
Abstract
Atazanavir (ATV) is widely used as anti-HIV agent having poor aqueous solubility needs to modulate novel drug delivery system to enhance therapeutic efficiency and safety. The main objective of the present work was to fabricate ATV-loaded nanostructured lipid carriers (NLCs) employing quality by design (QbD) approach to address the challenges of bioavailability and their safety after oral administration. Herein, the main objective was to identify the influencing variables for the production of quality products. Considering this objective, quality target product profile (QTPP) was assigned and a systematic risk assessment study was performed to identify the critical material attributes (CMAs) and critical process parameter (CPP) having an influence on critical quality attributes (CQAs). Lipid concentrations, surfactant concentrations, and pressure of high-pressure homogenizer were identified as CMAs and CPP. ATV-NLCs were prepared by emulsification-high pressure homogenization method and further lyophilized to obtain solid-state NLCs. The effect of formulation variables (CMAs and CPP) on responses like particle size (Y1), polydispersity index (Y2), and zeta potential (Y3) was observed by central composite rotatable design (CCRD). The data were statistically evaluated by ANOVA for confirmation of a significant level (p < 0.05). The optimal conditions of NLCs were obtained by generating design space and desirability value. The lyophilized ATV-NLCs were characterized by DSC, powder X-ray diffraction, and FT-IR analysis. The morphology of NLCs was revealed by TEM and FESEM. In vitro study suggested a sustained release pattern of drug (92.37 ± 1.03%) with a mechanism of Korsmeyer-Peppas model (r2 = 0.925, and n = 0.63). In vivo evaluation in Wistar rats showed significantly higher (p < 0.001) plasma drug concentration of ATV-NLCs as compared to ATV-suspension using chylomicron flow block model. The relative bioavailability of ATV-NLCs was obtained to be 2.54 folds. Thus, a safe and promising drug targeting system was successfully developed to improve bioavailability and avoiding first-pass effect ensures to circumvent the acute-toxicity of liver.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
To date, antiretroviral (ARV) drugs have been made remarkable progress in the treatment and control of human immunodeficiency virus (HIV) infection [1]. The combined therapy of ARV drugs so-called highly active antiretroviral therapy (HAART) is effective to extend the lifespan of individuals that suffer from HIV infection [2, 3]. However, they are inefficient to eradicate HIV completely from the host owing to their poor biodistribution in certain inaccessible compartments of the body such as CNS, genitals, and lymphatics [2]. Therefore, it is noteworthy to develop a novel strategy using efficient modalities for the treatment of HIV. Many scientists have believed that targeting lymphatics can be improved bioavailability of drug molecules [4].
Atazanavir (ATV) was selected as a model drug for the present investigation belongs to the category of azapeptide protease inhibitor. ATV falls under the category of BCS-II drugs exhibiting poor aqueous solubility and high permeability due to high log p value (log p = 4.11). It is the most recommended drug of choice in HAART regimen for treatment of HIV [5]. ATV is marketed as conventional therapeutic dose of 400 mg for adults. Regardless of promising molecule, ATV shows several biopharmaceutical and toxicological issues including rapid first-pass metabolism and P-gp efflux leads to a marked reduction in oral bioavailability to 60% [6]. Additionally, low absorption and frequent administration causes variation in plasma concentration and produces serious hepatic toxicity that limits clinical applications [7]. To overcome the aforementioned issues, there was a strong need to develop a novel strategy of ATV delivery to improve oral bioavailability and lowers hepatic toxicity.
Few works have been accounted for ATV to improve bioavailability including solid lipid nanoparticles (SNLs) [8, 9], solid dispersions [10], and nanoparticles of eudragit RL 100 [6]. However, no report has been addressed to target the lymphatics to improve bioavailability and subsequently safety. In this context, lipid-based delivery system holds the assurance to access the drugs into the intestinal lymphatics in spite of blood portal avoiding first-pass metabolism contributing to bioavailability improvement [11,12,13,14,15].
Nanostructured lipid carriers (NLCs) have been introduced as an option for the traditional carriers, such as liposomes, emulsions, microparticles, solid lipid nanoparticles (SLNs), and their polymeric counterparts due to numerous advantages [16, 17]. NLCs are referred to as next-generation and better than SLNs because of various benefits including higher drug entrapment, avoidance of drug expulsion, excellent biocompatibility, non-toxicity, shielding the drug from enzymatic degradation, controlled drug release, and industrial scalability [18]. NLCs are the combination of solid lipid and liquid lipid (oil) of GRAS (generally regarded as safe) status that makes the matrix system imperfect lattice arrangement to entrap a higher amount of drugs [17, 19, 20]. NLCs with smaller particle size < 200 nm reduce the hepatic uptake and extend blood circulation time [21]. More specifically smaller particle size and minimum polydispersity index (PDI) are the common attributes of NLCs for performing the targeting activity. Several researchers developed nanocarriers to target the lymphatics by anti-HIV drugs such as ritonavir, lopinavir, and efavirenz [1, 15, 22, 23].
The development of NLCs involves multistage production and multiple formulation variables including lipid concentrations, surfactant concentrations, amount of organic solvents, speed of high-speed homogenizer, and pressure of high pressure homogenizer. Controlling all the variables are difficult using several experiments. Therefore, a systematic approach is required to achieve the desired product. Thus, the application of quality by design (QbD) paradigm helps to identify, understand, and control the variables influencing the results [24]. QbD approach consigns the product quality, consistency in performance, and provides better control over the variables [25]. QbD-based development framework involves predefined quality target product profile (QTPP) describing desired product features. Identification and priority-based isolation of critical attributes were performed using Ishikawa diagram and risk assessment matrix (RAM) method. The individual and combined effect of variables were studied by employing central composite rotatable design (CCRD) in conjunction with response surface methodology (RSM) [26]. Further, optimized formulation was transformed into lyophilized NLCs. The dry NLCs were investigated for solid state characterizations, morphology, molecular interactions, and stability studies. Furthermore, NLCs were evaluated for drug dissolution, in vivo pharmacokinetic profile, and tissue distribution studies for bioavailability. Moreover, a toxicity study was conducted to ensure the safety of formulation using Wistar rats.
Materials and methods
Materials
Atazanavir (purity 99.0%) was obtained as a gift sample from Lupin research park (Aurangabad, India). Solid lipids Geleol™ Mono and Diglyceride NF (GMDG), and liquid lipid Capryol™ 90 were received as a gift sample from Gattefosse (Mumbai, India). Tween® 80 and Span® 20 were received from Hi-media Laboratories Pvt. Ltd. (Mumbai, India). D-mannitol (Purity ≥ 98%) was purchased from Sigma Aldrich Pvt. Ltd. (Bengaluru, India). A distilled water generated from Milli-Q Water Purification System (Millipore, USA) was used throughout the study. All other chemicals were received as an analytical reagent (AR) grade and used in experiments.
Screening of solid lipid, liquid lipid, and surfactants
The potential solid lipid and liquid lipid were screened using a reported method [27]. To the fixed quantity of ATV, the solid lipid was added with an increment of 500 mg at a temperature of 10 °C higher than the melting point of solid lipid. Various solid lipids such as Precirol® ATO5, Compritol® ATO 888, and Geleol™ Mono and Diglyceride (GMDG) and Compritol® HD5 ATO, Gellucire® 44/14, Palmitic acid, and Myristic acid were screened visually according to the solubilization behavior of ATV.
Likewise, the liquid lipid (oil) was screened by the continuous addition method. Briefly, an excess amount of ATV was added in a fixed amount (2 mL) of oil to achieve a saturation point. Each sample was centrifuged and analyzed by UV spectrophotometrically at 249 nm [28]. Thus, several oils such as Oleic acid, Captex® 200P, Labrafil® M 1944CS, Olive oil, Peceol™, Sunflower oil, Cottonseed oil, Maisine® CC, and Capryol™ 90 were tested in order to select appropriate liquid lipid suitable for the development of formulation.
The compatibility ratio of lipid mixture was selected based on phase separation parameter. The different mixture of solid lipid and liquid lipid in the proportion of 9:1, 8:2, 7:3, 6:4, and 5:5 was taken and heated at 5 °C above the melting point of solid lipid and was evaluated for the phase separation after 1 h, 12 h, and 24 h of solidification. No sign of phase separation was observed in the ratio of lipid mixture that was selected throughout the study [29]. The surfactant and co-surfactant were selected based on solubility and HLB values which lead to desired particle size, PDI, and stability [30].
QbD framework and its implementation
Assignment of QTPP and CQA
Implementation of QbD based concept was initiated by assigning the QTPP which could be achieved with the view of a patient-centric approach considering safety and efficacy of final formulation [31]. It was based on the recommendation of ICH Q8 guidelines, experimental judgment, and literature [32]. The essential parameters concerned with the route of administration, strength, dosage form, pharmacokinetics, and release profile were especially focused on the production of targeted product.
Moreover, the significant CQAs were identified that had a greater impact on the quality of product. The identified CQAs were monitored and controlled to ensure the desired quality of the product [33]. These attributes were further explored based on previous experience, experiments, and scientific knowledge gained through literature [34].
Risk assessment
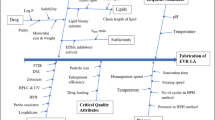
Risk assessment was performed to identify critical formulation variables like critical material attributes (CMAs) and critical process parameters (CPPs) which had a decisive effect on proposed product performance [34]. The Ishikawa diagram (Fig. 1) was constructed to enlist potential high-risk variables with their cause and effect to achieve the quality of the final product [35]. Furthermore, the risk assessment matrix (RAM) method was employed to find the effective and potential formulation variables. Thus, risk assessment of the proposed formulation was executed by combining the Ishikawa diagram and RAM method [36].
HPLC analysis
The ATV content present in formulation, plasma, spleen, and thymus was analyzed by the modified RP-HPLC method previously reported by Singh and Pai (2016) [6]. The mobile phase was prepared using potassium dihydrogen phosphate (pH 3.4) and acetonitrile in a ratio of (60:40, v/v). The sample (20 µL) for ATV analysis and bioanalytical sample (20 µL) was injected into Nucleosil C18 (4.6 mm I. D. × 250 mm) column at ambient temperature. The flow of 1.2 mL/min was adjusted for eluting ATV and was monitored at 249 nm by a UV detector. Furthermore, the developed method was validated as per the International Conference on Harmonization (ICH) guidelines, Q2 (R1) (2005) [37]. The standard curve was found to be linear (r2 = 0.991) over the range of 0.050–2.0 µg/mL.
Before analysis of sample, standard solutions of ATV and saquinavir (SAQ) Internal standard (IS) of strength 1000 μg/mL were prepared separately. A freshly collected rat plasma (blank) of approximately 0.5 mL was spiked into ATV and SAQ (IS) solutions of strength 1000 μg/mL. To this mixture, ethyl acetate was added and vortexed for 2 min in order to extract the drug content. Subsequently, the supernatant was obtained by centrifugation at 3000 rpm for 10 min and filtered through the 0.22-μm membrane filter. After evaporation, the dry residue was reconstituted with the mobile phase and the resulting solution of a 20-µL sample was injected into HPLC system.
Preparation of ATV-loaded NLCs
ATV-NLCs were prepared by the emulsification-hot high-pressure homogenization (HPH) technique [38]. Briefly, a measured quantity of GMDG (M. P. 54.0—64.0 °C), and Capryol™ 90 was added in 10 mL chloroform and melted on a water bath (Equitron water bath). To this phase, a measured amount of ATV (0.04%) and Span® 20 (co-surfactant) was dissolved. The organic solvent was completely evaporated by heating on a water bath at 70–75 °C. On the other hand, Tween® 80 (surfactant) was dissolved in distilled water and heated up to the same temperature as that of the lipid phase. After a clear and homogeneous lipid phase, the hot surfactant solution was dispersed into a melted lipid phase to obtain a pre-emulsion. This pre-emulsion was homogenized by high-speed homogenizer (Ultra-Turrax, T25 Basic, Ika Werke, Stanfer, Germany) at 10,000 rpm for 10 min. It was then passed through a hot high-pressure homogenizer (HPH) (PANDA 2 K, Niro Soavi, Italy) at 600–800 bar pressure and 5–7 cycles to produce the dispersion. The resultant dispersion was allowed to cool at room temperature to obtain NLCs and was stored in a cool place until characterizations [39].
Central composite rotatable design and statistical optimization
The CCRD was employed using Design Expert® version 12 (DX12) software to study the influence of different variables on responses. As summarized in Table 1, the formulation variables such as lipid concentrations (A), surfactant concentrations (B), and HPH pressure (C) were studied at five different levels as low (− 1), medium (0), high (+ 1), relatively low (− α), and relatively high (+ α). The responses were selected as particle size (Y1), PDI (Y2), and zeta potential (Y3). Thus, 20 experimental runs including 2 axial points (± α), 8 factorial design (± 1), and 10 central points (0) were studied for evaluation of variables as shown in Table 2. Thereafter, the responses were examined for interactions and 3D response plots were generated.
To achieve optimal formulation, the graphical and numerical analysis was performed based on constraints like minimum particle size, minimum PDI, and optimum zeta potential values [40]. A compromised solution for optimization was obtained using a desirability value that lies between 0 and 1 [41]. The observed result was compared with the predicted values, and bias (%) was determined in order to validate the selected experimental domain.
Size, PDI, and zeta potential analysis
A zeta sizer (Nano ZS90, Malvern, Worcestershire, UK) was used to measure particle size and PDI of NLCs [42]. The formulation was dispersed in Milli-Q water (aqueous medium) in the ratio of 1:100 and analyzed at 25 °C in triplicate. Zeta potential was measured by electrophoretic mobility of particles using zeta sizer [43]. NLCs were diluted 100 times using distilled Milli-Q water. The measurements were repeated in triplicate at 25 °C to obtain the average zeta potential value.
Determination of percent entrapment efficiency and drug loading
The percent drug entrapment of NLCs was determined by centrifugation method (a premier tabletop ultracentrifuge (Optima Max-XP, Beckman Coulter Inc., Brea, USA)). Briefly, a measured amount of dispersion of ATV-NLCs was centrifuged at 10,000 × g at 4 °C for 15 min. to separate the supernatant and nanocarrier pellets. The supernatant (1 mL) was suitably diluted and analyzed by spectrophotometrically (UV-1900, Shimadzu, Tokyo, Japan) at 249 nm. The percentage of drug entrapment was obtained in triplicates, and results were reported accordingly.
EE (%) and drug loading (% DL) was calculated by mathematical equations:
Lyophilization process
The optimized dispersion was lyophilized (Virtis-Bench Top Lyophilizer, Spinco Biotech Pvt. Ltd.) at a condition of − 74 °C temperature, 35 mT vacuum pressure for 42 h [44]. The different quantities of mannitol (2%, 2.5%, 3%, and 3.5%) were tested to find the optimal level to obtain a free-flowing solid powder and prevent lysis of NLCs.
Drug content (%)
An accurate 100 µg/mL sample of ATV-NLC was prepared and diluted suitably using the mobile phase. The sample was sonicated followed by filtering through a 0.22-µ membrane filter. Subsequently, the sample was injected into the HPLC system. The content was calculated by the following mathematical equation.
Solid state characterizations
Differential scanning calorimetry
Thermal behaviors of the ATV (pure drug), GMDG (solid lipid), physical mixture (PM) of ATV + GMDG, and optimized NLC formulation were recorded by DSC (Mettler Toledo, Italy) enabled with Mettler STARe V8.10 software. The test sample (2 mg) was sealed in an aluminum pan, whereas an empty pan was used as a reference [45]. The onset temperature, end set, and peak temperature were recorded at a temperature range of 40 to 200 °C using a heating rate of 10 °C per min in a nitrogen environment.
Powder X-ray diffraction
Diffraction patterns of crystalline substances were determined using PXRD study. This method was used to detect alterations in crystalline forms during development. In this study, the samples of ATV, GMDG (solid lipid), ATV + GMDG (PM), and optimized NLCs were analyzed through the X-ray diffractometer (Brucker AXS D8 advance®, Karlsruhe, Germany). Each sample was scanned at a 2θ scale with scanning range from 2 to 50 °C using voltage 40.0 kV and 35 mA current intensity.
Fourier-transform infrared spectroscopy
The IR spectra of pure ATV, GMDG (solid lipid), ATV + GMDG (PM), and optimized NLCs were recorded using FTIR spectrometer-430 (Shimadzu 8400S, Tokyo, Japan). Samples were physically mixed with dried potassium bromide (KBr) and applied a pressure to obtain a thin pellet. The wavelength for the scanning of KBr pellets was varied from 4000 to 500 cm−1.
Transmission electron microscopy and field emission scanning electron microscope
For surface morphology, the sample was prepared using negatively stained by phosphotungstic acid (2 wt%) and positively stained by uranyl acetate (1 wt%) with NLC dispersion on a copper grid. Following the air dry of sample, the image was captured using a TEM (TEM, Jeol/JEM 2100) instrument at a voltage 200 kV [46].
The surface structure of optimized nanocarriers was studied by FESEM (Quanta 250 FEG; FEI, Switzerland). The aqueous suspension of the NLCs (20 µL) was spread on clean glass slides and allowed to air-dry overnight. The dried glass slides were sputtered with 2-nm-thick gold and palladium layer at 20 mA before imaging [47].
Accelerated stability studies
The optimized ATV-NLCs were subjected to accelerated stability study for 90 days as per ICH guidelines at 4 °C/75% RH and 25 °C ± 2 °C/60% RH ± 5% RH. The lyophilized sample was sealed in a glass vial and stored in a stability chamber (CHM-10S, REMI Instruments Ltd., Mumbai, India) and examined for particle size, PDI, zeta potential, and % EE at 0th, 45th, and 90th days, respectively [48].
In vitro study and mechanism of release kinetics
In vitro drug release study for optimized ATV-NLCs and ATV-suspension was performed using the dialysis bag (soaked overnight in PBS solution to activate before use). A fixed volume of ATV-NLCs and ATV-suspension (containing 0.5% carboxymethyl cellulose) were sealed in the dialysis bag separately. The bag was hanged in 200-mL simulated gastric fluid (pH 1.2) for the first 2 h followed by simulated intestinal gastric fluid (pH 6.8) for a period of 24 h with a stirrer at 50 rpm at 37 °C maintaining a sink condition. Samples were withdrawn at various points of interval and analyzed spectrophotometrically at 249 nm after suitable dilution [49].
To investigate the drug release kinetics, the data were fitted to different models such as zero-order: % R = k0t, first order: log % unreleased = k1t/2.303, Higuchi-matrix: %R = kmt0.5, Korsmeyer–Peppas: % R = kptn, and Higuchi-Crowell: (% unreleased)1/3 = kht [50]. The correlation coefficient (r2) values were calculated to choose the best-fitted model and its associated mechanism of release.
Chylomicron flow block model for bioavailability and tissue distribution study
The experiment was designed as per the protocol approved by the Institutional Animal Ethics Committee (IAEC) of R. C. Patel Institute of Pharmaceutical Education and Research Shirpur (Ref. No. IAEC/RCPIPER/2018–19/29) with strictly followed the ethics and regulations of experiments. All the experiments were followed by the ARRIVE guidelines and performed in accordance with the UK Animals (Scientific Procedures) Act, 1986, and associated guidelines, EU Directive 2010/63/EU for animal experiments.
Healthy Wistar male rats weighing in the range of 220–230 ± 5 g were randomly divided into three cohorts (each having 5 rats in a cage) as follows:
-
Cohort-I was given oral aqueous ATV-suspension,
-
Cohort-II was administered ATV-NLCs, and.
-
Cohort-III was administered ATV-NLCs after the cycloheximide treatment.
A single oral dose to be administered to animals was calculated from dose conversion formula “Animal dose (mg/Kg) = human equivalent dose (HED) ÷ (animal weight in kg/human weight in kg)0.33” [51]. Since HED was used for the development of NLCs 1.16 mg/kg, the animals were administered a single oral dose of 7.2 mg/kg of their body weight using oral feeding needle [6]. The cohort-III was pre-treated with an intraperitoneal injection of cycloheximide (3 mg/kg) dissolved in saline solution (pH 7.4) (3 mg/mL) 1 h before the experiment. The cycloheximide, a chylomicron flow blocker, prevents the entry of a drug into the lymphatic system. Rats were anesthetized by intraperitoneal injection of 1 mL/kg of ketamine-xylazine (9%:1%) solution respectively [52]. Blood samples (approximately 200 µL) were collected from retro-orbital puncture under anesthesia at each sampling time point in the eppendorf tube containing 10% EDTA solution at a predetermined time interval of 0.5, 1, 2, 4, 8, 12 h, and 24 h [53]. Afterward, plasma was separated by centrifugation at 3500 rpm for 15 min and analyzed for ATV content using saquinavir (SAQ) as an internal standard (IS) by RP-HPLC with modifications in the reported method [6].
To investigate the tissue biodistribution, the rats were sacrificed by cervical dislocation technique at each time point. Surgical procedures were undertaken as described by Ahammed et al. [23]. The organs of our interest such as the spleen and thymus (containing a maximum amount of lymphocyte) were isolated and cut into pieces. The homogenate was prepared in methanol using a high-speed homogenizer (Ultra-Turrax, T25 Basic, Ika Werke, Stanfer, Germany). These samples were stored at − 20 °C until analysis.
For organ samples, the homogenate sample was subjected to the extraction process (described above for the plasma sample). Further, this sample was centrifuged at 15,000 × g for 15 min at 4 °C to obtain a supernatant. The obtained supernatant was evaporated to get the dry residue and analyzed for drug content by the estimated RP-HPLC method.
Calculation of pharmacokinetic parameters, relative bioavailability, and statistical analysis
The pharmacokinetic parameters were established using a Microsoft excel based PK solver 2.0 add-in program. Non-compartmental analysis was performed to determine the pharmacokinetic parameters. The AUC0–t (t = 24) was established by the trapezoidal method, and relative bioavailability was calculated using the following equation [54, 55].
where RBA is the relative bioavailability and AUC is area under curve. The various parameters such as Cmax (max. plasma concentration), tmax (time of occurrence), AUC0-t (area under the curve, t = 24), and MRT (mean residence time) were also calculated.
GraphPad Prism 6.0 (GraphPad Software Inc., California, USA) was used for statistical data evaluation. All the results were reported in mean ± S.D. (n = 3). The statistical analysis was performed by Student’s t-test and one-way ANOVA (analysis of variance) followed by Dunnett’s multiple comparisons. The data were considered to be statistically significant (p < 0.05).
Oral sub-acute toxicity study and serum analysis
The oral sub-acute toxicity study was performed on selected male Wistar rats. They were divided into control and ATV-NLCs administration cohorts. Animals were administered a low, medium, and high dose for 28 days as per the guidelines of OECD with pre-set time. They were under continuous observation for a behavioral sign, mortality rate, and body weight. Blood sampling followed by centrifugation at 1600 rpm for 20 min was done and freeze at − 40 °C until further study. The liver function tests were performed in order to analyze the changes in enzyme activity and ensure safety.
Histopathology studies
The animals were administered ATV-NLCs and standard ATV-suspension formulation over a period of 28 days for toxicity study. Animals were sacrificed, and liver was isolated according to the procedure. It was placed in a 10% formalin solution at 4 °C temperature for further analysis. The isolated tissues were washed with alcohol, xylene, and finally embedded in paraffin. These tissues were further sliced, dewaxed, and stained with hematoxylin and eosin for microscopic examination [56].
Results and discussion
Screening of solid lipid, liquid lipid, and selection of surfactants
Among the solid lipids screened, the GMDG was selected as potential solid lipid based on their highest solubilizing capacity for ATV, i.e., GMDG > Compritol® ATO 888 > Precirol® ATO5 > Gellucire® 44/14 > Compritol® HD 5 > Palmitic Acid > Myristic Acid (Supplementary Table S1). GMDG is a biocompatible and biodegradable lipid in combination with other ingredients. It was consistent with our previous publication that showed the highest solubility for ATV [57].
The liquid lipid, i.e., oil, is the crucial excipient for NLC production. The presence of oil can make the imperfect lattice arrangement to entrap the highest amount of drug. Among the tested oils, Capryol™ 90 was able to dissolve ATV to an appreciable extent (Fig. S1) and hence, was selected for the preparation purpose [58].
The appropriate concentration of surfactant or surfactant blend reduces the interfacial tension of formulation, and they are required to optimize the size of formulation. The surfactant’s HLB values lie between 8 and 20 are able to form a stable formulation. Therefore, based on the HLB value and solubility studies (Fig. S2), surfactant blend of Tween 80 and Span 20 (2:1) were used to minimize the particle size and PDI, and hence, this blend was selected for development of NLCs [30].
Lipid mixture compatibility
Based on the evaluation of phase separation parameter, the ratio of 7:3 (solid lipid: liquid lipid) lipid mixture was preferred throughout the study. This ratio could be constructed the imperfect lattice arrangement of carrier which results in desired particle size and maximum drug entrapment.
QbD framework and its implementation
QTPP, CQA, and risk assessment
In the QbD framework, initially, QTPP was assigned for the fabrication of NLCs achieving safe and efficacious formulation. The production of NLCs with planning from the initial stage was significant because a robust and reproducible production technique was required to meet the desired quality attributes. The identification of CQAs was linked with QTPP of expected formulation [33]. They were particle size (Y1), PDI (Y2), and zeta potential (Y3) and were presented as critical characteristics of formulation variables based on understanding and scientific knowledge [59]. For the desired quality product, the QTPP and CQA are summarized with their justifications in Table 3.
To perform the risk assessment, we plotted the Ishikawa diagram (Fig. 1) displaying the various root causes and effects of formulation variables [36]. Employing the risk assessment matrix (RAM) method, the formulation variables had an impact on CQAs that were isolated. In this method, we highlighted the different risk levels (high for red, medium for yellow, and low for green) of material attributes and process parameters (Table 4) [20]. The variables having a low or negligible impact on responses were neglected. RAM method suggested the high-risk variables such as CMAs, i.e., lipid concentrations, surfactant blend concentrations, and CPPs, i.e., HPH pressure based on the significant influence on critical attributes.

Ishikawa diagram illustrating the cause-and-effect relationship of CQAs of ATV-NLCs
Experimental design and statistical data analysis
In the present study, the CCRD was selected based on science and literature to observe the influence of formulation variables (lipid conc., surfactant conc., and HPH pressure) on their responses. As shown in Table 2, all the measurements were replicated three times to produce mean value (mean ± S.D., n = 3). The responses such as average particle size (Y1), PDI (Y2), and zeta potential (Y3) were ranged from 82.96 ± 1.23 to 127.7 ± 1.82 nm, 0.116 ± 1.93 to 0.486 ± 1.04, and − 7.1 ± 0.21 to − 13.7 ± 1.05, respectively (Table 2). As shown in Table 5, ANOVA suggested the fitted models were quadratic for responses and found to be significant (p < 0.05) with an insignificant lack of fit p-value (p > 0.10) (Table 6). The influence of formulation variables on the particle size and PDI of NLCs are illustrated in Eqs. (4) and (5), respectively, in association with 3D response surface plot (Fig. 2A and B) and found statistically significant for particle size (p < 0.0104, r2 = 0.808) and PDI (p < 0.013, r2 = 0.805).

3D response surface plots depicting effect of formulation variables on particle size (A) and PDI (B)
Influence of formulation variables on particle size and PDI
The particle size (Y1) and PDI (Y2) were the significant responses of formulation that determine the characteristics of NLCs. From Eq. 4, it was observed a positive relationship with lipid concentration (A), since an increase in lipid concentration gradually increases the particle size and PDI. These variables were attributed to several experimental trials and well in agreement with our previous finding [57]. The lower concentration of lipid was significant to produce smaller particles and uniform distribution. On the other hand, larger particle size was the result of a higher concentration of lipid, i.e., more than 3% [38]. The surfactant concentration (B) was the main ingredient in the formulation to stabilize the NLC system. It was revealed that appropriate surfactant and their optimal concentrations lead to a decrease in mean particle size and PDI [60]. Moreover, owing to hydrophilic and lipophilic domains of Tween 80, they generate strong repulsion by adsorbing on particles during storage to stabilize the system [61]. The combination of the surfactant (Tween 80) and co-surfactant (Span 20) was responsible for lowering particle size and PDI of the formulations. This was due to the reduction in interfacial tension and providing complete surface coverage to particles and provides steric repulsion.
The pressure of HPH significantly reduced the particle size during formulation processing. The generation of heat in homogenizer might be a contributing factor for the reduction of particle size due to the cavitational force produced in the homogenization gap. Beyond the optimal pressure, particle size increases gradually which could be due to possible collision and aggregation. Besides, the generated heat affects the particles that could alter the entrapment efficiency [62, 63].
Analysis of zeta potential (Y3)
Zeta potential (Y3) is a significant characteristic that develops on the particle surface offering stability through electrostatic repulsion [64]. In general, zeta potential of ± 30 is the desired value for good stability [65], but developed NLC showed a smaller zeta potential value as depicted in Table 2. This was due to the combined effect of non-ionic Tween 80 and Span 20 offering steric stabilization and avoiding particle aggregations [66, 67]. This indicates the particles were completely covered by the surfactants. Thus, the reduced zeta potential was an indicator of stabilized NLCs. These findings are well in agreement with other authors [25, 68].
Percent entrapment efficiency and drug loading
EE (%) of ATV-NLCs was found to be from 85.5 ± 0.24 to 96.23 ± 2.1. EE is essential for retention of drug molecules in lipid matrix. Higher solubility of the drug in lipid is the function of higher entrapment of drug. The content of oil in formulation offers an additional benefit to hold a high amount of drug in imperfect lattice arrangement. Thus, the combined effect of solid lipid and oil prevents drug leakage and improves drug entrapment [57]. Moreover, drug loading capacity was found to be 1.92%. The enhanced drug loading was due to the presence of components such as GMDG and Capryol 90 offering space to lodge the drugs due to imperfections in lipid matrix [69].
Formulation optimization and validation of experimental design
Design space was developed by obtaining a contour plot (Fig. 3A) and overlay plot (Fig. 3B) of formulation variables with given concentrations to meet the desired response. As can be seen in overlay plot (Fig. 3B), the colored region indicates all the desired criteria of responses. The working experimental optimal condition of NLC development was selected based on highest desirability value. Using suggested formulation values, the ATV-NLC was freshly prepared and responses so obtained were compared with predicted values, and percent (%) bias was calculated as 2.05 for particle size, 16.66 for PDI, and 0.019 for zeta potential, respectively (Table 7).

Contour plot (A) and overlay plot (B) depicting design space for optimization of formulation
Lyophilization
Mannitol as a cryoprotectant offers a significant role in the lyophilization process as it covers the particles to prevent aggregations. The optimal concentration of 3% was found to have free-flowing characteristics of the powder [45]. The dry powder can be redispersed in distilled Milli-Q water for further analysis after short sonication [70].
Drug content
Drug content was calculated as 97.24 ± 0.42% of optimized NLCs. The drug content indicated that the percentage of the total amount of actual ATV present in the formulation [71]. This was due to combining the solid lipid and liquid lipid domain that provides both mechanical stability and higher loading efficiency [72].
Solid state characterizations
Fourier-transform infrared spectroscopy
We investigated the interactions among the drug and main components of NLCs using FTIR analysis (Fig. 4). The broadbands at 3395.79 and 3266.56 were corresponding to N–H and O–H stretching. The bands at 2956.01, 1699.43, and 1243.16 were attributed to C–H stretching, C = C alkene (stretching (strong)), and C–O–C stretching confirming the structure of ATV (Fig. 4A) [73]. The spectra of GMDG (Fig. 4B), PM of GMDG, Capryol 90, and ATV (Fig. 4C) were not observed any variation indicating the lack of molecular interactions [74]. The spectrum of optimized NLC was shown the absorption bands (Fig. 4D) at 3285.85 associated with OH- stretching. Notably, the bands of O–H stretching shifted to a higher number (3285.85) during the NLC processing. This was observed because of hydrogen bond formation between the drug and the lipid molecule. The change in position of the absorption bands might be higher solubilization potential of drug molecules towards the lipid.

FTIR spectra of ATV (A), GMDG (B), physical mixture of ATV–GMDG (C), and optimized ATV-NLC (D)
DSC study
Thermograms behavior of pure ATV, GMDG, PM, and optimized ATV-NLC are depicted in Fig. 5. Pure ATV showed a specific broad endotherm at 197 °C (Fig. 5A) which indicates the crystalline nature of drug [75]. The GMDG displayed the sharp endotherm at 60.93 °C (Fig. 5B) which is corresponding to their melting point. The disappearance of the ATV peak at its melting range in the PM was due to the complete solubilization of drug in the lipid matrix (Fig. 5C). This was due to the conformational polymorphism during processing and indicates that ATV was molecularly dispersed in the lipid matrix that suggested an amorphous nature of drug present in PM [76].

DSC thermograms of ATV (A), GMDG (B), physical mixture of ATV–GMDG (C), and optimized ATV-NLC (D)
Thermogram of ATV-NLCs (Fig. 5D) showed the missing peak of ATV indicating the drug molecule was completely solubilized in lipid matrix and was in amorphous state. It was also noted that disappearance of peak associated with Kelvin effect, according to which particles in the nano range could melt at a temperature lower than the melting temperature of bulk material [54].
Importantly, as shown in Fig. 5, both exothermic and endothermic processes were occurred due to cold crystallization process by rapid cooling followed by crystalline melt in the heating pan. Exothermic events can be observed due to the increase in heat flow in the sample. Moreover, the temperature experienced by the sample was higher than that of the reference pan [77].
Powder X-ray diffraction
The diffraction patterns of pure ATV, GMDG, PM, and optimized NLCs are depicted in Fig. 6. The distinctive peaks of diffraction pattern were attributed to 2θ value in association with d-value (Å). Pure ATV showed the diffraction peaks at 2θ values of 3.102, 6.159, 9.23, and 12.307 (Fig. 6A) which were associated with d-values 28.45, 14.33, 9.57, and 7.18. These are the characteristic peaks of ATV indicating crystalline nature of ATV [75].

X-ray diffraction patterns for ATV (A), GMDG (B), physical mixture of ATV–GMDG (C), and optimized ATV-NLC (D)
GMDG (Fig. 6B) depicted the intense peaks at 2θ values of 5.4, 19.6, 22.9, and PM (Fig. 6C) of ATV, and GMDG showed the reduced intense peaks at 3.102, 6.159, 9.23, and 12.307 indicates that the ATV was incorporated and dispersed in the lipid matrix. The diffraction pattern of optimized NLCs (Fig. 6D) was revealed that the decline in degree of peaks at 2.69, 13.72, 17.36, 19.91, and 21.38 associated with d-value of 9.11, 6.44, 5.10, 4.45, and 4.15, respectively. This clearly indicated the phase transition behavior of ATV from crystalline to the amorphous state during formulation processing and affected complete molecular solubility in lipid matrix [42, 78, 79].
TEM and FESEM photomicrograph
TEM (Fig. 7B) of a spherical shape with a smooth surface and FESEM (Fig. 7C) depicted the discrete nanoparticles with uniform distribution of NLC which was a close agreement with DLS results shown in Fig. 7A [46]. FESEM is an advanced imaging technique to evaluate the morphology of particles.

Image depicting particle size (A), TEM analysis (B), and FESEM analysis (C)
Stability studies
As can be seen in Table 8, the small variations in particle size and PDI values after storage condition of 90 days was observed (p < 0.05). The zeta potential and EE values were almost remain unchanged on evaluation. These values were acceptable for the purpose of targeting delivery. The presence of the surfactant blend (Tween 80: Span 20) in NLCs provides steric stabilization strengthening the product enhancing the physical and chemical stability. Moreover, the presence of cryoprotectant prevents aggregation by covering the particles significantly [80].
In vitro drug dissolution and mechanism of kinetics
The comparative drug release profiles from optimized ATV-NLC and ATV-suspension are shown in Fig. 8. The cumulative drug release 92.37 ± 1.03% and 36.13 ± 0.06% were obtained from optimized ATV-NLC and ATV-suspension over a period of 24 h, respectively (Fig. 8). The observed difference in the release pattern was due to the sustained release behavior of drugs from the encapsulated NLCs. It was found that the ATV-NLCs had an initial burst release of around 60–70% at the end of 4 h, while the remaining amount of drug was released slowly over the period of 24 h [81]. The initial burst release from the optimized formulation for the first 4 h was attributed to the combined result of unentrapped ATV and adsorbed ATV on the surface of the nanocarriers [82]. It was also noted that the external portion of the nanocarrier was soft due to the presence of oil that contributed to the initial burst release [83]. The blend of lipids was not allowed to release the encapsulated drug particle equally due to the imperfect matrix structure. Therefore, the subsequent controlled release of ATV from the lipid carrier was shown due to the diffusion of drug through the lipid matrix. In addition, the presence of Tween 80 (hydrophilic surfactant) was contributed to modulate drug release from the lipid matrix [38]. Thus, considering all evidence, it was revealed that NLCs could be the potent carriers for the sustained delivery of ATV. The Korsmeyer-Peppas release kinetics was found to be best fit model with the highest correlation coefficient (r2) value of 0.925 (Table 9). The release exponent (n) was found to be 0.63 (n = 0.5–1.0) suggesting the non-fickian (anomalous) diffusion.

In vitro drug release profiles from an optimized ATV-NLC and ATV-suspension
In vivo pharmacokinetic study and tissue distribution studies
Despite the conventional surgical procedure for evaluation of lymphatic transport, we used the method from the published article [30]. In this method, no surgical procedure was carried out for the establishment of pharmacokinetic profile. The common protein synthesis inhibitor, i.e., Cycloheximide was used for lymphatic flow blocking approach. It works by inhibition of production of chylomicrons and inhibition of phagocytic activity of M cells thus block the pathway of lymphatic uptake of xenobiotics [1, 30]. In addition, no interference of other absorption pathways and no death or other side effect was observed in cycloheximide treated rats during study.
Figure 9A shows the pharmacokinetic profiles of ATV-suspension, ATV-NLCs, and ATV-NLC + Cycloheximide. The corresponding pharmacokinetic parameters are summarized in Table 9. The plasma drug concentration in cohort-III (i.e., ATV-NLC + Cycloheximide) was found significantly lower (p < 0.001), the Cmax value of 0.475 ± 0.45 μg/mL in comparison to cohort-II (i.e., ATV-NLCs), and the Cmax value of 0.765 ± 0.85 μg/mL. The lower AUC value of ATV-NLC (for Cycloheximide treated cohort-III) was due to the blocking of chylomicron flow in lymph by the pre-treatment of cycloheximide. Thus, blocking causes only entry in the blood portal which might further attempts to cause the first pass effects in liver [84]
On the other hand, significantly higher (p < 0.001) plasma concentration of Cmax and AUC0−t was observed in cohort-II receiving ATV-NLCs without treating cycloheximide. Because xenobiotics might be entered in lymphatic pathways avoiding reaching the liver for metabolism. This mechanism might be caused by the enhanced bioavailability of drug.
Herein, the key purpose of cycloheximide is that it only blocks the lymphatic uptake pathway and no interference of another absorption pathway. Additionally as stated earlier in the literature, the drug molecule should possess the log P value more than 5. Thus, in the present study, ATV has log p value = 4.1 and have inability to reach in lymphatics by conventional mean [1]. Therefore, to achieve the main objective, we modulate novel lipid-based NLC formulation to enhance the bioavailability. Several researchers have reported the lipid-based formulations that they have achieved the bioavailability using the chylomicron flow block model approach [30, 84,85,86].
From the results, ATV-suspension receiving cohort-I, it was observed that free drug suspension has not to access the lymphatic uptake, and hence, the AUC level was much lower (Table 10). Thus, pharmacokinetic study showed a significant (p < 0.001) improvement in relative bioavailability of ATV in plasma 2.54 folds post-oral administration of ATV-NLCs when compared to ATV-suspension (Table 10). This was due to the incorporation of liquid lipid (oil) in the formulation and subjected to entrap higher drug payload in the imperfect structure of lipid matrix. Here, component of lipid matrix, i.e., Capryol 90 and GMDG had key contribution of transporting the drug through the lymphatics by continuous secretion of triglyceride-rich chylomicron [4]. This secretion of chylomicron released by the endoplasmic reticulum of intestinal cell allows entry of NLCs into the lymph ducts through enterocytes. Such sequestration of NLCs might occur through major pathway of lipid uptake, i.e., through transcellular mechanism [87]. Pokharkar et al. have reported that lipids enhance bile secretion and subsequently combined with NLCs to form miscalls leading to absorption of intact NLC into lymphatic vessels which avoid the first pass effect [88].
The concentrations of ATV in suspension (Fig. 9B) and optimized NLCs (Fig. 9C) were estimated in lymphoid tissues such as thymus, spleen, and plasma (Table 10). The optimized ATV-NLCs in cohort-II after 4 h shows a significantly higher (p < 0.001) concentrations of drug in the spleen (0.95 ± 0.13 μg/ml) and thymus (0.82 ± 0.23 μg/ml) in comparison to plasma (0.76 ± 0.85 μg/ml). These results obtained due to the formulation components of the NLC that facilitated transport through the lymphatic pathway leads to rapid absorption into lymphatic organs [84, 89]. The GMDG was assisted to stimulate the intestinal lymphatic absorption because the long-chain lipid is known as drug transporter in lymphatic as reported by Shete et al.(2013) [87]. The non-ionic surfactant, Tween 80, was known as a P-gp inhibitor that facilitates to stimulate the secretion of chylomicrons [85, 90]. Nevertheless, Capryol 90 is effective in loosening tight junctions (TJs) and increased membrane fluidity of the intestinal epithelium that improves the lymphatic uptake [91]. As reported in Table 10, Tmax value for ATV-suspension and ATV-NLCs were found to be 2 h and 4 h, respectively. At the same time, the higher MRT values of optimized ATV-NLCs indicated a longer residence time of the drug in the spleen as compared to plasma and thymus. This was well in agreement with previous findings reported in the article [23].

Plasma concentration profiles of ATV in rats post-oral administration of ATV-suspension, ATV-NLCs, ATV-NLCs (cycloheximide treated) (A) and lymphoid tissues concentration of ATV in rat post-oral administration of ATV-suspension (B) and ATV-NLCs (C)
Oral dose toxicity and histopathology study
Animals were treated for 28 days with a repeated dose of control, and ATV-NLCs comprising of low, medium, and high dose. No significant variations in the weights of animals under study were found. However, two animals were died because of high dose and the remaining was survived throughout the study. Table 11 shows the results of the biochemical examination of liver function tests like aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase (ALP). Slight variations were observed in low and medium dose administered cohorts as compared to high-dose administered cohorts. As can be seen in Table 11, the high-dose cohorts showed an increased levels of AST and ALT and ALP, i.e., 2.3, 3.32, and 1.4 folds as compared to control cohorts. The low and medium dose values are normal in range for ATV-NLC cohort when compared to the control. An increased levels of AST and ALT were responsible for cell damage and the production of toxic species in the liver.
Histopathological examination was observed the normal size of vacuolation and sinusoidal space (Fig. 10A) for control cohorts, whereas suspension administered cohorts were found the liver injury that showing distortion in the liver with various degrees of sinusoidal and vacuolation spaces (Fig. 10B). In contrast, ATV-NLC administered cohort shows slight alterations (Fig. 10C) when compared to the control and suspension cohort. Thus, liver toxicity could be reduced by reduced particle size of nanocarriers and avoiding first-pass metabolism.

Histopathological photomicrograph of liver after 28 day repeated dosing to control cohort (A), suspension administered (B), and ATV-NLCs administered cohort (C) (magnification × 45)
Conclusion
The implementation of a QbD concept throughout the product lifecycle implies the time and cost-saving factor to ensure quality products. In the present study, we successfully developed the ATV-NLCs using hot high-pressure homogenization technique. The risk assessment tools such as the Ishikawa diagram and RAM method were implemented to identify the formulation variables. Optimized ATV-NLCs were investigated physicochemically and revealed the existence of the amorphous state of ATV and its compatibility with lipid carriers. This was observed due to the complete molecular solubility and reduced particle of the formulation. In vitro study revealed the sustained release of ATV could be tailored to meet desired results. The enhanced bioavailability of ATV-NLCs was obtained to be 2.54 folds as compared to a pure drug suspension. The higher accessibility of ATV by the transcellular mechanism in lymphatics avoids the portal circulation. Thus, ATV-NLCs ensure lower or no toxicity associated with a low and medium dose of the drug that could be the promising carrier for the effective oral delivery of lipophilic drugs.
Availability of data and materials
Data available on request from the authors.
References
Makwana V, Jain R, Patel K, Nivsarkar M, Joshi A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: elucidation of mechanism of uptake using chylomicron flow blocking approach. Int J Pharm [Internet]. Elsevier B.V.; 2015;495:439–46. https://doi.org/10.1016/j.ijpharm.2015.09.014.
Giacalone G, Hillaireau H, Fattal E. Improving bioavailability and biodistribution of anti-HIV chemotherapy. Eur J Pharm Sci [Internet]. Elsevier B.V.; 2015;75:40–53. https://doi.org/10.1016/j.ejps.2015.04.011.
Shaligram Mahajan H, Patil PH. Central composite design-based optimization of lopinavir vitamin E-TPGS Micelle: In Vitro Characterization and In Vivo Pharmacokinetic Study. Colloids Surfaces B Biointerfaces [Internet]. Elsevier B.V.; 2020;111149. https://doi.org/10.1016/j.colsurfb.2020.111149.
Dahan A, Hoffman A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur J Pharm Sci. 2005;24:381–8.
Singh G, Pai RS. Optimized self-nanoemulsifying drug delivery system of atazanavir with enhanced oral bioavailability: in vitro/in vivo characterization. Expert Opin Drug Deliv. 2014;11:1023–32.
Singh G, Pai RS. Atazanavir-loaded Eudragit RL 100 nanoparticles to improve oral bioavailability: Optimization and in vitro/in vivo appraisal. Drug Deliv. 2016;23:532–9.
Devi K, Pai R. Antiretrovirals: need for an effective drug delivery. Indian J Pharm Sci. 2006;68:1–6.
Chattopadhyay N, Zastre J, Wong HL, Wu XY, Bendayan R. Solid lipid nanoparticles enhance the delivery of the HIV protease inhibitor, atazanavir, by a human brain endothelial cell line. Pharm Res [Internet]. 2008;25:2262–71. https://doi.org/10.1007/s11095-008-9615-2.
Singh B, Diwan A. Effect of process parameters on formulation of solid lipid nanoparticles of protease inhibitor. Atazanavir Pharma Res. 2012;7:1–15.
Fukushima K, Terasaka S, Haraya K, Kodera S, Seki Y, Wada A, et al. Pharmaceutical approach to HIV protease inhibitor atazanavir for bioavailability enhancement based on solid dispersion system. Biol Pharm Bull. 2007;30:733–8.
Gohla S, Mader K, Muller RH. Solid lipid nanoparticles ( SLN ) for controlled drug delivery ± a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–77.
Müller RH, Freitas C, zur Mühlen A, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery. Eur J Pharm Sci [Internet]. 1996;4:S75. http://www.sciencedirect.com/science/article/pii/S0928098797862434.
Nasirizadeh S, Malaekeh-Nikouei B. Solid lipid nanoparticles and nanostructured lipid carriers in oral cancer drug delivery. J Drug Deliv Sci Technol [Internet]. Elsevier; 2020;55:101458. https://doi.org/10.1016/j.jddst.2019.101458.
Sanjula B, Shah FM, Javed A, Alka A. Effect of poloxamer 188 on lymphatic uptake of carvedilol-loaded solid lipid nanoparticles for bioavailability enhancement. J Drug Target. 2009;17:249–56.
Kumar S, Narayan R, Ahammed V, Nayak Y, Naha A, Nayak UY. Development of ritonavir solid lipid nanoparticles by Box Behnken design for intestinal lymphatic targeting. J Drug Deliv Sci Technol [Internet]. Elsevier B.V.; 2018;44:181–9. https://doi.org/10.1016/j.jddst.2017.12.014.
Cirri M, Bragagni M, Mennini N, Mura P. Development of a new delivery system consisting in ‘“ drug – in cyclodextrin – in nanostructured lipid carriers ”’ for ketoprofen topical delivery. Eur J Pharm Biopharm [Internet]. Elsevier B.V.; 2012;80:46–53. https://doi.org/10.1016/j.ejpb.2011.07.015.
Jaiswal P, Gidwani B, Vyas A. Nanostructured lipid carriers and their current application in targeted drug delivery. Artif Cells, Nanomedicine, Biotechnol [Internet]. 2016;44:27–40. https://doi.org/10.3109/21691401.2014.909822.
Shevalkar G, Vavia P. Solidified nanostructured lipid carrier (S-NLC) for enhancing the oral bioavailability of ezetimibe. J Drug Deliv Sci Technol [Internet]. Elsevier; 2019;53:101211. https://doi.org/10.1016/j.jddst.2019.101211.
Naseri N, Valizadeh H, Zakeri-Milani P. Solid lipid nanoparticles and nanostructured lipid carriers: structure preparation and application. Adv Pharm Bull [Internet]. 2015;5:305–13. https://doi.org/10.15171/apb.2015.043.
Garg NK, Sharma G, Singh B, Nirbhavane P, Tyagi RK, Shukla R, et al. Quality by Design (QbD)-enabled development of aceclofenac loaded-nano structured lipid carriers (NLCs): an improved dermatokinetic profile for inflammatory disorder(s). Int J Pharm [Internet]. Elsevier B.V.; 2017;517:413–31. https://doi.org/10.1016/j.ijpharm.2016.12.010.
Tsai MJ, Wu PC, Huang Y Bin, Chang JS, Lin CL, Tsai YH, et al. Baicalein loaded in tocol nanostructured lipid carriers (tocol NLCs) for enhanced stability and brain targeting. Int J Pharm [Internet]. Elsevier B.V.; 2012;423:461–70. https://doi.org/10.1016/j.ijpharm.2011.12.009.
Aji Alex MR, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42:11–8.
Ahammed V, Narayan R, Paul J, Nayak Y, Roy B, Shavi G V., et al. Development and in vivo evaluation of functionalized ritonavir proliposomes for lymphatic targeting. Life Sci [Internet]. Elsevier Inc.; 2017;183:11–20. https://doi.org/10.1016/j.lfs.2017.06.022.
Bonde S, Bonde CG, Prabhakar B. Quality by design based development and validation of HPLC method for simultaneous estimation of paclitaxel and vinorelbine tartrate in dual drug loaded liposomes. Microchem J [Internet]. Elsevier; 2019;149:103982. https://doi.org/10.1016/j.microc.2019.103982.
Shekhawat P, Pokharkar V. Risk assessment and QbD based optimization of an Eprosartan mesylate nanosuspension: in-vitro characterization, PAMPA and in-vivo assessment. Int J Pharm [Internet]. Elsevier; 2019;567:118415. https://doi.org/10.1016/j.ijpharm.2019.06.006.
Kincl M, Turk S, Vrečer F. Application of experimental design methodology in development and optimization of drug release method. Int J Pharm. 2005;291:39–49.
Singh A, Neupane YR, Mangla B, Kohli K. Nanostructured lipid carriers for oral bioavailability enhancement of exemestane: formulation design, In Vitro, Ex Vivo, and In Vivo Studies. J Pharm Sci [Internet]. Elsevier Ltd; 2019;108:3382–95. https://doi.org/10.1016/j.xphs.2019.06.003.
Jazuli I, Annu, Nabi B, moolakkadath T, Alam T, Baboota S, et al. Optimization of nanostructured lipid carriers of lurasidone hydrochloride using Box-Behnken design for brain targeting: in vitro and in vivo studies. J Pharm Sci [Internet]. Elsevier Ltd; 2019;108:3082–90. https://doi.org/10.1016/j.xphs.2019.05.001.
Alam T, Khan S, Gaba B, Haider MF, Baboota S, Ali J. Adaptation of quality by design-based development of isradipine nanostructured–lipid carrier and its evaluation for in vitro gut permeation and in vivo solubilization fate. J Pharm Sci [Internet]. Elsevier Ltd; 2018;107:2914–26. https://doi.org/10.1016/j.xphs.2018.07.021.
Rangaraj N, Pailla SR, Shah S, Prajapati S, Sampathi S. QbD aided development of ibrutinib-loaded nanostructured lipid carriers aimed for lymphatic targeting: evaluation using chylomicron flow blocking approach. Drug Deliv Transl Res. Drug Delivery and Translational Research; 2020;
Shailesh S. Chalikwar , Sanjay J. Surana , Sameer N. Goyal KKC& PVD. Solid self-microemulsifying nutraceutical delivery system for hesperidin using quality by design: Assessment of biopharmaceutical attributes and shelf-life. J Microencapsul [Internet]. Taylor & Francis; 2020;0:000. https://doi.org/10.1080/02652048.2020.1851788.
ICH guideline Q8(R2). ICH harmonised tripartite guideline pharmaceutical development. Curr Step 4 version. 2009;1–24.
Pallagi E, Ambrus R, Szabó-Révész P, Csóka I. Adaptation of the quality by design concept in early pharmaceutical development of an intranasal nanosized formulation. Int J Pharm [Internet]. Elsevier B.V.; 2015;491:384–92. https://doi.org/10.1016/j.ijpharm.2015.06.018.
Manteghi R, Pallagi E, Olajos G, Csóka I. Pegylation and formulation strategy of anti-microbial peptide (AMP) according to the quality by design approach. Eur J Pharm Sci [Internet]. Elsevier B.V.; 2020;144:105197. https://doi.org/10.1016/j.ejps.2019.105197.
Dangre PV, Phad RD, Surana SJ, Chalikwar SS. Quality by Design ( QbD ) Assisted fabrication of fast dissolving buccal film for clonidine hydrochloride : exploring the quality attributes. Adv Polym Technol. 2019;2019:1–13.
Luo T, Wu C, Duan L. Fishbone diagram and risk matrix analysis method and its application in safety assessment of natural gas spherical tank. J Clean Prod [Internet]. Elsevier Ltd; 2018;174:296–304. https://doi.org/10.1016/j.jclepro.2017.10.334.
ICH guideline Q2(R1). ICH harmonised tripartite guideline, validation of analytical procedures: text and methodology. Curr. Step 4 version Parent Guidel. dated 27 Oct. 1994 2005 p. 13.
Kumbhar DD, Pokharkar VB. Engineering of a nanostructured lipid carrier for the poorly water-soluble drug, bicalutamide : physicochemical investigations. Colloids Surfaces A Physicochem Eng Asp [Internet]. Elsevier B.V.; 2013;416:32–42. https://doi.org/10.1016/j.colsurfa.2012.10.031.
Chalikwar SS, Belgamwar VS, Talele VR, Surana SJ, Patil MU. Formulation and evaluation of Nimodipine-loaded solid lipid nanoparticles delivered via lymphatic transport system. Colloids Surfaces B Biointerfaces [Internet]. Elsevier B.V.; 2012;97:109–16. https://doi.org/10.1016/j.colsurfb.2012.04.027.
Ferreira M, Chaves LL, Lima SAC, Reis S. Optimization of nanostructured lipid carriers loaded with methotrexate: a tool for inflammatory and cancer therapy. Int J Pharm. 2015;492:65–72.
Abbou A, Kadri N, Dahmoune F, Chergui A, Remini H, Berkani F, et al. Optimising functional properties and chemical composition of Pinus halepensis Mill. Seeds protein concentrates. Food Hydrocoll [Internet]. Elsevier Ltd; 2020;100:105416. https://doi.org/10.1016/j.foodhyd.2019.105416.
Shete H, Patravale V. Long chain lipid based tamoxifen NLC. Part I: Preformulation studies, formulation development and physicochemical characterization. Int J Pharm [Internet]. Elsevier B.V.; 2013;454:573–83. https://doi.org/10.1016/j.ijpharm.2013.03.034.
Das S, Ng WK, Kanaujia P, Kim S, Tan RBH. Formulation design, preparation and physicochemical characterizations of solid lipid nanoparticles containing a hydrophobic drug: effects of process variables. Colloids Surfaces B Biointerfaces [Internet]. Elsevier B.V.; 2011;88:483–9. https://doi.org/10.1016/j.colsurfb.2011.07.036.
Liu H, Rivnay B, Avery K, Myung JH, Kozak D, Landrau N, et al. Optimization of the manufacturing process of a complex amphotericin B liposomal formulation using quality by design approach. Int J Pharm [Internet]. Elsevier B.V.; 2020;585:119473. https://doi.org/10.1016/j.ijpharm.2020.119473.
Karakucuk A, Celebi N, Teksin ZS. Preparation of ritonavir nanosuspensions by microfluidization using polymeric stabilizers: I. A Design of Experiment approach. Eur J Pharm Sci [Internet]. Elsevier B.V.; 2016;95:111–21. https://doi.org/10.1016/j.ejps.2016.05.010.
Neupane YR, Srivastava M, Ahmad N, Kumar N, Bhatnagar A, Kohli, Kanchan. Lipid based nanocarrier system for the potential oral delivery of decitabine: formulation design, characterization, ex vivo, and in vivo assessment. Int J Pharm [Internet]. Elsevier B.V.; 2014;477:601–12. https://doi.org/10.1016/j.ijpharm.2014.11.001.
Kumar R, Singh A, Garg N, Siril PF. Solid lipid nanoparticles for the controlled delivery of poorly water soluble non-steroidal anti-inflammatory drugs. Ultrason Sonochem [Internet]. 2018;40:686–96. https://doi.org/10.1016/j.ultsonch.2017.08.018.
Kaur R, Ajitha M. Transdermal delivery of fluvastatin loaded nanoemulsion gel: preparation, characterization and in vivo anti-osteoporosis activity. Eur J Pharm Sci [Internet]. Elsevier; 2019;136:104956. https://doi.org/10.1016/j.ejps.2019.104956.
Li HL, Zhao X Bin, Ma YK, Zhai GX, Li LB, Lou HX. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J Control Release [Internet]. Elsevier B.V.; 2009;133:238–44. https://doi.org/10.1016/j.jconrel.2008.10.002.
Garg NK, Singh B, Sharma G, Kushwah V, Tyagi RK, Jain S, et al. Development and characterization of single step self-assembled lipid polymer hybrid nanoparticles for effective delivery of methotrexate. RSC Adv [Internet]. 2015;5:62989–99. http://xlink.rsc.org/?DOI=C5RA12459J.
U.S. Department of Health and Human Services. Guidance for industry: characterization and qualification of cell substrates and other biological starting materials used in the production of viral vaccines for the prevention and treatment of infectious diseases. Biotechnol Law Rep. 2005;1–27.
Dahan A, Mendelman A, Amsili S, Ezov N, Hoffman A. The effect of general anesthesia on the intestinal lymphatic transport of lipophilic drugs: comparison between anesthetized and freely moving conscious rat models. Eur J Pharm Sci. 2007;32:367–74.
Patil-Gadhe A, Pokharkar V. Montelukast-loaded nanostructured lipid carriers : Part I Oral bioavailability improvement. Eur J Pharm Biopharm [Internet]. Elsevier B.V.; 2014;88:160–8. https://doi.org/10.1016/j.ejpb.2014.05.019.
Tiwari R, Pathak K. Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int J Pharm [Internet]. Elsevier B.V.; 2011;415:232–43. https://doi.org/10.1016/j.ijpharm.2011.05.044.
Pardeshi CV, Belgamwar VS. Improved brain pharmacokinetics following intranasal administration of N,N,N-trimethyl chitosan tailored mucoadhesive NLCs. Mater Technol [Internet]. Taylor & Francis; 2020;35:249–66. https://doi.org/10.1080/10667857.2019.1674522.
Nassimi M, Schleh C, Lauenstein HD, Hussein R, Hoymann HG, Koch W, et al. A toxicological evaluation of inhaled solid lipid nanoparticles used as a potential drug delivery system for the lung. Eur J Pharm Biopharm [Internet]. Elsevier B.V.; 2010;75:107–16. https://doi.org/10.1016/j.ejpb.2010.02.014.
Gurumukhi VC, Bari SB. Fabrication of efavirenz loaded nano-formulation using quality by design (QbD) based approach: exploring characterizations and in vivo safety. J Drug Deliv Sci Technol [Internet]. Elsevier; 2020;56:101545. https://doi.org/10.1016/j.jddst.2020.101545.
Gokhale JP, Mahajan HS, Surana SS. Quercetin loaded nanoemulsion-based gel for rheumatoid arthritis: in vivo and in vitro studies. Biomed Pharmacother [Internet]. Elsevier; 2019;112:108622. https://doi.org/10.1016/j.biopha.2019.108622.
Amasya G, Aksu B, Badilli U, Onay-Besikci A, Tarimci N. QbD guided early pharmaceutical development study: production of lipid nanoparticles by high pressure homogenization for skin cancer treatment. Int J Pharm [Internet]. Elsevier B.V.; 2019;563:110–21. https://doi.org/10.1016/j.ijpharm.2019.03.056.
Fangueiro JF, Andreani T, Egea MA, Garcia ML, Souto SB, Silva AM, et al. Design of cationic lipid nanoparticles for ocular delivery: development, characterization and cytotoxicity. Int J Pharm [Internet]. Elsevier B.V.; 2014;461:64–73. https://doi.org/10.1016/j.ijpharm.2013.11.025.
Shah B, Khunt D, Bhatt H, Misra M, Padh H. Application of quality by design approach for intranasal delivery of rivastigmine loaded solid lipid nanoparticles: effect on formulation and characterization parameters. Eur J Pharm Sci [Internet]. Elsevier B.V.; 2015;78:54–66. https://doi.org/10.1016/j.ejps.2015.07.002.
Wissing SA, Kayser O, Müller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug Deliv. Rev. 2004. p. 1257–72.
Bhise K, Kashaw SK, Sau S, Iyer AK. Nanostructured lipid carriers employing polyphenols as promising anticancer agents: quality by design (QbD) approach [Internet]. Int. J. Pharm. Elsevier B.V.; 2017. p. 506–15. https://doi.org/10.1016/j.ijpharm.2017.04.078.
Shegokar R, Singh KK, Müller RH. Production & stability of stavudine solid lipid nanoparticles—from lab to industrial scale. Int J Pharm [Internet]. Elsevier B.V.; 2011;416:461–70. https://doi.org/10.1016/j.ijpharm.2010.08.014.
Khosa A, Reddi S, Saha RN. Nanostructured lipid carriers for site-specific drug delivery. Biomed Pharmacother [Internet]. Elsevier; 2018;103:598–613. https://doi.org/10.1016/j.biopha.2018.04.055.
Han F, Li S, Yin R, Liu H, Xu L. Effect of surfactants on the formation and characterization of a new type of colloidal drug delivery system: nanostructured lipid carriers. Colloids Surfaces A Physicochem Eng Asp. 2008;315:210–6.
Shah R, Eldridge D, Palombo E, Harding I. Lipid nanoparticles: production, characterization and stability. 2015;11–23. https://doi.org/10.1007/978-3-319-10711-0.
Espinosa-Olivares MA, Delgado-Buenrostro NL, Chirino YI, Trejo-Márquez MA, Pascual-Bustamante S, Ganem-Rondero A. Nanostructured lipid carriers loaded with curcuminoids: physicochemical characterization, in vitro release, ex vivo skin penetration, stability and antioxidant activity. Eur J Pharm Sci [Internet]. Elsevier; 2020;155:105533. https://doi.org/10.1016/j.ejps.2020.105533.
Üstündaǧ-Okur N, Gökçe EH, Bozbiyik DI, Eǧrilmez S, Özer Ö, Ertan G. Preparation and in vitro-in vivo evaluation of ofloxacin loaded ophthalmic nano structured lipid carriers modified with chitosan oligosaccharide lactate for the treatment of bacterial keratitis. Eur J Pharm Sci. 2014;63:204–15.
Fan H, Liu G, Huang Y, Li Y, Xia Q. Development of a nanostructured lipid carrier formulation for increasing photo-stability and water solubility of Phenylethyl Resorcinol. Appl Surf Sci [Internet]. Elsevier B.V.; 2014;288:193–200. https://doi.org/10.1016/j.apsusc.2013.10.006.
Iqbal R, Ahmed S, Jain GK, Vohora D. Design and development of letrozole nanoemulsion: a comparative evaluation of brain targeted nanoemulsion with free letrozole against status epilepticus and neurodegeneration in mice. Int J Pharm [Internet]. Elsevier B.V.; 2019;565:20–32. https://doi.org/10.1016/j.ijpharm.2019.04.076.
Dan N. Nanostructured lipid carriers: effect of solid phase fraction and distribution on the release of encapsulated materials. Langmuir. 2014;30:13809–14.
Chitturi SR, Somannavar YS, Peruri BG, Nallapati S, Sharma HK, Budidet SR, et al. Gradient RP-HPLC method for the determination of potential impurities in atazanavir sulfate. J Pharm Biomed Anal [Internet]. Elsevier B.V.; 2011;55:31–47. https://doi.org/10.1016/j.jpba.2011.01.002.
Prathyusha M, Manjunath K, Tippanna S, Shivanandappa B. Bixin loaded solid lipid nanoparticles for enhanced hepatoprotection—preparation, characterisation and in vivo evaluation. Int J Pharm. 2014;473:485–92.
Sherwood WJ, D. Atmurr S. Patent Application Publication ( 10 ) Pub . No .: US 2005 / 0131113 A1. 2005;1:6–9.
Mendes AI, Silva AC, Catita JAM, Cerqueira F, Gabriel C, Lopes CM. Miconazole-loaded nanostructured lipid carriers ( NLC ) for local delivery to the oral mucosa: improving antifungal activity. Colloids Surfaces B Biointerfaces [Internet]. Elsevier B.V.; 2013;111:755–63. https://doi.org/10.1016/j.colsurfb.2013.05.041.
EAG Laboratories. Characterization of polymers using differential scanning calorimetry (DSC) importance of characterizing thermal characterization of polymers using differential scanning calorimetry (DSC). 2017;1–5. f: file:///C:/Users/hamil/Downloads/white-paper-characterization-of-polymers-using-differential-scanning-calorimetry-dsc-m-012816.pdf
Bose S, Michniak-kohn B. Preparation and characterization of lipid based nanosystems for topical delivery of quercetin. Eur J Pharm Sci [Internet]. 2013;48:442–52. https://doi.org/10.1016/j.ejps.2012.12.005.
Madan J, Pandey RS, Jain V, Katare OP, Chandra R, Katyal A. Poly ( ethylene )-glycol conjugated solid lipid nanoparticles of noscapine improve biological half-life, brain delivery and efficacy in glioblastoma cells. Nanomedicine Nanotechnology, Biol Med [Internet]. Elsevier Inc.; 2013;9:492–503. https://doi.org/10.1016/j.nano.2012.10.003.
Kim B Do, Na K, Choi HK. Preparation and characterization of solid lipid nanoparticles (SLN) made of cacao butter and curdlan. Eur J Pharm Sci. 2005;24:199–205.
Kumbhar DD, Pokharkar VB. Physicochemical investigations on an engineered lipid – polymer hybrid nanoparticle containing a model hydrophilic active, zidovudine. Colloids Surfaces A Physicochem Eng Asp [Internet]. Elsevier B.V.; 2013;436:714–25. https://doi.org/10.1016/j.colsurfa.2013.07.044.
Jose S, Anju SS, Cinu TA, Aleykutty NA, Thomas S, Souto EB. In vivo pharmacokinetics and biodistribution of resveratrol-loaded solid lipid nanoparticles for brain delivery. Int J Pharm [Internet]. Elsevier B.V.; 2014;474:6–13. https://doi.org/10.1016/j.ijpharm.2014.08.003.
Chen Y, Yang X, Zhao L, Almásy L, Garamus VM, Willumeit R, et al. Preparation and characterization of a nanostructured lipid carrier for a poorly soluble drug. Colloids Surfaces A Physicochem Eng Asp [Internet]. Elsevier B.V.; 2014;455:36–43. https://doi.org/10.1016/j.colsurfa.2014.04.032.
Shrivastava S, Gidwani B, Kaur CD. Development of mebendazole loaded nanostructured lipid carriers for lymphatic targeting: optimization, characterization, in-vitro and in-vivo evaluation. Part Sci Technol [Internet]. Taylor & Francis; 2020;0:1–11. https://doi.org/10.1080/02726351.2020.1750515.
Xing Q, Song J, You X, Xu D, Wang K, Song J, et al. Microemulsions containing long-chain oil ethyl oleate improve the oral bioavailability of piroxicam by increasing drug solubility and lymphatic transportation simultaneously. Int J Pharm [Internet]. Elsevier B.V.; 2016;511:709–18. https://doi.org/10.1016/j.ijpharm.2016.07.061.
Fang G, Tang B, Chao Y, Zhang Y, Xu H, Tang X. Improved oral bioavailability of docetaxel by nanostructured lipid carriers: in vitro characteristics, in vivo evaluation and intestinal transport studies. RSC Adv [Internet]. 2015;5:96437–47. http://xlink.rsc.org/?DOI=C5RA14588K.
Shete H, Chatterjee S, De A, Patravale V. Long chain lipid based tamoxifen NLC. Part II: pharmacokinetic, biodistribution and in vitro anticancer efficacy studies. Int J Pharm [Internet]. Elsevier B.V.; 2013;454:584–92. https://doi.org/10.1016/j.ijpharm.2013.03.036.
Pokharkar V, Patil-Gadhe A, Kaur G. Physicochemical and pharmacokinetic evaluation of rosuvastatin loaded nanostructured lipid carriers: influence of long- and medium-chain fatty acid mixture. J Pharm Investig. Springer Netherlands; 2018;48:465–76.
Permana AD, Tekko IA, McCrudden MTC, Anjani QK, Ramadon D, McCarthy HO, et al. Solid lipid nanoparticle-based dissolving microneedles: a promising intradermal lymph targeting drug delivery system with potential for enhanced treatment of lymphatic filariasis. J Control Release [Internet]. Elsevier; 2019;316:34–52. https://doi.org/10.1016/j.jconrel.2019.10.004.
Nerurker MM, Burton PS, Borchardt RT. The use of surfactants to enhance the permeability of peptides through Caco-2 cells by inhibition of an apically polarized efflux system. Pharm. Res. 1996. p. 528–34.
Ukai H, Iwasa K, Deguchi T, Morishita M, Katsumi H, Yamamoto A. Enhanced intestinal absorption of insulin by capryol 90, a novel absorption enhancer in rats: Implications in oral insulin delivery. Pharmaceutics. 2020;12:1–16.
Acknowledgements
The authors are thankful to Lupin Research Park, Aurangabad, for providing a gift sample of pure Atazanavir. The authors are grateful to Management and Principal of R. C. Patel Institute of Pharmaceutical Education and Research (RCPIPER), Shirpur, for providing various research facilities.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All the animal experiments were performed according to the ARRIVE guidelines and UK Animals (Scientific Procedures) Act, 1986, and associated guidelines.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gurumukhi, V.C., Bari, S.B. Quality by design (QbD)–based fabrication of atazanavir-loaded nanostructured lipid carriers for lymph targeting: bioavailability enhancement using chylomicron flow block model and toxicity studies. Drug Deliv. and Transl. Res. 12, 1230–1252 (2022). https://doi.org/10.1007/s13346-021-01014-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-021-01014-4