconditioning detected by Western blot. D Predicted specific binding site between miR-24 and KEAP1 by TargetScan. E miR-24 binding to KEAP1 verified by dual luciferase reporter assay. F Expression of KEAP1 in HL-1 cardiomyocytes transfected with NC-mimic or miR-24-mimic detected by RT-qPCR. G Protein expression of KEAP1 in HL-1 cardiomyocytes transfected with NC-mimic or miR-24-mimic detected by Western blot. In the graph, for RT-qPCR, data of Keap1 relative expression are normalized to GAPDH expression and relative to the NC-mimic group; for dual luciferase reporter assay, data of relative luciferase activity are normalized to Renilla luciferase activity and relative to the NC-mimic group. * p < 0.05. Data were expressed as mean ± standard deviation. Comparison between two groups was performed by unpaired t-test, and multi-group data comparison was performed by one-way ANOVA. Cell experiments were repeated three times
miR-24 inhibits cardiomyocyte apoptosis and M1 polarization of macrophages through the KEAP1/Nrf2/HO-1 signaling
A previous study has shown that KEAP1 can activate the Nrf2/HO-1 signaling pathway and aggravate myocardial I/R injury [32]. Meanwhile, the above results that miR-24 could bind to the 3′UTR of KEAP1 allowed us to speculate that miR-24 delivered by hUC-MSC-EVs may regulate the Nrf2/HO-1 signaling pathway by targeting KEAP1. In order to further clarify the role of KEAP1 in miR-24-regulated cardiomyocyte apoptosis and M1 macrophage polarization, we designed two shRNA sequences targeting KEAP1 and used RT-qPCR to determine the silencing efficiency. sh-KEAP1-1 showed the superior silencing efficiency (Supplementary Fig. 3A, B) and was thus used for subsequent experiments. In addition, Western blot results presented a decline in the expression of KEAP1, Nrf2, and HO-1 proteins in HL-1 and RAW264.7 cells overexpressing miR-24, which could be reversed by additional treatment with overexpression (oe)-KEAP1. Moreover, an enhancement was found in the expression of KEAP1, Nrf2, and HO-1 proteins in HL-1 and RAW264.7 cells transfected with miR-24 inhibitor, the effect of which was abolished by further treatment with sh-KEAP1 (Fig. 5A, Supplementary Fig. 3C). This result indicated that miR-24 can target KEAP1 and reduce its expression to downregulate the Nrf2/HO-1 signaling.

miR-24 arrests H/R-induced cardiomyocyte apoptosis and M1 polarization of macrophages via the KEAP1/Nrf2/HO-1 signaling. A KEAP1, Nrf2 and HO-1 protein expression in HL-1 and RAW264.7 cells treated with miR-24-mimic and/or oe-KEAP1 detected by Western blot. B The analysis of the apoptosis rate of H/R-exposed HL-1 cells transfected with miR-24-mimic and/or oe-KEAP1 by flow cytometry. C The detection of apoptosis-related proteins by Western blot in H/R-exposed HL-1 cells transfected with miR-24-mimic and/or oe-KEAP1. D After LPS induction, Western blot was used to detect the protein expression of iNOS and Arg-1 in RAW264.7 cells transfected with miR-24-mimic and/or oe-KEAP1. E The positive rates of CD86 and CD206 in LPS-induced RAW264.7 cells transfected with miR-24-mimic and/or oe-KEAP1 detected by flow cytometry. In the graph, for Western blot, data are normalized to GAPDH expression. n.s. p > 0.05. * p < 0.05. Data were expressed as mean ± standard deviation. Comparison between two groups was performed by unpaired t-test, and multi-group data comparison was performed by one-way ANOVA. Cell experiments were repeated three times
To determine the role of miR-24/KEAP1/Nrf2/HO-1 axis in myocardial I/R injury, we designed two siRNA sequences targeting Nrf2 sh-Nrf2-1 and sh-Nrf2-2. RT-qPCR, and Western blot data revealed the superior silencing efficiency of sh-Nrf2-1 (Supplementary Fig. 3D, E). Then we performed flow cytometry to evaluate the effect of Nrf2 on the apoptosis of H/R-exposed HL-1 cells. The results exhibited that the apoptosis rate of H/R-exposed HL-1 cells was increased in response to Nrf2 overexpression, while overexpression of miR-24 could abolish the effect. Relative to miR-24 overexpression alone, combination with Nrf2 overexpression contributed to higher apoptosis rate of HL-1 cells. In addition, apoptosis rate of HL-1 cells was repressed following Nrf2 silencing while it was augmented following miR-24 inhibition. Concomitant silencing of miR-24 and Nrf2 led to reduced cell apoptosis rate compared to miR-24 silencing alone (Fig. 5B; Supplementary Fig. 3F). Meanwhile, Western blot results found that the protein expression of pro-apoptosis protein Bax and cleaved caspase-3 was elevated and that of anti-apoptosis protein Bcl-2 was diminished in response to Nrf2 overexpression; conversely, overexpression of miR-24 abrogate the effect of Nrf2 overexpression. In addition, the protein expression of Bax and cleaved caspase-3 was limited, while Bcl-2 was up-regulated following Nrf2 silencing. However, miR-24 inhibition caused contrary results. Dual inhibition of miR-24 and Nrf2 led to higher Bax and cleaved caspase-3 expression yet lower Bcl-2 expression than individual inhibition of miR-24 (Fig. 5C; Supplementary Fig. 3G).
Next, we investigated the changes of macrophage M1 and M2 polarized molecular indicators and protein level in RAW264.7 cells. It was evident that the M1 polarized protein iNOS was increased, and M2 polarized protein Arg-1 was reduced in LPS-treated RAW264.7 cells overexpressing Nrf2, the effect of which was reversed by miR-24 overexpression. Besides, iNOS expression was decreased and Arg-1 expression was increased upon Nrf2 silencing, the effect of which was abrogated by miR-24 silencing. Moreover, simultaneous silencing of miR-24 and Nrf2 restored the effect of Nrf2 silencing on the expression of the two proteins (Fig. 5D; Supplementary Fig. 3H). Furthermore, we conducted flow cytometric analysis to determine the positive rate of macrophage surface molecules CD86 and CD206. The results exhibited increased CD86+CD206− cells and decreased CD86−CD206+ cells upon Nrf2 overexpression, the effect of which was rescued by overexpression of miR-24. Moreover, simultaneous overexpression of miR-24 and Nrf2 restored the effect of Nrf2 overexpression. In the absence of Nrf2, decreased CD86+CD206− cells and increased CD86−CD206+ cells were found, while these results were undermined by miR-24 inhibition. However, dual suppression of miR-24 and Nrf2 restored the effect of Nrf2 suppression alone (Fig. 5E; Supplementary Fig. 3I). Altogether, the above results demonstrate that miR-24 can downregulate the activity of Nrf2/HO-1 signaling pathway by inhibiting the expression of KEAP1, thereby reducing M1 macrophage polarization and cardiomyocyte apoptosis caused by H/R.
Discussion
In the model of acute I/R injury, many treatments have been tried and proven to have strong cardioprotective effects. However, the effect in clinical application was still unsatisfactory. The main reason is that I/R injury is multifactorial, leading to cardiomyocyte death through multiple mechanisms [33]. In this study, the factors related to myocardial injury were adjusted in various ways to achieve the purpose of protecting cardiac function. Collectively, we confirmed that after DEX preconditioning, hUC-MSC-EVs effectively inhibited myocardial injury and promoted myocardial function recovery after I/R. hUC-MSC-EVs mainly inhibited the apoptosis and inflammation of cardiomyocytes, inhibited M1 polarization of macrophages, and promoted M2 polarization of macrophages to exert its protective effect. Meanwhile, we found that hUC-MSC-EVs delivered miR-24 which targeted KEAP1 and downregulate the Nrf2/HO-1 pathway to improve myocardial I/R injury after DEX preconditioning.
Initially, our results showed decreased miR-24 expression after I/R injury, and that miR-24 presented high expression in hUC-MSC-EV-treated cardiomyocytes. In addition, miR-24 in hUC-MSC-EVs inhibited cardiomyocyte apoptosis and inflammatory response after DEX preconditioning and participated in the macrophage polarization. It has been highly documented that DEX preconditioning can improve the cardiac function of I/R hearts and reduced myocardial infarction [34]. Similarly, in a mouse model of AMI from other published study, miR-24 was found to inhibit cardiomyocyte apoptosis, reduce the infarct size, and ameliorate cardiac dysfunction [35]. Besides, prior research unfolded that miR-24 could prevent against myocardial I/R injury via regulation of the NF-κB/TNF-α pathway [12]. These reports can support our finding on the protective function of miR-24 against myocardial I/R injury.
In the following experiments, we found that miR-24 could target KEAP1, and KEAP1 contributed to the activation of the Nrf2/HO-1 pathway to promote cardiomyocyte apoptosis, inflammation response as well as macrophages M2 polarization. Besides, miR-24 delivered by hUC-MSC-EVs could inhibit the Nrf2/HO-1 signaling pathway by down-regulation of KEAP1 expression, thereby suppressing cardiomyocyte apoptosis and macrophage M1 polarization. miR regulation on Nrf2 has been suggested as an important approach to modulating I/R injury [36]. KEAP1 is able to regulate Nrf2 activity [37]. More importantly, the KEAP1/Nrf2 system has been widely used in the development of protein–protein interaction inhibitors to stabilize Nrf2 [38]. Intriguingly, the inactivated KEAP1/Nrf2 signaling pathway by treatment with Yiqi Huoxue recipe could suppress cardiomyocyte apoptosis induced by heart failure [14]. Moreover, down-regulation of HO-1 by DEX treatment aids in ameliorating sepsis-triggered myocardial cellular injury [39]. However, these reports fail to explore the upstream miR regulatory mechanism of the KEAP1/Nrf2/HO-1 axis.
Importantly, MSC-EVs bear great responsibility in the modulation of cardiac function, and we confirmed that miRNA transfer was mainly through MSC-EVs. For example, MSC-derived exosomes containing miR-125b-5p improve cardiac function by inhibiting cell autophagy and death and down-regulating p53-Bnip3 signal transduction to reduce infarct size and apoptosis [40]. Additionally, MSC-derived exosomes participate in macrophage immune regulation and cardiac injury repair after myocardial I/R injury, and moreover, MSC-derived exosomes change macrophage polarization through miR-182 in mice [11]. In our study, by establishing a mouse model of myocardial I/R injury, the effect of EVs containing miR-24 on myocardial I/R injury was verified in vivo. miR-24 in hUC-MSC-EVs inhibited inflammation and apoptosis, repressed the macrophage M1 type polarization, and relieved the degree of myocardial cell damage and fibrosis.
In conclusion, miR-24 could be transferred by hUC-MSC-EVs into cardiomyocytes where miR-24 enhanced the inhibiting effect of DEX preconditioning on cell apoptosis and promoting effect on cell viability. In addition, miR-24 shuttled by hUC-MSC-EVs could promote M2-type polarization of macrophages and suppress M1-type macrophage polarization. Thus, the alleviating effect of DEX preconditioning on myocardial I/R injury was increased. The mechanism was associated with disruption of the KEAP1/Nrf2/HO-1 signaling. The pathogenesis of myocardial I/R injury was further elucidated in this study, which laid a theoretical foundation for the treatment of myocardial I/R injury. To further confirm the aforementioned results, further research is warranted to explore how DEX specifically acts on the KEAP1/Nrf2/HO-1 signaling.
Data availability
Data will be made available on request.
References
Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, Hausenloy DJ. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. 2018;186:73–87. https://doi.org/10.1016/j.pharmthera.2018.01.001.
Li Y, Chen B, Yang X, Zhang C, Jiao Y, Li P, Liu Y, Li Z, Qiao B, Bond Lau W, Ma XL, Du J. S100a8/a9 signaling causes mitochondrial dysfunction and cardiomyocyte death in response to ischemic/reperfusion injury. Circulation. 2019;140:751–64. https://doi.org/10.1161/CIRCULATIONAHA.118.039262.
Fan Q, Tao R, Zhang H, Xie H, Lu L, Wang T, Su M, Hu J, Zhang Q, Chen Q, Iwakura Y, Shen W, Zhang R, Yan X. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation. 2019;139:663–78. https://doi.org/10.1161/CIRCULATIONAHA.118.036044.
Xiong W, Zhou R, Qu Y, Yang Y, Wang Z, Song N, Liang R, Qian J. Dexmedetomidine preconditioning mitigates myocardial ischemia/reperfusion injury via inhibition of mast cell degranulation. Biomed Pharmacother. 2021;141:111853. https://doi.org/10.1016/j.biopha.2021.111853.
Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38:187–97. https://doi.org/10.1093/eurheartj/ehw002.
Frangogiannis NG. Emerging roles for macrophages in cardiac injury: cytoprotection, repair, and regeneration. J Clin Invest. 2015;125:2927–30. https://doi.org/10.1172/JCI83191.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28. https://doi.org/10.1038/nrm.2017.125.
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–83. https://doi.org/10.1083/jcb.201211138.
Martellucci S, Orefice NS, Angelucci A, Luce A, Caraglia M, Zappavigna S. Extracellular vesicles: new endogenous shuttles for miRNAs in cancer diagnosis and therapy? Int J Mol Sci. 2020;21: https://doi.org/10.3390/ijms21186486.
Fu S, Zhang Y, Li Y, Luo L, Zhao Y, Yao Y. Extracellular vesicles in cardiovascular diseases. Cell Death Discov. 2020;6:68. https://doi.org/10.1038/s41420-020-00305-y.
Zhao J, Li X, Hu J, Chen F, Qiao S, Sun X, Gao L, Xie J, Xu B. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc Res. 2019;115:1205–16. https://doi.org/10.1093/cvr/cvz040.
Li C, Fang M, Lin Z, Wang W, Li X. MicroRNA-24 protects against myocardial ischemia-reperfusion injury via the NF-kappaB/TNF-alpha pathway. Exp Ther Med. 2021;22:1288. https://doi.org/10.3892/etm.2021.10723.
Tan H, Qi J, Fan BY, Zhang J, Su FF, Wang HT. MicroRNA-24-3p attenuates myocardial ischemia/reperfusion injury by suppressing RIPK1 expression in mice. Cell Physiol Biochem. 2018;51:46–62. https://doi.org/10.1159/000495161.
Hu L, Xu Y, Wang Q, Liu M, Meng L, Yan D, Hu H, Xiao M, Yin Z, Li Y, Kang X. Yiqi Huoxue recipe inhibits cardiomyocyte apoptosis caused by heart failure through Keap1/Nrf2/HIF-1alpha signaling pathway. Bioengineered. 2021;12:969–78. https://doi.org/10.1080/21655979.2021.1900634.
Zhang H, Liu Y, Cao X, Wang W, Cui X, Yang X, Wang Y, Shi J. Nrf2 Promotes inflammation in early myocardial ischemia-reperfusion via recruitment and activation of macrophages. Front Immunol. 2021;12:763760. https://doi.org/10.3389/fimmu.2021.763760.
Yu H, Chen B, Ren Q. Baicalin relieves hypoxia-aroused H9c2 cell apoptosis by activating Nrf2/HO-1-mediated HIF1alpha/BNIP3 pathway. Artif Cells Nanomed Biotechnol. 2019;47:3657–63. https://doi.org/10.1080/21691401.2019.1657879.
Abd Alkhaleq H, Kornowski R, Waldman M, Levy E, Zemel R, Nudelman V, Shainberg A, Miskin R, Hochhauser E. Leptin modulates gene expression in the heart and cardiomyocytes towards mitigating ischemia-induced damage. Exp Cell Res. 2020;397:112373. https://doi.org/10.1016/j.yexcr.2020.112373.
Clough E, Barrett T. The gene expression omnibus database. Methods Mol Biol. 2016;1418:93–110. https://doi.org/10.1007/978-1-4939-3578-9_5.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. https://doi.org/10.1093/nar/gkv007.
Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4: https://doi.org/10.7554/eLife.05005.
Vejnar CE, Blum M, Zdobnov EM. miRmap web: comprehensive microRNA target prediction online. Nucleic Acids Res. 2013;41:W165–8. https://doi.org/10.1093/nar/gkt430.
Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, Sirota-Madi A, Olender T, Golan Y, Stelzer G, Harel A, Lancet D. GeneCards Version 3: the human gene integrator. Database (Oxford). 2010;2010:baq020. https://doi.org/10.1093/database/baq020.
Zhang J, Li JH, Wang L, Han M, Xiao F, Lan XQ, Li YQ, Xu G, Yao Y. Glucocorticoid receptor agonist dexamethasone attenuates renal ischemia/reperfusion injury by up-regulating eNOS/iNOS. J Huazhong Univ Sci Technolog Med Sci. 2014;34:516–20. https://doi.org/10.1007/s11596-014-1308-y.
Du J, Xu Z, Zhen J, Liu J, Yang D, Zheng EL, Leng JY. Dexmedetomidine attenuates myocardial ischemia/reperfusion injury through regulating lactate signaling cascade in mice. Eur Rev Med Pharmacol Sci. 2019;23:3527–32. https://doi.org/10.26355/eurrev_201904_17721.
Cao Y, Xu Y, Auchoybur ML, Chen W, He S, Qin W, Su C, Huang F, Qiu Z, Li L, Chen X. Regulatory role of IKKa in myocardial ischemia/reperfusion injury by the determination of M1 versus M2 polarization of macrophages. J Mol Cell Cardiol. 2018;123:1–12. https://doi.org/10.1016/j.yjmcc.2018.08.021.
Chen Z, Ding T, Ma CG. Dexmedetomidine (DEX) protects against hepatic ischemia/reperfusion (I/R) injury by suppressing inflammation and oxidative stress in NLRC5 deficient mice. Biochem Biophys Res Commun. 2017;493:1143–50. https://doi.org/10.1016/j.bbrc.2017.08.017.
Ou H, Teng H, Qin Y, Luo X, Yang P, Zhang W, Chen W, Lv D, Tang H. Extracellular vesicles derived from microRNA-150–5p-overexpressing mesenchymal stem cells protect rat hearts against ischemia/reperfusion. Aging (Albany NY). 2020;12:12669–83. https://doi.org/10.18632/aging.102792.
Yang C, Dou R, Wei C, Liu K, Shi D, Zhang C, Liu Q, Wang S, Xiong B. Tumor-derived exosomal microRNA-106b-5p activates EMT-cancer cell and M2-subtype TAM interaction to facilitate CRC metastasis. Mol Ther. 2021;29:2088–107. https://doi.org/10.1016/j.ymthe.2021.02.006.
Ma C, Qi X, Wei YF, Li Z, Zhang HL, Li H, Yu FL, Pu YN, Huang YC, Ren YX. Amelioration of ligamentum flavum hypertrophy using umbilical cord mesenchymal stromal cell-derived extracellular vesicles. Bioact Mater. 2023;19:139–54. https://doi.org/10.1016/j.bioactmat.2022.03.042.
Wei Q, Wang Y, Ma K, Li Q, Li B, Hu W, Fu X, Zhang C. Extracellular vesicles from human umbilical cord mesenchymal stem cells facilitate diabetic wound healing through MiR-17-5p-mediated enhancement of angiogenesis. Stem Cell Rev Rep. 2022;18:1025–40. https://doi.org/10.1007/s12015-021-10176-0.
Rurik JG, Aghajanian H, Epstein JA. Immune cells and immunotherapy for cardiac injury and repair. Circ Res. 2021;128:1766–79. https://doi.org/10.1161/CIRCRESAHA.121.318005.
Xiao X, Lu Z, Lin V, May A, Shaw DH, Wang Z, Che B, Tran K, Du H, Shaw PX. MicroRNA miR-24-3p reduces apoptosis and regulates Keap1-Nrf2 pathway in mouse cardiomyocytes responding to ischemia/reperfusion injury. Oxid Med Cell Longev. 2018;2018:7042105. https://doi.org/10.1155/2018/7042105.
Davidson SM, Ferdinandy P, Andreadou I, Botker HE, Heusch G, Ibanez B, Ovize M, Schulz R, Yellon DM, Hausenloy DJ, Garcia-Dorado D, Action CC. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol. 2019;73:89–99. https://doi.org/10.1016/j.jacc.2018.09.086.
Chen Y, Cao S, Chen H, Yin C, Xu X, Yang Z. Dexmedetomidine preconditioning reduces myocardial ischemia-reperfusion injury in rats by inhibiting the PERK pathway. Arq Bras Cardiol. 2021;117:1134–44. https://doi.org/10.36660/abc.20200672.
Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–60. https://doi.org/10.1084/jem.20101547.
Padmavathi G, Ramkumar KM. MicroRNA mediated regulation of the major redox homeostasis switch, Nrf2, and its impact on oxidative stress-induced ischemic/reperfusion injury. Arch Biochem Biophys. 2021;698:108725. https://doi.org/10.1016/j.abb.2020.108725.
Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015;88:93–100. https://doi.org/10.1016/j.freeradbiomed.2015.06.006.
Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. 2015;88:101–7. https://doi.org/10.1016/j.freeradbiomed.2015.05.034.
Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang CF, Li Y, Zhang Z. Dexmedetomidine alleviated sepsisinduced myocardial ferroptosis and septic heart injury. Mol Med Rep. 2020;22:175–84. https://doi.org/10.3892/mmr.2020.11114.
Xiao C, Wang K, Xu Y, Hu H, Zhang N, Wang Y, Zhong Z, Zhao J, Li Q, Zhu D, Ke C, Zhong S, Wu X, Yu H, Zhu W, Chen J, Zhang J, Wang J, Hu X. Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of miR-125b. Circ Res. 2018;123:564–78. https://doi.org/10.1161/CIRCRESAHA.118.312758.
Funding
This work was supported by the Excellent Youth Foundation of Hunan Provincial Department of Education (19B477), Project of Hunan Provincial Health Commission (20200018, 20200037), Natural Science Foundation of Hunan Province (2021JJ70043), and Guiding Project of Hengyang Science and Technology Bureau (202121034637).
Author information
Authors and Affiliations
Contributions
ZXH planned the experiments; FRY AND KMC performed the experiments, prepared the Figs. and analyzed the data; YXW performed some of the experiments; JQ was responsible for the collection of the clinical samples; FL contributed to drafting the manuscript All authors have read and approved the final submitted manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was implemented in the light of the Declaration of Helsinki and the guidelines issued by National Institutes of Health (Bethesda, MA) and ratified by the clinical ethics committee of The First Affiliated Hospital, Hengyang Medical School, University of South China. Human umbilical cords were obtained from infants with parental consent. Animal experiments were implemented in the light of the institutional animal care and use committee of The First Affiliated Hospital, Hengyang Medical School, University of South China.
Consent for publication
Consent for publication was obtained from the participants.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hou, Z., Yang, F., Chen, K. et al. hUC-MSC-EV-miR-24 enhances the protective effect of dexmedetomidine preconditioning against myocardial ischemia–reperfusion injury through the KEAP1/Nrf2/HO-1 signaling. Drug Deliv. and Transl. Res. 14, 143–157 (2024). https://doi.org/10.1007/s13346-023-01388-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-023-01388-7