
Abstract
The objective of the present study was to develop a pH-sensitive drug delivery system by using polymethylmethacrylate-grafted gellan gum (PMMA-g-GG). PMMA-g-GG was synthesized by free radical polymerization reaction by using redox initiator ceric ammonium nitrate (CAN), and a series of graft copolymers were prepared with varying concentrations of methylmethacrylate (MMA) and CAN. Grafting parameters such as the percentage and efficiency of grafting were calculated, and the effect of monomer as well as initiator concentration was studied on the grafting yield. Optimization was done by one optimal response surface methodology. The batch with a better percentage grafting and grafting efficiency was selected and characterized by elemental analysis (CHN), FT-IR, DSC, PXRD, 1H-NMR, and SEM. Furthermore, acute oral toxicity study and histopathological analysis suggested non-toxic and biocompatible nature of the grafted gum. Metformin hydrochloride pellets were prepared using PMMA-g-GG, characterized in detail, and assessed for biocompatibility and efficacy. PMMA-g-GG-based formulation (M4) exhibited a pH-sensitive as well as sustained release of the drug over the period of 12 h and the release profile followed Peppas model. In vivo efficacy studies indicated a promising antidiabetic potential of the prepared formulation. Thus, PMMA-g-GG-based formulations can be implicated as novel drug delivery systems for facilitated antidiabetic therapy in the near future.

Graphical abstract
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biodegradability, easy availability, low cost, and non-toxicity are the factors which make natural polymers more preferable over the synthetic ones and are attracting great deal of interest in research in recent time [1]. However, they have certain disadvantages such as uncontrolled hydration rate, microbial load or contamination, and also the viscosity reduction throughout the storage, and their amalgamation with the synthetic monomers (or polymers) modifies their properties. Recently, polymers and their applications in controlled and/or sustained drug delivery systems have become sophisticated by overextending the period for effective drug release for any particular drug. As an instance, smart polymers not only determine where the drug is to be delivered but also provide the information regarding when and at which time interval it will be released [2,3,4,5]. The graft copolymer possesses macromolecular chain with the single or more species of the block as a side chain(s) attached to the main chain, and hence, it can be known to have the general structure, wherein polymer backbone (main chain) has branches of other polymeric chain (side chains) originating from diverse points along its length [6,7,8]. In simple words, modification by graft copolymerization, which is also referred to as grafting, is an irreversible process which involves covalent attachment of monomer to the polymer backbone [5, 6]. Grafting introduces specific characteristics such as hydrophobicity, enhanced stability, and the steric bulkiness that significantly protects the matrix as well as polymer backbone to hold up the release of drug [7,8,9].
Gellan gum (hereafter referred to as GG) is one of the natural polymers which has recently attracted a huge interest of researchers, and has been used in diverse drug delivery systems including oral, opthalmic, controlled, sustained, and floating drug delivery. GG is an anionic, deacetylated exopolysaccharide having a high molecular weight (1,000,000–2,000,000) and is generally produced by fermenting species Sphingomonas elodea (formerly Pseudomonas elodea, Gram negative, aerobic, and non-pathogenic bacterium). It is composed of the tetrasaccharide repeating unit of the one α-l,1,3-rhamnose and one β-1,4,d-glucuronic acid along with the two β-1,4,d-glucose units [10,11,12,13,14,15,16,17]. Despite being of bacterial origin, GG is commercially supplied, endotoxin free, and is extensively used for applications in drug delivery and therapeutics.
By definition, the graft copolymers possess the long sequence of polymer backbone along with the single or more branches (also called as grafts) of other monomer or polymer. The graft copolymer synthesis process begins with the preformed polymer, where the external agent may be used for creating free radical sites on the backbone of preformed polymer. For this process, the external agent to be used should be capable of creating the necessary free radical sites without disrupting the structural integrity of preformed polymer backbone. After formation of free radical sites, other monomer or polymer can be attached to the polymer backbone via chain propagation, thereby forming the grafted chains. Diverse methods for synthesizing the graft copolymers differ in the mechanism required for generating free radical sites on preformed polymer backbone. Ceric ammonium nitrate (CAN)-induced free radical polymerization reaction is an efficient, less time consuming, and reproducible synthesis method for developing graft copolymers, which can be used to fabricate pH-sensitive as well as sustained release polymers for desired drug delivery systems [5,6,7,8].
The pH-sensitive drug delivery systems propose an elevated therapeutic effectiveness along with patient compliance and are gaining importance with passing days as they are promising for various conditions like diabetes [18,19,20], fungal infections [21, 22], peptic ulcers [23, 24], asthma [25,26,27], hypertension [28, 29], cardiovascular diseases [30, 31], and cancer [32,33,34,35], to list a few. For the treatment involving an adequate dose, the variations not only in disease state but also in plasma drug concentration must be considered at the time of designing the drug delivery systems, as drug pharmacokinetics may also be pH sensitive [36].
Pellets are meant to be administered orally and can be defined as the agglomerates of bulk drugs along with the excipients, consisting small, solid, and free flowing spherical units, which characteristically vary from 0.5 to 1.5 mm. This multiparticulate dosage form consists of small independent subunits with active ingredient. Pellets being free flowing in nature, easy to pack, and resulting in reproducible as well as uniform fill weight of capsules are preferred over tablets. The technique primarily used for developing the pellets with spherical shape and homogenous surface is the extrusion-spheronization pelletization (ESP) which involves five unit operations starting from blending followed by wet massing, extrusion, spheronization, and drying [37, 38].
Metformin HCl is the most widely used biguanide derivative and is prescribed to nearly 120 million people globally for managing the type 2 diabetes as a first-line therapy. The drug plays three main different roles such as slowing down the sugar absorption into small intestine, stopping liver to convert stored sugar into the blood sugar, and finally helping the body to utilize the natural insulin in an extra efficient manner. As a result, it reduces the blood glucose levels and does not induce overt hypoglycemia. Being hydrophilic in nature, it gets absorbed slowly and partially from the gastrointestinal tract. Post oral administration of a single dose of 500 mg, it is primarily absorbed from the small intestine and has a relatively low bioavailability (absolute bioavailability is 50–60%). In addition, this drug has comparatively short plasma elimination half-life (2–4 h) and thus multiple doses are required for effective treatment which leads to non-compliance in patients. Metformin HCl formulation with sustained release drug delivery, maintaining the plasma levels of drug for at least 8–12 h, may be adequate for once a day dosing. Metformin HCl develops the hepatic as well as peripheral tissue sensitivity for the insulin. The major barrier for successful exploitation of metformin HCl therapy is higher frequency of gastrointestinal-associated symptoms like abdominal discomfort, increased flatulence, anorexia, diarrhea, cramps, nausea, vomiting, weight loss, and gustatory disturbances [39, 40]. Thus, metformin HCl formulations wherein release can be sustained are required to extend and enhance its duration of action as well as the patient compliance.
Therefore, for designing such formulations, the polymer modification or grafting could be insightful. Hence, in the present research vocation, we have attempted to develop a novel pH-sensitive polymethylmethacrylate-grafted gellan gum (PMMA-g-GG)-based formulation that would be useful to improve drug bioavailability, reduce the dosing frequency, enhance patient compliance, and decrease gastrointestinal side effects and toxicity. Additionally, the proposed system may be helpful for an effective combination therapy and management of type 2 diabetes in the near future.
Materials
Metformin HCl was received from Salius Pharma, Mumbai, India. Gellan gum was a kind gift by Sigma-Aldrich, USA. Methylmethacrylate (MMA) was bought from Rankem India Pvt. Ltd., Mumbai, India. Ceric ammonium nitrate (CAN) was obtained from the Merck Specialties Pvt. Ltd., Mumbai, India. Hypromellose, microcrystalline cellulose, and talc were procured from the Loba Chemie Pvt. Ltd., Mumbai, India. Methacrylic acid copolymer (Eudragit® NE30D) was gifted by Rohm GmbH Chemische Fabrik, Darmstadt, Germany. All chemicals were of reagent grade and were used as procured.
Methods
Synthesis of polymethylmethacrylate-grafted gellan gum
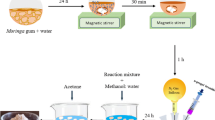
To synthesize PMMA-g-GG by free radical polymerization reaction, various ratios of gellan gum:methylmethacrylate were used. Two grams of GG was taken and dissolved in a beaker containing 100 ml of double distilled water and transferred into the flask. The flask was heated at 80 °C and required quantity of methylmethacrylate (MMA) and ceric ammonium nitrate (CAN) was added to the flask containing GG solution. Polymerization for about 1 h was carried out and resulting solution was cooled at an ambient temperature. The product was poured and kept in the excess amount of methanol for 24 h. The obtained copolymer was filtered, washed by methanol repetitively, and finally dried at 50 °C overnight in a hot air oven (UNB400, Memmert Pvt. Ltd., India). Alkaline hydrolysis of PMMA-g-gellan copolymer was carried out by dissolving the required amount in 100 ml solution of 0.9 M NaOH, followed by stirring for 60 min at 75 °C in a thermostatic water bath. At the termination of reaction, the solution was kept for cooling and then poured in an excess of methanol. Hydrolyzed copolymer was then separated via filtration and washed by methanol repeatedly, followed by drying in a hot air oven at 50 °C overnight [17, 38,39,40]. The percent grafting (% G) and percent grafting efficiency (% GE) for PMMA-g-GG were calculated using respective equations [22,23,24]. Table 1 represents the details of synthesis for the preparation of PMMA-g-GG.
W0, W1, and W2 respectively represent weight of native gum, grafted copolymer, and MMA.
Characterization of PMMA-g-GG
Elemental analysis
Elemental analysis for GG, MMA, and PMMA-g-GG was done using elemental analyzer (Thermo Finnigan, FLASH EA 1112 Series, Italy) for determining carbon, hydrogen, and nitrogen content [41, 42].
Fourier transform infrared spectroscopy
Fourier transform infrared spectroscopy (FT-IR) analysis of GG and PMMA-g-GG was carried out using FT-IR spectrophotometer (8400S, Shimadzu, Japan) for analyzing probable alterations in functional groups of native gum with respect to its grafted product. The material to be analyzed was mixed with KBr in a small amount to compress into a thin film pellet and was scanned in the range 500–4000 cm−1.
Differential scanning calorimetry
Thermal analysis of GG and PMMA-g-GG was done using differential scanning calorimeter (DSC-60, Shimadzu, Japan) under the N2 at a flow rate of 50 mL/min, at 10 °C/min heating rate.
Powder X-ray diffraction
Powder X-ray diffraction (PXRD) of GG and PMMA-g-GG was carried out via bench top X-ray diffractometer (Rigaku, Bruker AXS D8 Advance; Germany).
Proton nuclear magnetic resonance analysis
Proton nuclear magnetic resonance (1H-NMR) analysis of GG and PMMA-g-GG was performed to confirm the grafting by using NMR spectrophotometer (Agilent 400MHz FT-NMR spectrophotometer, CA, USA). The spectra recorded were in the chemical shift (δ) range 0–7 ppm. Solvents used were deuterium oxide (D2O) for native gum and dimethyl sulfoxide (DMSO) for PMMA-g-GG [43,44,45].
Scanning electron microscopy
Morphology of GG and PMMA-g-GG was examined via scanning electron microscopy (SEM) system (Smart SEM 5.05, Zeiss EVO LS 15; Germany). For this study, samples were coated by gold for enhancing electron beams conductivity. An accelerating voltage of 15 kV with a working distance of 9 mm was implied for the analysis [46].
Effect of monomer (MMA) concentration on grafting
Effect of MMA concentration on grafting (% G and % GE) was evaluated by varying the MMA concentration ratios from 1:2 to 1:14 at different CAN concentrations.
Effect of initiator (CAN) concentration on grafting
Effect of redox initiator (CAN) concentration on grafting (% G and % GE) was studied by varying the CAN concentration in range of 100–400 mg [47, 48].
Acute oral toxicity
This study was carried out for synthesized PMMA-g-GG according to the Guidelines of Organization of Economic Co-operation and Development (OECD). OECD 425 Guidelines adopted on December 17, 2001, for testing the chemicals were followed for this study. The protocol was approved in advance by Institutional Animal Ethics Committee (IAEC) of JSS College of Pharmacy, Mysuru, India (CPCSEA Approval No. P17247/2017).
Six nulliparous, non-pregnant female mice (Swiss albino species) of 8 weeks age were selected for the toxicity study. Mice were kept for housing with provision of adequate food and mineral free water at a temperature of 19–25 °C with a relative humidity of 40–70% and a 12-h light on/off cycle. One animal was selected randomly and kept as control with no dose administration or treatment. Single 2000 mg/kg body weight dose of PMMA-g-GG was administered to first test animal from the set via gavage using stomach tube. The same dose of PMMA-g-GG was administered to the four test animals remaining in the set, after survival of first test animal. Post dosing, all the test animals were observed occasionally for almost 4 h, followed by inspection at predetermined intervals that continued up to 14 days. Any sign, symptoms of toxicity and mortality rate was reported visually [17, 39, 40].
Histopathological analysis
One random control animal which survived the toxicity test was taken, euthanized by diethyl ether, and subsequently sacrificed. Brain, kidney, lung, liver, and heart were then isolated and cleaned with 0.9% (w/v) NaCl, followed by fixation in 10% formalin. Each isolated organ was dehydrated serially with 50% and 80% followed by absolute alcohol, and then blocked in paraffin. Thin sections of isolated organs were obtained by cutting the fixed block with microtome and mounted on the glass slides for staining with hematoxylin-eosin. At last, examination of permanent slides was done via light microscope (Motic, B1 Series, India) with a camera (Magnus, MITS, India) at × 40 magnification. Photomicrographs of test organs were compared with control organs [49, 50].
Preparation of metformin HCl pellets
For the preparation of pellets, various concentrations of optimized PMMA-g-GG, microcrystalline cellulose (MCC) as pelletization aid, hypromellose as binder, talc as glidant-cum-lubricant, and drug (metformin HCl) were taken and homogeneously mixed together for 10 min. A total of 6 batches were prepared (M1-M6), and the total bulk of each batch was 1150 mg, taken as 100%. All the six batches included 500 mg drug, 550–500 mg MCC (with a difference of 10 mg per batch), 40 mg hypromellose, 50 mg talc, and 10–60 mg PMMA-grafted gum (with a difference of 10 mg per batch). The required volume of mixture of water:isopropyl alcohol was added in a dry blend of each batch for making wet mass having appropriate consistency. Later, the wet mass was passed through EXT 65/037 rotating roller extruder (R.R. Enterprises, Thane, India) followed by SPH 150/010 spheronizer (R.R. Enterprises, Thane, India) at 1600 rpm in order to get the pellets. Finally, the obtained pellets were collected and dried in hot air oven at 40 °C (UNB400, Memmert Pvt. Ltd., India) for 10 h. The obtained drug loaded pellets were then film coated with a polymer blend (chitosan: Eudragit® NE30D in 4:6 ratio, resulting in 10% coat loading) to overcome any possibility of pellets aggregation, and surface erosion and burst drug release in stomach. After coating, the pellets were dried for 1 h in coating chamber to evaporate the residual solvent in polymeric coating and finally collected and stored [38].
On the basis of the outcomes of various characterization parameters such as angle of repose, tapped density, Carr’s index, particle size, and friability, the batch M4 was found to be superior, and hence, considered as an optimized and selected for the further evaluation parameters. The composition of optimized batch M4 was 500 mg drug, 520 mg MCC, 40 mg hypromellose, 50 mg talc, and 40 mg PMMA-g-GG.
Evaluation of pellets
In vitro drug release and release kinetics
For obtaining in vitro dissolution profile, pellets sample equivalent to 500 mg of metformin HCl was filled in the hard gelatin capsule (size 00) and subjected to dissolution. The USP- Method A was adopted and dissolution study was initially performed at the gastric pH using 0.1 N HCl pH 1.2 for 2 h, followed by dissolution at intestinal pH (in PBS pH 6.8) for 10 h. The dissolution apparatus used for the study was USP Type-II apparatus (Electrolab, Mumbai, India) assembled with the paddles. Freshly prepared 0.1 N HCl and PBS (as per the specifications of the USP Edition 36) were taken as dissolution media and dissolution test was carried out at 37 ± 1 °C temperature with 50 rpm paddle rotation. For the acid stage dissolution, 750 ml of 0.1 N HCl was placed in the vessels and allowed to equilibrate to 37 ± 1 °C. Later, one dosage unit (capsule) was placed in each vessel, covered with polyacrylic lids, and operated. On predetermined specific time intervals, 10-ml samples were withdrawn from the dissolution jar (from a zone midway of the media surface and top of rotating paddle blade by keeping > 1 cm distance from vessel wall) and replaced immediately with an equal amount of fresh media for retaining sink condition. For the buffer stage, 250 ml of 0.20 M tribasic sodium phosphate equilibrated to 37 ± 1 °C was added to each vessel (already containing 750 ml of acidic media) and pH of the dissolution media was checked. If needed, pH was adjusted to 6.8 ± 0.05 using 2 N hydrochloric acid or 2 N sodium hydroxide solutions. All these buffer additions and pH adjustments were done within 5 min and apparatus operation was continued for further 10 h with in line sampling. Finally, all the aliquots were filtered through 0.45 μ membrane, and amount of drug released was quantified via UV spectrophotometry at 232 nm. The cumulative drug release was calculated using the standard formula and drug release profile was plotted. Furthermore, drug release data was fitted in diverse kinetic models to ascertain the release kinetics. The release profile of prepared pellet formulation (optimized batch) was compared with the release profile of marketed formulation Glycomet SR 500 mg tablet as a product performance assessment tool [51,52,53].
In vivo antidiabetic activity
For in vivo antidiabetic activity, the prepared study protocol approved earlier by Institutional Animal Ethics Committee (IAEC), JSS College of Pharmacy, Mysuru, India (CPCSEA Approval No. P17247/2017) was followed. Wistar albino rats (male) weighing in range 250–300 g were selected for the study and maintained in the polypropylene cages at the controlled room temperature. Rats were fed with normal laboratory food and water, but the food (except water) was taken out 24 h prior to experiment. For inducing diabetes, animal subjects, i.e., rats, were initially injected with a single i.p. dose of 110 mg/kg body weight nicotinamide in physiological saline and post 15 min streptozotocin (STZ) at 60 mg/kg i.p. dose was injected. For the dosing purpose, nicotinamide and STZ were dissolved in physiological saline and 0.5 ml of 0.1 M citrate buffer of pH 4.5, respectively. After 2–4 days of nicotinamide-STZ treatment, the extent of diabetic induction was checked on the basis of blood glucose levels. The blood glucose level higher than 250 mg/dl was taken as basal level of diabetes [50,51,52]. The complete study procedure was carried out by dividing the rats into four groups, each containing six rats, and the treatment was as follows:
Group I: Normal control (non-diabetic): rats administered p.o. with 0.5% sodium CMC.
Group II: Diabetic control: rats wherein hyperglycemia was induced by nicotinamide-STZ method and vehicle was administered i.p.
Group III: Diabetic standard: hyperglycemia was induced by nicotinamide-STZ method, and marketed formulation (Glycomet SR 500 mg tablet) equivalent to 500 mg/kg body weight in sodium CMC solution was administered p.o.
Group IV: Diabetic + formulation: hyperglycemia was induced via nicotinamide-STZ method, and metformin pellets (optimized batch M4) prepared by using PMMA-g-GG equivalent to 500 mg/kg body weight in sodium CMC solution were administered p.o.
0.5% w/v sodium CMC solution was used as a suspending agent to prepare the suspension of pellets for oral administration.
After this treatment, under mild anesthesia, 0.2 ml samples of blood were withdrawn from the retro-orbital plexus of each rat, and blood glucose levels were checked on 0, 2, 4, 6, 8, 10, and 12 h by using glucometer (Accu-Check Active Glucometer, Roche Diabetes Care India Pvt. Ltd., Mumbai, India). After 14 days, blood samples were collected in micro-centrifuge tube presaturated with heparin, and subjected to centrifugation for 20 min at 3000 rpm. The obtained serum was separated and used immediately for the estimation of biochemical parameters such as total cholesterol, total protein, and triglyceride levels by using diagnostic kits (Merck Pvt. Ltd., Mumbai, India).
Results and discussion
Synthesis of polymethylmethacrylate-grafted gellan gum
Synthesis of PMMA-g-GG was done via free radical polymerization reaction. CAN is generally used to initiate the free radical graft copolymerization. Earlier, grafting of diverse monomers excluding MMA onto gellan gum was done via ceric (IV)-induced free radical graft copolymerization [23, 24, 26]. The general mechanism of reaction involves attack of ceric ion of redox initiator CAN on gellan gum macrochains, thereby forming gellan gum-ceric complex. Later within the complex, these ceric (IV) ions are reduced to ceric (III) ions via oxidizing hydrogen atom, which leads to the generation of free radicals onto gellan gum backbone. Hence, the redox initiator like CAN is required in a threshold amount in order to form the free radical on polymer backbone.
The grafting of monomer, i.e., MMA, onto gellan gum backbone was initiated by reaction between free radicals formed and MMA. In the presence of MMA, gellan gum free radical gets coupled covalently to monomer unit, resulting into the chain propagation subsequently producing polymethylmethacrylate (PMMA) graft chains [17, 39,40,41]. Figure 1 represents the schematic representation of the reaction for synthesis of PMMA-g-GG. Table 2 covers synthesis outcomes of ceric induced graft copolymerization, % G as well as % GE of grafted gellan gum, and elemental analysis.

Schematic hypothetical presentation for the synthesis of PMMA-g-GG
Characterization of PMMA-g-GG
Elemental analysis
On the basis of % G and % GE, batch G11 was selected for elemental analysis to confirm the grafting reaction. The outcomes of an elemental analysis are quoted in Table 2. In case of batch G11, a relatively higher percentage of carbon and hydrogen was observed as compared to the native GG. This was possible only if MMA chains were successfully grafted onto the gellan gum backbone to form PMMA-g-GG, thereby confirming the successful grafting reaction. Hence, the selected batch was subjected to further characterization.
Fourier transform infrared spectroscopy
Overlay FT-IR spectra of pure metformin HCl, GG, PMMA-g-GG, physical mixtures, and prepared formulation, i.e., metformin HCl pellets prepared by using PMMA-g-GG, are shown in Fig. 2. In the course of FT-IR studies, it has been observed that all the characteristic peaks were present in FT-IR spectra of metformin HCl (Fig. 2a) and GG (Fig. 2b), which confirmed the purity of procured drug and polymer. In case of pure metformin HCl, peaks were observed at 3371 cm−1 (N-H asymmetric stretching), 3155 to 3309 cm−1 (N-H symmetric stretching), 2938 cm−1 (CH3 asymmetric stretching), 2816 cm−1 (CH3 symmetric stretching), 1581 cm−1 (C=N stretching), and 1473 cm−1 (CH3 asymmetric deformation). In case of pure GG, peaks were observed at 1604 cm−1 (carbonyl group C-O stretching), 3425 cm−1 (O-H stretching), 2924–3300 cm−1 (C-H stretching), 1411 cm−1 (methyl C-H bonding), and 810 cm−1 (C-O stretching for alkyl ether). In case of PMMA-g-GG (Fig. 2d), peaks were observed at 1612 cm−1 (C=O stretching), 1023 cm−1 (C–O stretching), and 1005 cm−1 (O–H bending). Additional peaks due to grafted PMMA chains were traced which confirmed the grafting. Moreover, in the FT-IR spectrum of physical mixtures (Fig. 2c and e) and prepared pellet formulation (Fig. 2f), characteristic peaks of drug and polymer (PMMA-g-GG) were retained and there were no appearance of new peaks and/or disappearance of the existing peaks thereby indicating no interaction in between drug and polymer. All characteristic peaks of metformin HCl were observed in the FT-IR spectrum of physical mixture during the investigation of compatibility study. Hence, FT-IR spectroscopy results showed that the drug was compatible with native GG as well as grafted GG.

Overlain FT-IR spectra of (a) metformin HCl, (b) gellan gum, (c) physical mixture 1 (metformin HCl and gellan gum), (d) PMMA-g-GG, (e) physical mixture 2 (metformin HCl and PMMA-g-GG), and (f) formulation
Differential scanning calorimetry
Differential scanning calorimetry (DSC) thermograms of pure metformin HCl, GG, PMMA-g-GG, physical mixtures, and prepared formulation were recorded as shown in Fig. 3. DSC thermogram of pure metformin HCl (Fig. 3a) showed a sharp melting endotherm at temperature 230.86 °C. However, GG and PMMA-g-GG depicted exothermic peaks at 252.44 °C and 254.51 °C, respectively (Fig. 3b and d). The melting endotherm of drug was also observed in thermograms of physical mixtures at 231.78 °C and 228.62 °C (Fig. 3c and e) thereby indicating the absence of any interaction between drug and polymers. The melting endotherm of physical mixture was not that sharp as of pure metformin HCl, which can be attributed to the presence of GG. The endothermic drug peak and exothermic PMMA-g-GG peak was traced in thermogram of prepared pellet formulation (Fig. 3f).

Overlain DSC thermograms of (a) metformin HCl, (b) gellan gum, (c) physical mixture 1 (metformin HCl and gellan gum), (d) PMMA-g-GG, (e) physical mixture 2 (metformin HCl and PMMA-g-GG), and (f) formulation
Powder X-ray diffraction
Figure 4 depicts X-ray diffractogram of GG (Fig. 4b) and PMMA-g-GG (Fig. 4c). The diffractogram of GG has exhibited the broad band located at the low diffraction angle (2θ) values, indicating the amorphous structure of the sample. In PMMA-g-GG sample, an overall increase in the peak intensity suggested the convergence of GG towards the more ordered arrangement after grafting with PMMA. Hence, the grafting of GG with PMMA resulted in transformation to crystalline nature as expected and evident from earlier studies [39, 54]. Moreover, X-ray diffractograms of pure metformin HCl, physical mixture, and prepared pellet formulation were also represented. As per the diffractograms, drug was crystalline in nature both in its pure form (Fig. 4a), in physical mixture (Fig. 4d), and also in prepared pellet formulation (Fig. 4e), as evident from several sharp peaks instead of humps and less noise. Furthermore, intensities at the different 2θ values for the pure drug were found to be retained in the prepared pellet formulation, thereby indicating no incompatibility in between the drug and formulation components.

XRD diffractogram of (a) metformin HCl, (b) gellan gum, (c) PMMA-g-GG, (d) physical mixture of metformin HCl and PMMA-g-GG, and (e) formulation
Proton nuclear magnetic resonance analysis
In case of GG (Fig. 5a), the protons of tetrasaccharide repeating units formed by glucose, glucuronic acid, and rhamnose units have appeared in the 3.16–3.91 δ region. –CH of rhamnose appeared at 5.27 δ, –CH of glucuronic acid appeared at 4.99 δ, –CH of glucose appeared at 4.39 δ, and –CH3 of rhamnose appeared at 1.16 δ. In case of 1H-NMR spectrum of PMMA-g-GG (Fig. 5b), the distinctive peaks of –CH3, –CH2, and –OCH3 of MMA appeared at 1.19 δ, 2.46 δ, and 3.37 δ, respectively. The signals traced in both the pure native GG and PMMA-g-GG spectra were verified against formerly published data and were found to be consistent with the literature [17, 38, 39]. Thus, 1H-NMR spectroscopy outcomes were coherent with FT-IR spectroscopy findings, thereby testifying the grafting of MMA on GG [23,24,25,26,27,28,29,30].

1H-NMR spectra of (a) gellan gum and (b) PMMA-g-GG
Scanning electron microscopy
Grafting introduces morphological changes with respect to the size and surface of polysaccharide particles. SEM analysis of GG (Fig. 6a) and its grafted product PMMA-g-GG (Fig. 6b) indicated the reflective morphological changes as a result of the transition from granular to lobular aggregate structure with higher heterogeneity owing to the grafting of PMMA chains on the GG backbone. In a nutshell, as the result of grafting, granular morphology of GG was lost and changed to lobular morphology [39,40,41,42,43].

SEM images of (a) gellan gum and (b) PMMA-g-GG
Effect of monomer (MMA) concentration on grafting
Effect of MMA concentration on grafting is reported in Table 2. In case of % G, it was found that % G increased with an increase in the MMA concentration up to a ratio of 1:10, ahead of which it leveled off. This outcome can be attributed to the fact that, with an increase in the monomer concentration, the complexation between monomer and ceric (IV) of CAN is also increased. Moreover, a large number of grafting polymeric chains formed at higher concentration of monomer are involved in the generation of additional active sites onto grafted GG via chain transfer reaction. Conversely, decreased % GE with an increased MMA concentration can be attributed to the possibility that the grafted chains may have acted as diffusion barriers hindering the diffusion of monomer to the polymer backbone. This consequently leads to the poor availability of monomer for the grafting and results in the homopolymer (PMMA) formation [42, 43, 49].
Effect of initiator (CAN) concentration on grafting
By varying concentration of redox initiator, i.e., CAN from 100 to 400 mg, effect of CAN concentration on grafting was studied, and the results are presented in Table 2. As per the outcomes, % G and % GE were increased with increasing concentration in ceric ion up to 300 mg and reached the maximum values of 89.7% and 8.97%, respectively. Above 300 mg of redox initiator concentration, the values of % G and % GE were decreased. The preliminary increase in the grafting with respect to the CAN concentration was due to the availability of adequate number of ceric (IV) ions in reaction mixture that had been consumed ultimately for the generation of active sites onto GG backbone, thereby facilitating graft copolymerization of the monomer (MMA) on GG backbone. The decrease observed in grafting at higher concentration of CAN (i.e., above 300 mg) can be attributed to the excessive radical concentration present in polymerization medium, which may have enhanced the termination rate of the growing grafted chains leading to the lower grafting. Moreover, the formation of homopolymer (PMMA) at higher concentration of redox initiator, which may compete with the reaction of grafting for available monomer (MMA), could also result in the decrease in % G and % GE [43, 49].
Acute oral toxicity
Post dosing no mortality was observed up to a period of 14 days. As per the Guidelines 425 of OECD, adopted on December 17, 2001, for test of chemicals, the LD50 value was found to be higher than 2000 mg/kg dose of PMMA-g-GG. Moreover, according to Globally Harmonized System (GHS), if the value is higher than 2000 mg/kg dose, the test product falls under category 5 indicating zero toxicity rating. Hence, our synthesized PMMA-g-GG comes under category 5 with zero toxicity rating [17, 39, 40].
Histopathological analysis
Figure 7 represents the micrographs (× 40) of different organs of control and test animals. The micrographs of brain tissue from both the control and test animals have depicted the similar granular cells morphology. Additionally, both the micrographs (control as well as test) of lung have shown alveoli, inter-alveolar septa, and the connective tissue sheets with capillary bed with no sign of morphological change. In case of heart morphology, the test micrograph has shown the similar patterns as that of the control micrograph by providing an overview of cardiac myocytes with nucleus located at center, intercalated disks connecting further cardiac myocytes and the Purkinje fibers. Furthermore, both the micrographs of kidney have shown similar morphology for the cuboidal epithelial cells of loop of Henle as well as the collecting tubules. In case of liver, both the micrographs have shown normal hepatocytes, large polygonal cells, and Kupffer cells. In conclusion, the histopathological analysis has established the physiological compatibility of synthesized PMMA-g-GG.

Micrographs of (a) control brain, (b) test brain, (c) control lung, (d) test lung, (e) control heart, (f) test heart, (g) control kidney, (h) test kidney, (i) control liver, (j) test liver
Preparation of metformin HCl pellets
Extrusion-spheronization technique has been carried out in this study to formulate the metformin HCl-loaded pellets by using PMMA-g-GG. The preliminary extrudates broke into small particles of cylindrical shape as a result of friction from spheronizer plate. The process further went through few shape changing steps of broken extrudates, such as cylindrical with round ends followed by dumbbells, ellipsoidal, and finally spheroidal particles [38].
Evaluation of pellets
In vitro drug release and release kinetics
The drug dissolution profile of optimized pellet formulation (batch M4) prepared by using PMMA-g-GG is shown in Fig. 8. It has been observed that increasing concentration of polymer caused significant decrease in drug release. Sustained drug release from PMMA-g-GG-based pellets was observed, owing to hydrophobic barrier limiting the access of water and thereby the drug dissolution. Moreover, as reported in several former studies, drug release from dosage form containing cellulosic and/or hydrophobic matrix follows three steps: starting with penetration of dissolution medium into dosage form, i.e., hydration, followed by matrix erosion, and finally transportation of dissolved drug to surrounding dissolution medium via eroded area of dosage form or from the hydrated matrix [46].

In vitro dissolution profiles of metformin HCl pellets (M4) prepared by using PMMA-g-GG and marketed formulation (M) in pH 1.2 and pH 6.8 buffer media
Study outcomes have shown that optimized pellet formulation (batch M4) did not release significant amount of drug in acidic pH (1.2), while it has released the drug at a higher pH (6.8). This could be attributed to the possibilities that lower pH keeps the carboxyl groups of PMMA-g-GG-based metformin HCl pellets almost in unionized state, and hydrogen bonding can be formed between −COOH groups of PMMA-g-GG in acidic environment, thereby making the polymer segments rigid, which consequently hinders the water uptake and lowers the swelling extent. At higher pH, carboxyl groups of PMMA-g-GG-based metformin HCl pellets get ionized, thereby dissociating hydrogen bonding between −OH and −COOH groups. This fosters matrix erosion and loaded drug dissolution. Additionally, repulsion amongst the similarly charged COO− group increases the chain relaxation that further endorses the pellets to swell dramatically [49, 52, 53].
The drug release from batch M4 was enormously significant, and cumulative amount of drug released was 96.27 ± 2.86% at end of 12 h, whereas the drug release from marketed formulation Glycomet SR 500 mg tablet was 97.52 ± 2.61% at the end of 12 h. However, it is worth mentioning that marketed formulation (M) has shown drug release in acidic pH 1.2 (11.23 ± 1.93%), with respect to which, drug release from optimized M4 batch was noted to be quite negligible (2.09 ± 1.57%). These outcomes can be attributed to pH-dependent structural changes in the PMMA-g-GG as aforementioned, and it testified our intent of developing a pH-sensitive delivery system with advantage over marketed formulation. The drug release data was subjected to the diverse release kinetic models, which indicated that both the formulations (optimized M4 as well as marketed formulation M) followed the Peppas model with r2 value of 0.982 and 0.974 for M4 and M formulation, respectively.
In vivo antidiabetic activity
Inducing experimental diabetes in male adult rats was indeed the first step to decrease nicotinamide-adenine dinucleotide in pancreas islet beta cells, thereby causing histopathological effects in beta cells probably intermediating diabetes induction [50, 51]. Nicotinamide at 110 mg/kg dose and streptozotocin (STZ) at 60 mg/kg dose were injected intraperitoneally for inducing diabetes in rats. STZ was used as it is claimed and reported to possess advantages of greater specificity and lower toxicity over alloxan [50].
After 2–4 days of nicotinamide-STZ treatment, the extent of diabetic induction was checked on the basis of blood glucose levels. The blood glucose level higher than 250 mg/dl was taken as basal level of diabetes. Figure 9 shows the antidiabetic activity of marketed formulation Glycomet SR 500 mg tablet (diabetic standard) and optimized metformin HCl pellet formulation (batch M4) prepared by using PMMA-g-GG. The blood glucose levels in diabetic control group were significantly elevated as compared to the normal group. The normal group has shown the blood glucose levels in the range of 68.48 ± 2.84 to 72.52 ± 3.2 mg/dl. The diabetic control group has shown the blood glucose levels in the range of 249.87 ± 3.82 to 263.74 ± 2.5 mg/dl. The diabetic standard group, i.e., marketed formulation, has shown a significant decline in blood glucose levels within 2–6 h owing to the higher drug release as compared to the optimized formulation (M4) which depicted comparatively less hypoglycemic effect. As a conclusion, optimized formulation M4 was found to possess notable antidiabetic efficacy.

Blood glucose levels in nicotinamide-STZ–induced diabetic rats treated with marketed formulation (M) and optimized formulation (M4)
Furthermore, diabetes leads to the various metabolic deviations in animal such as increased blood glucose and cholesterol level and decreased protein content. There was noteworthy decrease in plasma protein content of diabetic control group as compared to the normal group. The optimized pellet formulation (M4) and marketed formulation (M) were significantly able to correct the metabolic disturbances. As mentioned in Table 3, the values of biochemical parameters obtained for optimized as well as marketed formulation were found close to the values obtained for the normal group and also were well within the normal range.
Conclusions
Polymethylmethacrylate-grafted gellan gum (PMMA-g-GG) was synthesized by using ceric ammonium nitrate (CAN)-induced free radical polymerization reaction for the grafting of methylmethacrylate (MMA) monomer on gellan gum (GG) backbone. Higher levels of CAN and MMA have shown higher grafting up to a certain concentration. The grafting of MMA onto GG backbone was evidenced by elemental analysis, FT-IR, DSC, PXRD, 1H-NMR, and SEM analysis. Moreover, toxicity study of PMMA-g-GG at 2000 mg/kg body weight dose was done by following OECD 425 Guidelines, and there was no mortality observed during the study period. Additionally, histopathological examination in Swiss albino mice has shown no significant differences when test photomicrographs of different organs were compared with control photomicrographs, thereby indicating biocompatibility of synthesized PMMA-g-GG. Pellets of metformin HCl were formulated (M4) by using PMMA-g-GG as a pH-sensitive as well as sustained release polymer along with MCC as pelletization aid. The prepared metformin HCl pellet formulation exhibited sustained release over the period of 12 h and the release profile followed Peppas model. Furthermore, metformin HCl pellets (M4) have shown good antidiabetic efficacy. To conclude, ceric ammonium nitrate (CAN)-induced free radical polymerization reaction is an efficient, less time consuming, and reproducible synthesis method for developing graft copolymers, which can be used to fabricate pH-sensitive as well as sustained release polymers for desired drug delivery systems.
References
Bhardwaj TR, Kanwar M, Lal R, Gupta A. Natural gums and modified natural gums as sustained-release carriers. Drug Dev Ind Pharm. 2000;26:1025–38.
Soppimath KS, Aminabhavi TM, Dave AM, Kumbar SG, Rudzinski WE. Stimulus responsive smart hydrogels as novel drug delivery systems. Drug Dev Ind Pharm. 2002;28:957–74.
Zohuriaan-Mehr MJ. Advances in chitin and chitosan modification through graft copolymerization: a comprehensive review. Iran Polym J. 2005;14:235–65.
Singh V, Sethi R, Tewari A, Srivastava V, Sanghi R. Hydrolysis of plant seed gums by microwave irradiation. Carbohydr Polym. 2003;54:523–5.
Bhosale RR, Osmani RAM, Moin A. Natural gums and mucilages: a review on multifaceted excipients in pharmaceutical science and research. IJPPR. 2014;6:901–12.
Bhosale RR, Osmani RAM, Moin A. Review on natural polysaccharide based particulate drug delivery systems: an inimitable tactic in novel drug delivery systems. IJCPRR. 2015;5:138–55.
Bhosale RR, Gangadharappa HV, Moin A, Gowda DV, Osmani RAM. A review on grafting modification of polysaccharides by microwave irradiation- distinctive practice for application in drug delivery. IJCPRR. 2015;6:8–17.
Bhosale RR, Gangadharappa HV, Moin A, Gowda DV, Osmani RAM. Grafting technique with special emphasis on natural gums: applications and perspectives in drug delivery. NPJ. 2015;5:124–39.
Matricardi P, Cencetti C, Ria R, Alhaique F, Coviello T. Preparation and characterization of novel gellan gum hydrogels suitable for modified drug release. Molecules. 2009;14:3376–91.
Agnihotri SA, Jawalkar SS, Aminabhavi TM. Controlled release of cephalexin through gellan gum beads: effect of formulation parameters on entrapment efficiency, size and drug release. Eur J Pharm Biopharm. 2006;63:249–61.
Kedzierewicz F, Lombry C, Rios R, Hoffman M, Maincent P. Effect of the formulation on the in vitro release of propranolol from gellan beads. Int J Pharm. 1999;178:129–36.
Miyazaki S, Aoyama H, Kawasaki N, Kubo W, Attwood D. In situ-gelling gellan formulations as vehicles for oral drug delivery. J Control Release. 1999;60:287–95.
Deogade UM, Deshmukh VN, Sakarkar DM. Natural gums and mucilage in NDDS: application and recent approaches. Int J Pharmtech Res. 2012;4:799–814.
Kumar R, Setia A, Mahadevan N. Grafting modification of the polysaccharide by the use of microwave irradiation- a review. Int J Rec Adv Pharm Res. 2012;2:45–53.
Mishra S, Mukul A, Sen G, Jha U. Microwave assisted synthesis of polyacrylamide grafted starch (St-g-PAM) and its applicability as flocculant for water treatment. Int J Biol Macromol. 2011;48:106–11.
Mishra S, Rani U, Sen G. Microwave initiated synthesis and application of polyacrylic acid grafted carboxymethyl cellulose. Carbohydr Polym. 2012;87:2255–62.
Rani P, Sen G, Mishra S, Jha U. Microwave assisted synthesis of polyacrylamide grafted gum ghatti and its application as flocculant. Carbohydr Polym. 2012;89:275–81.
Sung HW, Sonaje K, Liao ZX, Hsu LW, Chuang EY. pH-responsive nanoparticles shelled with chitosan for oral delivery of insulin: from mechanism to therapeutic applications. Acc Chem Res. 2012;45(4):619–29.
Souto EB, Souto SB, Campos JR, Severino P, Pashirova TN, Zakharova LY, et al. Nanoparticle delivery systems in the treatment of diabetes complications. Molecules. 2019;24(23):4209.
Xie J, Li A, Li J. Advances in pH-sensitive polymers for smart insulin delivery. Macromol Rapid Commun. 2017;38(23):1700413.
Lombardo D, Kiselev MA, Caccamo MT. Smart nanoparticles for drug delivery application: development of versatile nanocarrier platforms in biotechnology and nanomedicine. J Nanomater. 2019;2019:1–26.
Cao Z, Li W, Liu R, Li X, Li H, Liu L, et al. pH-and enzyme-triggered drug release as an important process in the design of anti-tumor drug delivery systems. Biomed Pharmacother. 2019;118:109340.
El-Mahrouk GM, Aboul-Einien MH, Makhlouf AI. Design, optimization, and evaluation of a novel metronidazole-loaded gastro-retentive ph-sensitive hydrogel. AAPS PharmSciTech. 2016;17(6):1285–97.
Wells CM, Harris M, Choi L, Murali VP, Guerra FD, Jennings JA. Stimuli-responsive drug release from smart polymers. J Funct Biomater. 2019;10(3):34.
Lavakumar V, Sowmya C, Venkateshan N, Ravichandiran V, Leela KV, Harikrishanan N, et al. Microcapsule-based chronomodulated drug delivery systems of montelukast sodium in the treatment of nocturnal asthma. Int J Pharm Investig. 2018;8(1):24–32.
Singh B. Design of microparticulated salbutamol sulfate pH sensitive pulsatile delivery system for chronotherapy of nocturnal asthma. Asian J Pharm. 2019;12(04):S1215–22.
Liu G, Zhao X, Zhang Y, Xu J, Xu J, Li Y, et al. Engineering biomimetic platesomes for ph-responsive drug delivery and enhanced antitumor activity. Adv Mater. 2019;31(32):1900795.
Alam T, Khan S, Gaba B, Haider MF, Baboota S, Ali J. Nanocarriers as treatment modalities for hypertension. Drug Deliv. 2017;24(1):358–69.
Fancher IS, Rubinstein I, Levitan I. Potential strategies to reduce blood pressure in treatment-resistant hypertension using food and drug administration–approved nanodrug delivery platforms. Hypertension. 2019;73(2):250–7.
Men W, Zhu P, Dong S, Liu W, Zhou K, Bai Y, et al. Layer-by-layer pH-sensitive nanoparticles for drug delivery and controlled release with improved therapeutic efficacy in vivo. Drug Deliv. 2020;27(1):180–90.
Zhang R, Liu R, Liu C, Pan L, Qi Y, Cheng J, et al. A pH/ROS dual-responsive and targeting nanotherapy for vascular inflammatory diseases. Biomaterials. 2020;230:119605.
Li Y, Wang S, Song FX, Zhang L, Yang W, Wang HX, et al. A pH-sensitive drug delivery system based on folic acid-targeted HBP-modified mesoporous silica nanoparticles for cancer therapy. Colloids Surf A Physicochem Eng Asp. 2020;25:124470.
Niu S, Williams GR, Wu J, Wu J, Zhang X, Chen X, et al. A chitosan-based cascade-responsive drug delivery system for triple-negative breast cancer therapy. J Nanobiotechnology. 2019;17(1):1–8.
Lv Y, Hao L, Hu W, Ran Y, Bai Y, Zhang L. Novel multifunctional pH-sensitive nanoparticles loaded into microbubbles as drug delivery vehicles for enhanced tumor targeting. Sci Rep. 2016;6:29321.
Cai X, Luo Y, Zhang W, Du D, Lin Y. pH-sensitive ZnO quantum dots–doxorubicin nanoparticles for lung cancer targeted drug delivery. ACS Appl Mater Interfaces. 2016;8(34):22442–50.
Balamuralidhara V, Pramodkumar TM, Srujana N, Venkatesh MP, Gupta V, Krishna KL, et al. pH sensitive drug delivery systems: a review. Am J Drug Discov Dev. 2011;1:24–48.
Gajdos B. Rotary granulators- evaluation of process technology for pellet production using factorial design. Drugs Made Ger. 1984;27:30–6.
Gowrav MP, Hani U, Shivakumar HG, Osmani RAM, Srivastava A. Polyacrylamide grafted guar gum based glimepiride loaded pH sensitive pellets for colon specific drug delivery: fabrication and characterization. RSC Adv. 2015;5:80005–13.
Vijan V, Kaity S, Biswas S, Isaac J, Ghosh A. Microwave assisted synthesis and characterization of acrylamide grafted gellan, application in drug delivery. Carbohydr Polym. 2012;90:496–506.
Kaity S, Isaac J, Mahesh Kumar P, Bose A, Wong TW, Ghosh A. Microwave assisted synthesis of acrylamide grafted locust bean gum and its application in drug delivery. Carbohydr Polym. 2013;98:1083–94.
Mishra S, Sen G. Microwave initiated synthesis of polymethylmethacrylate grafted guar (GG-g-PMMA), characterizations and applications. Int J Biol Macromol. 2011;48:688–94.
Giri TK, Verma P, Tripathi DK. Grafting of vinyl monomer onto gellan gum using microwave: synthesis and characterization of grafted copolymer. Adv Compos Mater. 2014;24:531–43.
Karthika JS, Vishalakshi B. Novel stimuli-responsive gellan gum-graft-poly(DMAEMA) hydrogel as adsorbent for anionic dye. Int J Biol Macromol. 2015;81:648–55.
Karthika JS, Vishalakshi B, Naik J. Gellan gum-graft-polyaniline-an electrical conducting biopolymer. Int J Biol Macromol. 2016;82:61–7.
Trivedi JH, Thaker MD, Trivedi HC. Photo-induced synthesis and characterization of poly(methylmethacrylate) grafted sodium salt of partially carboxymethylated guar gum. Chin J Polym Sci. 2014;32:1690–703.
Nandi G, Patra P, Priyadarshini R, Kaity S, Ghosh LK. Synthesis, characterization and evaluation of methacrylamide grafted gellan as sustained release tablet matrix. Int J Biol Macromol. 2015;72:965–74.
Wong TW, Chan LW, Lee HY, Heng PW. Release characteristics of pectin microspheres prepared by an emulsification technique. J Microencapsul. 2002;19:511–22.
Chaudhari MP, Chaudhari DP. Formulation and evaluation of multiparticulate system for chronotherapeutic delivery of salbutamol sulphate. J Pharm Bioallied Sci. 2012;4:71–3.
Kumar P, Ganure AL, Subudhi BB, Shukla S. Preparation and characterization of pH-sensitive methyl methacrylate-g-starch/hydroxypropylated starch hydrogels: in vitro and in vivo study on release of esomeprazole magnesium. Drug Deliv Transl Res. 2015;5:243–56.
Niharika K, Nanjappaiah HM, Naikawadi AA, Kulkarni RV. Composite microspheres of rosin gum and ethyl cellulose for controlled release of an anti-diabetic drug: in-vitro and in-vivo assessment. Drug Deliv Lett. 2014;4:227–35.
Balamurugan K, Nishanthini A, Mohan VR. Antidiabetic and antihyperlipidaemic activity of ethanol extract of Melastoma malabathricum Linn. leaf in alloxan induced diabetic rats. Asian Pac J Trop Biomed. 2014;4:S442–8.
Osmani RAM, Aloorkar NH, Ingale DJ, Kulkarni PK, Hani U, Bhosale RR, et al. Microsponges based novel drug delivery system for augmented arthritis therapy. Saudi Pharm J. 2015;23:562–72.
Bhosale RR, Osmani RAM, Padmaja C, Moin A. Formulation and evaluation of sustained release dosage form using modified cashew gum. Int J Pharm Pharm Sci. 2015;7:141–50.
Srivastava A, Gowda DV, Hani U, Shinde CG, Osmani RA. Fabrication and characterization of carboxymethylated bael fruit gum with potential mucoadhesive applications. RSC Adv. 2015;5(55):44652–9.
Acknowledgments
The author(s) express their gratitude for the financial support provided by the Department of Science and Technology, Government of India, New Delhi, India, under grant SR/SO/HS-0107/2016. Author(s) express deep sense of gratitude towards Central Food Technological Research Institute (CFTRI), Mysuru (a constituent laboratory of CSIR, New Delhi) and JSS Academy of Higher Education and Research (JSSAHER), Mysuru, for provision of obligatory facilities to carry out present research work. The author(s) are profusely thankful to Dr. Ekta Singh, Department of Biosciences and Bioengineering (BSBE), Indian Institute of Technology Bombay (IITB), Mumbai, for proof reading and constructive suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 292 kb).
Rights and permissions
About this article

Cite this article
Bhosale, R.R., Gangadharappa, H.V., Osmani, R.A.M. et al. Design and development of polymethylmethacrylate-grafted gellan gum (PMMA-g-GG)-based pH-sensitive novel drug delivery system for antidiabetic therapy. Drug Deliv. and Transl. Res. 10, 1002–1018 (2020). https://doi.org/10.1007/s13346-020-00776-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-020-00776-7