
Abstract
The current studies investigate the application of quality by design-enabled type III self-emulsifying delivery system (Type III-SEDDS) of sorafenib tosylate (SFN) in improving its biopharmaceutical attributes. Initially, lipidic and emulsifying excipients were selected by carrying out solubility and phase titration experiments. After screening studies using Taguchi OA design, Type III-SEDDS were further optimised using D-optimal mixture design. The prepared formulations were assessed for globule size, zeta potential and percent of drug release. Following graphical optimisation, the optimum formulation was earmarked and further supersaturated to form saturated Type III-SEDDS (Sat-Type III-SEDDS) using a combination of HPMC and PVP to improve the stability of the formulation for a prolonged period. In vitro drug release of Type III-SEDDS study indicated approximately 8-fold improvement in dissolution rate over the pure powder drug. Cell uptake studies demonstrated higher uptake of dye-loaded Type III-SEDDS formulations in Caco-2 cells vis-à-vis plain dye. Cytotoxicity assay on Hep G2 cells revealed significant reduction in cell growth with Type III- and Sat-Type III-SEDDS vis-à-vis the pure drug. Furthermore, in situ permeation studies carried out using Wistar rats exhibited nearly 8.3- to 10.2-fold augmentation in permeation and absorption parameters of the drug from the Type III- and Sat-Type III-SEDDS, respectively, vis-à-vis the pure drug. Pharmacokinetic studies indicated nearly 3.98- and 3.62-fold improvement in AUC0–72, and 8.01- and 5.42-fold in Cmax, along with 0.25-fold decrease in Tmax of the drug from Type III- and Sat-Type III-SEDDS, respectively, in comparison with the SFN suspension. Furthermore, high degree of level A linear correlation was established between fractions of drug dissolved (in vitro) and of drug absorbed (in vivo) at the corresponding time points for Sat-Type III-SEDDS and pure drug, whereas the Type III-SEDDS exhibited a nonlinear relationship. Stability studies indicated the robustness of Sat-Type III-SEDDS, when stored at 25 °C for 3 months. Overall, the manuscript documents the successful systematic development of SFN-loaded Sat-Type III-SEDDS with distinctly improved biopharmaceutical performance.

Graphical abstract
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) or malignant hepatoma is one of the most commonly diagnosed cancers across the globe and is ranked second and sixth foremost cause of cancer deaths in men and women, respectively [1, 2]. Since it has a long dormant period, when diagnosed first, it is already either in the intermediate or advanced stage, leaving the only option of treatment is chemotherapy. However, the conventional chemotherapeutic agents are associated with low response rate and severe side effects owing to their lack of selectivity.
Kinases, in this regard, have emerged out as the promising targets in cancer research, as these have critical involvement in cell signalling [3]. Sorafenib tosylate (SFN) [4], a tyrosine kinase inhibitor, has turned out to be the cornerstone in the medical treatment of various carcinomas including HCC, renal carcinoma, and thyroid cancer [5]. Lately, it has become the first-line drug for patients with advanced HCC. However, the clinical use of SFN is limited by its low (~ 8.43%) and variable oral bioavailability, ostensibly owing to its poor aqueous solubility, high presystemic hepatic metabolism and high efflux by permeability glycoprotein (P-gp) [6]. Nevertheless, with the advent of novel drug delivery technologies, the challenge to deliver such drugs as successful drug products has been successfully achieved. Several approaches investigated for effective delivery of SFN include inclusion in cyclodextrin complexes [7], encapsulation in micro/nanoparticles [8,9,10], solid dispersions [11], nanosuspensions [6], nanodiamonds [12] and various lipid-based systems, namely, solid lipid nanoparticles (SLNs) [13, 14], nanostructured lipidic carriers (NLCs) [15], self-microemulsifying delivery systems [16] and mixed micelles [17].
Lipid-based formulations, of late, have gained high interest among the researchers, as the carriers to deliver poorly water-soluble drugs (PWSDs), owing to the wide availability with apt regulatory and safety profiles of lipids [18, 19]. In this regard, lipid formulation classification system (LFCS) categorises the self-emulsifying formulations into four sub-categories, based on their ability to disperse in aqueous medium and aqueous solubility of the excipient(s) used [20]. Out of these, LFCS type III self-emulsifying drug delivery systems (Type III-SEDDS) have been established as the promising tool to augment oral bioavailability of PWSDs. The enormous potential of such systems is quite evident from the fact that most of the emulsifying products available in the market belong to Type III-SEDDS, such as cyclosporine (Neoral®), sirolimus (Rapamune®), ritonavir (Norvir®) and tipranavir (Aptivus®) [21, 22]. Furthermore, as none of the literature reports on delivery of SFN has investigated the incorporation of a unit human dose, i.e. 200 mg, in a single formulation to date, subsequent supersaturation of Type III-SEDDS(s) would be carried out for the purpose. Supersaturable systems have evolved as a vital modification of such type III systems for improving the biopharmaceutical performance of drugs with high doses. These tend to employ a suitable polymer as precipitation inhibitor (PPI) agent (e.g., cellulosic polymers), for maintaining supersaturated state of the drug after mixing with water (in vitro) and in biological fluids (in vivo) [22,23,24].
Attempts, therefore, were made to investigate the potential of Type III-SEDDS for ameliorating biopharmaceutical performance of SFN following oral intake. Furthermore, the systematic approach of quality by design (QbD), encompassing DoE principles, was employed to furnish the rational and holistic understanding of the possible relationship(s) among the input variables, while embarking upon the “the optimised” formulation and thus saving a significant volume of resources [25].
Materials and methods
SFN was provided as a generous gift sample by M/s Cipla Pvt. Ltd., Mumbai, India. Peceol, Labrasol, Captex, Capryol™ 90, Labrafac™ 1349, Labrafac PG, Maisine and Transcutol HP were provided ex-gratis by M/s Gattefosse, Saint-Priest, France. Various grades of Acrysol and Acconon were procured from M/s Abitec (Wisconsin, USA). Polyethylene glycol (PEG) 200, PEG 300, Span 20, Span 80, Tween 20, Tween 80 and oleylamine were purchased from M/s Merck (Mumbai, India). Wheat oil, sunflower oil, olive oil and polyvinyl pyrrolidine (PVP)-K25 were obtained from M/s BASF, Mumbai, India, while HPMC-E5 was obtained as gift samples from M/s Colorcon Asia Pvt. Ltd., Verna, India. M/s ACG Capsules, Mumbai, India, provided the gelatin capsule shells as gift samples. The remaining other chemicals employed throughout the current research were of analytical grade.
Preformulation studies
Solubility studies on SFN in various lipids, surfactants and cosolvents
SFN solubility was studied in several solvents like natural oils (wheat germ oil, sunflower oil and olive oil, etc.), synthetic lipids (i.e., Peceol, Captex, Capryol™ 90, Labrafac™ 1349, Labrafac PG, Maisine and Capmul derivatives), surfactants (Labrasol and various grades of Span and Tween) and co-surfactants (Transcutol HP, PEG-200 and PEG-300) using shake flask method. For the purpose, SFN in excess amount was placed in a screw-capped vial, which consisted 1 mL of the particular solvent. The resulting mixtures were stirred employing a vortex mixer (CycloMixer101, Remi, India) and then allowed to equilibrate at 37 ± 2 °C for 72 h in a water bath shaker at 100 rpm (Rivotek, India). The solutions, thus formed, were then centrifuged at 10000 rpm (9055g) for 15 min in order to allow sedimentation of the undissolved drug per se, followed by analysis of supernatant, employing UV spectrophotometer (UV 3000+, Labindia, India) at 265 nm, after appropriate dilution with methanol [26].
Ternary phase titration studies
The selected surfactant and cosolvent were mixed to prepare Smix in the ratios 1:0, 1:1 and 1:2, followed by their mixing with lipid in the ratios ranging between 1:9 and 9:1. These mixtures were subsequently titrated against water, to demarcate the nanoemulsion area in the pseudoternary phase diagrams, constructed using PCP Disso software [27]. The combination of ratios exhibiting maximal nanoemulsion region was chosen for further studies.
Physical compatibility studies
These studies were performed to rule out the probability of any interaction(s) between SFN and each of the chosen excipients, viz., lipid, surfactant and cosolvent, employing Fourier transform infrared (FTIR) spectrophotometer (M/s Perkin Elmer, Massachusetts, USA) [28]. FTIR spectra were recorded after adsorption of the samples on KBr disk.
Delineating quality target product profile and critical quality attributes
Initially, the quality target product profile (QTPP) was embarked upon defining all the requisite quality characteristics of the formulation to be developed for achieving maximal therapeutic efficacy of SFN [26]. Table S1 of Supplementary Data enumerates the elements of QTPP during development of Type III-SEDDS. In this regard, various quality attributes (QAs) like physical attributes of the drug delivery system, viz., globule size (i.e., Dnm is an indicator of release rate, along with subsequent potential to accumulation in the liver), zeta potential (i.e., for sufficient interactions with biological membranes and therefore tumour cells, and facilitated entrance into tumour cells for effective cytotoxic effect [29]), emulsification time (i.e., index of solubilization rate of the SFN), percent of drug release (i.e., measure of absorption rate) and dissolution parameters, i.e. mean dissolution time (MDT) and percent of dissolution efficiency (i.e., % DE is measure of dissolution performance). Table 1 enlists the various critical quality attributes (CQAs) which exhibit significant influence on the biopharmaceutical performance of the purposed formulations as well as their respective fitting explanation(s).
Formulation of Type III-SEDDS
SFN (i.e., 200 mg) was dissolved in the previously warmed lipid, i.e. Peceol at 37 ± 1 °C, in order to prepare a homogenous mixture. This was followed by the addition of the chosen surfactant, i.e. Labrasol and the cosolvent, i.e. Transcutol HP (T-HP) to the lipid-drug solution with continuous stirring. Later, oleylamine (0.5% w/w) was added to each prepared Type III-SEDDS of SFN in order to impart the cationic charge to the formulations.
Factor screening studies
During these studies, a first-order 7-variables-2-level Taguchi OA design with a total of 8 experimental runs was employed. The influence of material attributes (MAs) (i.e., amount of lipid, surfactant and cosolvent) and process parameters (PPs) (i.e., mixing type, stirring time, stirring speed and temperature) was investigated for selected CQAs, viz., Dnm, emulsification efficiency and percent of drug release at 60 min (% Rel60min). Table 2 represents the design matrix employed for factor screening. The highly influential variables from the various MAs/PPs were identified using half-normal plots and Pareto charts [30].
Characterisation of formulations
Emulsification time determination:
Each formulation(s) (1.0 g each) was supplemented separately to 0.1 M hydrochloric acid containing 1% w/v sodium lauryl sulphate (SLS) solution (500 mL) with constant stirring (50 rpm) at 37 ± 0.5 °C employing USP Apparatus 2 (Labindia, DS 8000, Mumbai, India). Time taken by the formulation to thoroughly and uniformly dispersed into the system was noted as the emulsification time in seconds [26].
Globule size analysis and zeta potential measurements:
Preconcentrate aliquots of the prepared formulations (1 mL each) were diluted and analysed for their globule(s) size employing dynamic laser scattering equipment (ZS 90, M/s Malvern, Worcestershire, UK).
In vitro drug dissolution studies:
Dissolution studies were conducted using USP Apparatus 2 (M/s Labindia, DS 8000, Mumbai, India) employing 900 mL of 0.1 M hydrochloric acid containing 1% w/v sodium lauryl sulphate (SLS) as the dissolution medium maintained at 37 ± 0.5 °C and 100 rpm [31]. Type III-SEDDS incorporating a unit dose of SFN, i.e. 200 mg, were put into a hard gelatin capsule and placed into the dissolution medium [32]. At pre-determined time intervals, an aliquot of 2 mL sample (each) was withdrawn with subsequent replacement of equivalent volumes of the medium. After suitable dilution(s), the samples were subjected to spectrophotometric analysis at λmax of 265 nm. Furthermore, % Rel60min was calculated after applying suitable correction factor [33].
Systematic optimisation
Systematic optimisation studies were conducted using a D-optimal mixture design (D-opt). Effect of various critical material attributes (CMAs) and critical process parameters (CPPs) were investigated on their CQAs like Dnm, zeta potential, % Rel60min, %DE and MDT. Table S2 of the Supplementary Data illustrates CMAs and CPPs and their corresponding low, mid and high levels used during the optimisation studies. The prepared formulations were characterised for selected CQAs, as described earlier in the “Characterisation of formulations” section.
Response surface methodology and search for the optimum formulation
Following data analysis of the experimental outcomes for each of the response variable (CQA), the optimised formulation was identified. Mathematical modelling was used to establish relationship(s) between the studied CMAs and CPPs, and the observed CQAs. Multiple linear regression technique was employed, followed by fitting of the data with an apt polynomial model. Interaction terms and aptness of model fitting were studied using statistical parameters. Response surface mapping was carried out by employing 2D and 3D graphs to understand the relationship(s) among the CMAs and studied CQAs [34]. The projection of optimum formulation was carried out by using desirability and design space as the functions of numerical and graphical optimisation, respectively.
Supersaturation of optimised Type III-SEDDS of SFN
Furthermore, HPMC-E5, PVP-K25 and their mixture combination (1:1), in the concentrations of 5%, were selected as PPI. These were individually added to the optimised Type III-SEDDS with thorough mixing to form a homogenous supersaturated system [35, 36]. The prepared formulation(s) were further evaluated for the degree of supersaturation (SS°).
Characterisation of supersaturated optimised LFCS type III system
Measurement of degree of supersaturation
Supersaturation test was carried out according to the protocol reported earlier [35] using in-house fabricated assembly consisting of a three-neck round-bottom flask and a digital mechanical stirrer was used for conducting the test. The saturated optimised Type III-SEDDS (Sat-Type III-SEDDS) containing 200 mg drug was seeded in a 250-mL round-bottom flask holding 100 mL (2 mg/mL) of 0.1 M hydrochloric acid containing SLS (1% w/v) at 37 ± 1 °C, with constant stirring at 100 rpm [31]. Samples (1 mL each) were taken out at 0.25, 0.5, 0.75, 1, 1.5, 2 and 3 h without replenishing the medium. Furthermore, the drug concentration of the samples was determined by subjecting these to analysis using spectrophotometer at 265 nm. Test conditions were maintained for one whole day (i.e., 24 h) to assess the equilibrium solubility (Seq) and SS° for each of the selected PPIs [36]. The complete protocol was repeated in triplicate for each PPI employed. This aided in the selection of apt PPI for supersaturating the optimised Type III-SEDDS. The concentrated drug residuals deposited on the base of RBF were collected and studied, as discussed under.
Globule size and zeta potential
The prepared formulations, viz., Type III-SEDDS and Sat-Type III-SEDDS, were evaluated for their Dnm and zeta potential, as described earlier in the “Characterisation of formulations” section.
Optical microscopy
Morphological structure of SFN from Type III-SEDDS and Sat-Type III-SEDDS was visualised using an optical microscope (M/s Radical, RXL-5, M No.B-1810398, India). A little amount of the desiccated precipitates was kept on a glass slide, and the image was captured at × 100 magnification.
Field emission scanning electron microscopy
Likewise, both formulations, i.e. Type III-SEDDS and Sat-Type III-SEDDS, were visualised using field emission scanning electron microscopy (FESEM) type II (Model-S-4800, M/s Hitachi, Tokyo, Japan). Samples were initially dried and positioned over aluminium stubs and platinum sputtering at 10 Torr vacuum was carried out before the visual examination.
Powder X-ray diffraction studies
Powder X-ray diffraction (PXRD) studies for SFN (i.e., pure drug) and residual precipitates (i.e., from supersaturated preconcentrates) were conducted using an automated X-ray diffractometer, X’Pert PRO (M/s PANalytical, Almelo, Netherlands). Cu Kα (PW3050/60, 1.54 Å) was used as the radiation source with 45 kV of applied voltage and 40 mA current supplies. Samples (10 mg each) were added to the sample holder while rotating the stage over the range of 2θ from 5° to 50° and change in values of 2θ was recorded for the respective samples.
Rheological studies
The rheological properties of the prepared system were evaluated using rheometer (RheolabQC SN000000; ID81946396, Anton Paar), equipped with concentric cylindrical geometry (CC10; gap = 0.8 mm) at 100 rpm at 25 °C ± 0.5 °C.
In vitro drug dissolution studies
In vitro drug release studies on both the formulations, i.e. Type III-SEDDS and Sat-Type III-SEDDS, were performed as described in the “Characterisation of formulations” section. Furthermore, to predict the drug release mechanism, the release data were fitted into Korsmeyer-Peppas model [37].
Cell culture studies
Caco-2 cell culture and qualitative cell uptake studies. Caco-2 cells (NCCS, Pune, India) were seeded in a 96-well plate (1 × 105 cells/well) and supplemented with nutrient consisting of 20% foetal bovine serum (FBS), Dulbecco’s Modified Eagle’s media (DMEM) and antibiotics in specified quantity (i.e., 100 μg mL−1 each). The treated cells were kept in tissue culture flasks (75 cm2), followed by incubation with 5% CO2 (Heracell™ 150 i, Thermo, USA) at 37 ± 0.5 °C.
Qualitative uptake of optimised formulation(s) by cells was studied using confocal laser scanning microscopy (CLSM; Nikon C2+, Japan). The cell culture medium was supplemented with coumarin 6 (C6)-loaded formulation(s) and kept aside in an incubator at 37 °C for 3 h. The cover slips were put on slides after washing these with PBS (six times) and examined under CLSM and images were taken subsequently. C6-loaded formulation(s) were prepared following a similar methodology, as described in the “Formulation of Type III-SEDDS” section, after replacing the drug with the dye.
Cytotoxicity assay. RPMI 1640 medium containing 10% foetal bovine serum and 2 mM l-glutamine was used to grow Hep G2 cells. Furthermore, cells were inoculated into a microtiter plate (96-well; approximately 1 × 105 cells/well) and incubated with 5% CO2 at apt standard conditions of temperature and humidity for 24 h, before the addition of the test formulation(s). Pure drug (SFN), Type III-SEDDS and Sat-Type III-SEDDS were initially solubilized in dimethyl sulfoxide (100 mg/mL) and diluted to 1 mg/mL, followed by their storage in the frozen state before use. At the time of testing, an aliquot of frozen concentrate of each of the test formulations (1 mg/mL) was thawed and diluted to 10, 20, 40 and 80 μg/mL SFN concentrations. In situ fixation of cells was carried out with gentle addition of cold trichloroacetic acid (30% w/v) and incubated for 60 min at 4 °C. The plates were then washed with tap water and air-dried, after discarding the supernatant. This was followed by the addition of 50 μL of sulforhodamine B (SRB) solution (0.4% w/v in acetic acid) to every well, and plates were kept for incubation at ambient temperature for another 25 min. Furthermore, the residual stain was washed with acetic acid (1%), and the plates were air-dried. Bound dye was eluted with Trizma base (10 mM), and then the absorbance was read on a plate reader at a wavelength of 540 nm with 690 nm reference wavelength.
Percent growth at each concentration of drug was calculated [38], using Eq. 1:
where Tz is growth at time zero, C is control growth, and Ti is test growth in the presence of drug at the four concentration levels.
In vivo characterisation
After obtaining the requisite permission from Institutional Animal Ethics Committee (IAEC) of the Panjab University, vide their letter no PU/IAEC/S/16/18, in vivo studies were carried out as per the approved experimental plan.
Single-pass intestinal perfusion (SPIP) studies. Wistar rats (unisex, 200–300 g) were randomly grouped, namely, group I for pure SFN suspension, group II for Type III-SEDDS and group III for Sat-Type III-SEDDS. Each group, consisting three animals, was fasted overnight. Experiments were performed, using an in-house fabricated assembly, as reported [39]. Animals were anaesthetized by injecting 50 mg/kg of ketamine (i.p.). Initially, the abdominal cavity was opened by a middle incision, and a small segment of the jejunum was cannulated using polyethylene tubing to create cannulated inlet and outlet. The cannulae were secured with a surgical suture. Furthermore, it was perfused with lukewarm Krebs Ringer Phosphate Buffer (KRB) solution, followed by perfusion at a rate of 0.2 mL/min with the pure drug, Type III-SEDDS and Sat-Type III-SEDDS at dose calculated for each rat based upon the human dose of 200 mg/70 kg for the per oral route [40]. An aliquot of 1 mL each was withdrawn after 30 min, with sample withdrawal every 15 min to 2 h. Furthermore, the perfusate was extracted, centrifuged (9055×g) and quantified for SFN concentration using UV spectrophotometer at a λmax of 265 nm. Afterwards, the permeability parameters, like effective permeability (Peff) and wall permeability (Pwall), and absorption parameters, like fraction absorbed (Fa) and absorption number (An), were computed [41].
In vivo pharmacokinetic studies. Pharmacokinetic studies were conducted on Wistar rats (unisex), following single-dose parallel design. Each group consisted of three animals. Group I was administered with SFN suspended in 0.2% CMC, and group II was administered with the Sat-Type III-SEDDS, while group III was administered with the Type III-SEDDS using an oral gavage. The dose of SFN was calculated for each rat based upon the human dose of 200 mg/70 kg for the per oral route [40]. About 200 μL of blood was withdrawn at 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 60 and 72 h from the retro-orbital plexus of rats. Blood samples were added to a heparin-containing microcentrifuge and centrifuged at 12,000 rpm (10,866×g) for 10 min to precipitate out the plasma proteins. Furthermore, the rat plasma was extracted and subsequently subjected to high-performance liquid chromatographic analysis to estimate SFN, employing a mobile phase consisting of acetonitrile and water (containing orthophosphoric acid, pH 4.1) (65:35 v/v) at the flow rate of 0.8 mL/min with UV detection at 265 nm using a C18 column, an analytical method previously reported by us [42]. Various pharmacokinetic parameters, like maximum plasma concentration (Cmax), time at maximum concentration (Tmax), area under curve (AUC), absorption rate constant (Ka), elimination rate constant (K) and half-life (t1/2) were calculated [36].
In vitro/in vivo correlations
Linear as well as nonlinear point-to-point level A in vitro/in vivo correlations (IVIVC) was attempted between in vitro percent drug released with in vivo percent drug absorbed at the corresponding time points for Type III-SEDDS, Sat-Type III-SEDDS and pure drug suspension, and the statistical significance of each discerned [36]. The values of cumulative relative fraction (or percent) absorbed of drug at particular time points were computed employing modified Wagner Nelson method, as the drug was observed to follow one-compartment open body model pharmacokinetics [43].
Stability studies
Type III-SEDDS and Sat-Type III-SEDDS were poured in glass containers and stability was evaluated employing humidity chamber (Newtronic Lifecare Equipment Pvt. Ltd., Mumbai, India). The samples were stored for a period of 3 months at three different conditions of temperature and humidity, i.e. 2–8 °C, 25–30 °C/60% RH 45–50 °C/75% RH to assess the stability of the developed systems [44]. Samples were withdrawn at 0, 30, 60 and 90 days, and tested for any change in Dnm, zeta potential and dissolution performance vis-à-vis the original data.
Statistical analysis
In this study, all data are expressed as means ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism software version 8 (GraphPad Holdings, LLC, USA).
Results and discussion
Preformulation studies
Solubility studies on SFN in lipids, surfactants and emulgents
Figures S1 a and b depict the solubility of SFN in various lipids, surfactants and cosolvents, respectively. Such drug solubility profiles are considered essential to facilitate the final selection of the various functional excipients prior to the development of the SEDDS formulation [45, 46]. Particularly in the current studies, as the dose of SFN is quite high (i.e., 200 mg), it will be ideal to choose the lipid, surfactant and cosolvent in which its solubility is quite adequate. Peceol, a monounsaturated fatty acid, showing maximum solubility potential for SFN, was considered as the lipid of choice for formulation development. Among various emulgents investigated, the highest solubility of SFN was observed in Labrasol. Furthermore, Labrasol owing to its nonionic nature would possibly produce only reversible changes in the intestinal mucosa [47, 48] and would be less toxic than the ionic ones after oral intake [49, 50]. Moreover, Labrasol has been identified as one of the most effective surfactants inhibiting the P-gp efflux too [51]. SFN was found to be maximally soluble in T-HP, among the different cosolvents investigated for the purpose. Hence, Peceol, Labrasol and T-HP were finally considered to delineate a stable microemulsion region for the preparation of Type III-SEDDS.
Phase titration studies
Construction of pseudoternary plots is a vital exercise to deduce the concentration ranges of MAs for the SEDDS, depending upon the spontaneity of emulsification in the physiological fluids and stability of resulting emulsion formed [52]. The pseudoternary phase diagrams, constructed employing Peceol, Smix of Labrasol and T-HP in the ratios of 1:0, 1:1, and 1:2, and water, showing a stable emulsion region, are portrayed in Fig. S2. The combination of Peceol and Smix of Labrasol and T-HP (1:1) was observed to depict higher water uptake capacity, with maximal emulsion region for the formulation of type III LFCS SEDDS (Fig. S2b). Significant reduction in this region was observed, while increasing the ratio of Smix to 1:2 (Fig. S2c), attributable to the availability of relatively lower concentration of Labrasol for decreasing the interfacial tension between the oil globules [45]. Additionally, on increasing the Smix concentration to 2:1, further reduction in the nanoemulsion region was observed vis-à-vis the concentration of 1:1, signifying the superior emulsification capacity of the latter [53]. Hence, as deciphered from the pseudoternary phase diagram, Smix concentration of Labrasol and T-HP in the ratio of 1:1 yielded maximal and stable microemulsion region; hence, this ratio was further considered in selecting the levels of the chosen material attributes during the subsequent factor screening studies.
Drug-excipient compatibility studies
Figure 1 portrays the FTIR spectra of SFN with each of the individual chosen excipients, i.e. Peceol, Labrasol, T-HP and their physical mixture thereof. In the current studies, the likelihood of any interaction between the drug and the functional or non-functional excipients is noticed by any change (i.e., appearance or disappearance of a peak corresponding to a particular functional group) in the characteristic peaks of the drug in the presence of each excipient and the physical mixture. The IR spectrum of the physical mixture of SFN with the excipients showed hardly any change in the characteristic peaks of SFN, as enumerated in Table S3 of Supplementary Data, thus ratifying the absence of any interaction between the drug and studied excipients.

FTIR spectra of SFN with (a) Peceol, (b) Labrasol, (c) T-HP and (d) physical mixture depicting the compatibility between SFN and the chosen excipients
Factor screening studies
The prime basis of this exercise is to identify potentially significant main effects, rather than the interaction effects [54]. In the present studies, screening of various PPs and MAs, potentially exhibiting their influence on the chosen CQAs, i.e. Dnm, emulsification time and % Rel60min (Table 3), was performed. A saturated Taguchi design (TGD) was employed for the purpose, as it required relatively less number (i.e., 8) of experimental runs for the studied large number (i.e., 7) of independent input variables. Screening studies are documented to help in identification of the most critical factors to facilitate subsequent thorough investigations involving minimal experimentation and consequently reducing the expenditure of time, effort and material resources [54, 55].
Preparation of the Type III-SEDDS
Type III-SEDDS formulations, prepared as described in the “Formulation of Type III-SEDDS” section, revealed distinct variation in the values of evaluated response variables Dnm, zeta potential and % Rel60min of the prepared formulations. This can plausibly be ascribed to the difference in the composition of these formulations in terms of lipid, surfactant and cosolvent.
Model generation
A total of seven polynomial coefficients (i.e., β1 to β7) were generated with β0 as the intercept, as per the polynomial relationship for the chosen design (Eq. 2).
Model analysis of half-normal plots and Pareto charts
Half-normal plots and Pareto charts were drawn to facilitate the identification of the influential factors, i.e. CMAs or CPPs, which lie either over the t value line or near the Bonferroni line, as shown in Fig. 2. As deciphered from the plots, lipid (i.e., Peceol), surfactant (i.e., Labrasol) and cosolvent (i.e., T-HP) exhibited the most remarkable effect on the selected CQAs, i.e., Dnm, emulsification time and % Rel60min per se, and these were accordingly chosen as the CMAs for the subsequent response surface optimisation studies.

Pareto charts and half-normal plots depicting the influence of MPs/PPs on chosen CQAs, i.e., (a) Dnm (b) Rel60min (c) Emulsification time, during factor screening studies
Systematic optimization
Based on the results of preformulation and screening studies, type III LFCS SEDDS formulations were optimised employing a 3-factor-3-level D-optimal mixture design. During development, the characteristics of the finished pharmaceutical product containing multiple functional excipients generally depend upon nature as well as amounts of these excipients. As the total of the proportions of all chosen excipients being unity here, D-optimal mixture design was preferred as the appropriate response surface optimisation design [25, 54, 56, 57].
A total of the 16 Type III-SEDDS formulations, including the quintuplicate runs, were prepared according to the D-optimal design matrix and characterised for CQAs, % Rel60, Dnm, MDT and % DE as enlisted in Table 4. Figure S3 of the Supplementary Data depicts the dissolution profile of all the formulations, prepared as per the D-optimal design.
Response surface optimization mapping
The coefficients of the second-order quadratic polynomial model equations, along with interaction terms, generated for all the chosen CQAs demonstrated excellent fitting to the obtained data as well as the insignificant lack of fit (p > 0.05) [58].
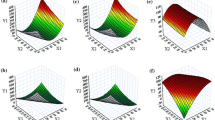
Figure 3 (A) illustrates the 3D and 2D response surface mapping for the chosen CMAs and CPPs. Figure 3 (A) depicts an increase in Dnm with increasing levels of Peceol, which can plausibly be ascribed to increased surface tension between the globules and is in good agreement with previous report [59]. However, while moving from low to high levels of T-HP, a declining drift in Dnm was observed, attributable to the reduction in the interfacial tension at higher concentration of the cosolvent [59]. However, the gradual increase in Labrasol showed a significant increase in size. This may be ascribable to the hydrophilic nature of Labrasol (i.e., HLB value 14), possibly leading to its partitioning in the aqueous phase, thus failing to reduce the size [60]. Similar inferences for change in the Dnm with changes in the levels of lipid, surfactant and cosolvent were quite apparent from the corresponding 2D plot.


(A) 2D contour and 3D response surface plots portraying the influence of Peceol, Labrasol and T-HP on (a) Dnm, (b) Rel60min, (c) MDT and (d) % DE during optimization studies. (B) Overlay plot depicting the design space (yellow region) for optimised Type III-SEDDS
An increasing linear trend in % Rel60 values was noticed with increasing levels of each of the three chosen CMAs, i.e. lipid, surfactant and cosolvent (Fig. 3 (A(b)). However, the increase was modest with increasing levels of Labrasol, which could be attributed to the interplay of simultaneous decrease in lipid as well as cosolvent levels. Analogous observations are fairly vivid from the corresponding 2D graph plot, indicating maximal SFN release at the highest levels of all the three CQAs.
An enigmatic relationship was observed between the three CMAs and % DE values. Escalation in lipid content resulted in an initial rise, followed by a dip in DE (Fig. 3 (A(c)). Rise in Labrasol and T-HP levels exhibited high DE at mid-levels, while dip in the values of DE was deciphered at their extreme high and low levels. Likewise, 2D graph also portrayed that the mid-levels of three CMAs were conducive for attaining high % DE.
MDT increases with an increase in levels of T-HP owing to the simultaneous decrease in surfactant (i.e., Labrasol) levels (Fig. 3 (A(d)). Furthermore, with increasing lipid levels, an increase in MDT was observed, ostensibly owing to the longer period of time required to emulsify high lipid content contained in the formulation. In the case of Labrasol, however, a decline in MDT values was noticed, followed by an increase with increasing levels of Labrasol. This could eventually be ascribed to attainment of critical micellar concentration at the intermediate levels of Labrasol after which no further reduction in surface tension was possible.
Search for the optimum formulation
The optimum formulation was earmarked employing numerical desirability function, by “trading off” various CQAs for achieving the required goals, i.e. maximization of % Rel60 (i.e., representing the rapidness absorption) and DE (i.e., demonstrating adequate drug release), and minimization of Dnm (i.e., necessary for absorption via lymphatic route) and MDT (indicating faster and completed drug release).
The constraints were further narrowed down to demarcate the design space in the overlay contour plot between the chosen three factors across 2D experimental domains, as shown in Fig. 3 (A(B). The latter depicts the desirable optimal design space region in the yellow colour, surrounded with the grey colour region, called the knowledge space.
Characterisation of supersaturated Type III-SEDDS
In vitro supersaturation test
Figure 4 illustrates the supersaturation effect of an individual as well as the combination of PPIs (1:1) for inhibition of the precipitation of SFN from the emulsifying delivery system. The effectiveness of the PPIs was evaluated based on degree of supersaturation (SS°). The obtained results for the studied HPMC-E5 was found to be superior in inhibiting the precipitation of drug to the studied PVP-K25 grade, as the SS° values for HPMC was found to be 37.5, whereas for PVP was 28.28. These observed results were found to be in close agreement with those reported in several earlier studies [61,62,63]. Interestingly, it was observed that the formulations containing HPMC and PVP in combination (1:1) were quite stable for longer periods of time than the formulations containing either of the individual polymers. The underlying reason could be the same that the PVP molecules, unlike HPMC, do not self-associate and tend to interact with HPMC through hydrogen bonding [64]. Thus, these HPMC-PVP interactions may be considered responsible for accumulating at the boundary layer around the drug crystals, leading eventually to growth inhibition and additional prolonged stability. This is also quite evident from the sharp declining trends in the drug precipitation rate during in vitro supersaturation test for the polymer blend. Moreover, this inference was also substantiated with high magnitudes of SS°, i.e. 56.93 observed for the PVP-HPMC blend, and therefore, this was finally selected for supersaturating Type III-SEDDS.

Apparent drug concentration versus time profile of Sat-Type III-SEDDS using HPMC, PVP and their mixture (1:1) as polymeric precipitation inhibitors, each at 5% w/w concentration (n = 3; ±SD)
Globule size and polydispersity index
The value of Dnm of the Sat-Type III-SEDDS (i.e., 434 nm) was found to be slightly higher than that of its corresponding unsaturated Type III-SEDDS (i.e., 407 nm) (Fig. 5). Higher globule size of Sat-Type III-SEDDS vis-à-vis Type III-SEDDS can be attributed to the presence of polymers in the supersaturated state, which might have increased the value of Dnm by virtue of their coating on the exterior of emulsion globules. These observations were found to be in agreement with several of the previous literature reports on supersaturated systems [27, 65, 66]. Besides, the values of PDI were also found to be less than 0.5 in all the formulations, vouching quite uniform distribution of globule size [46].

Globule size distribution and zeta potential distribution. (a) Type III-SEDDS. (b) Sat-Type III-SEDDS
Zeta potential
The values of zeta potential for the Type III-SEDDS and Sat-Type III-SEDDS were found to be 31.7 mV and 25.8 mV, respectively (Fig. 5). The positive charge of formulation(s) is attributable to presence of oleylamine (0.5%), which was selected as a cationic charge inducer on the basis of prior art [67]. The observed high magnitudes of zeta potential for both formulations indicated a thermodynamically stable characteristic of the prepared formulations [46, 68]. However, a modest decline in the zeta potential values for Sat-Type III-SEDDS vis-à-vis Type III-SEDDS could plausibly be owing to the presence of PVP. Although HPMC and PVP were added in the ratio of 1:1 as the precipitation inhibitors, yet only PVP contributed towards modest decline in the zeta potential values for Sat-Type III-SEDDS, possibly because HPMC is nonionic in nature [69], while PVP is documented to exhibit anionic surface charge [70].
Optical microscopy
Figure S4 of the Supplementary Data depicts the optical microscopic images of the drug precipitates obtained after supersaturation test and the optimised formulation. Type III-SEDDS showed the presence of SFN mostly as spear-shaped crystals, while negligible quantity of SFN precipitated out of the Sat-Type III-SEDDS [68, 71].
Field emission scanning electron microscopy
Figures 6(a) and (b) portray the surface photomicrographs of the Type III-SEDDS system as well as its saturated system, i.e. Sat-Type III-SEDDS, respectively. The presence of a vivid concentric layer around the spherically shaped globules can possibly be ascribed to the presence of the polymers imparting supersaturation to the developed formulation, i.e. Sat-Type III-SEDDS (Fig. 6(c)). However, naïve Type III-SEDDS were found to be devoid of any such layer.

FESEM images. (a) Type III-SEDDS and (b) Sat-Type III-SEDDS, while (c) represents Sat-Type III-SEDDS at high magnification with a clear concentric layer of polymers around the spherically shaped globules
Powder X-ray diffraction
Figure 7 depicts the diffractograms of the pure drug as well as the precipitates of the drug obtained from the Sat-Type III-SEDDS. The sharp peaks in thePXRD pattern of the pure drug are indicative of its highly crystalline nature. However, the saturated formulations revealed amorphous appearance of drug with the presence of single characteristic peak in PXRD pattern. The results, therefore, suggest that the supersaturated system was quite effective in controlling the crystal growth of SFN, while maintaining sufficient amount of drug in the solubilized form under supersaturated state. This is ascribable to an interplay of polymeric interactions between HPMC and PVP, along with oleylamine, in inhibiting the nucleation of drug, while maintaining the supersaturation state [72, 73]. The results obtained were found to be in close agreement with the prior literature reports, accentuating the use of polymers for inhibition of precipitation of the drugs during supersaturated state [35, 71, 74, 75].

X-Ray diffractogram of (a) pure drug and (b) precipitates obtained from Sat-Type III-SEDDS depicting loss of crystallinity in the developed SEDDS formulation
Rheological studies
Figure S5 of the Supplementary Data illustrates a linear relationship between shear stress (0 to 10 Pa) and shear rate (0 to 100 s−1) for both of the tested formulations, thus unequivocally indicating their Newtonian flow characteristics. Moreover, the viscosity of both formulations, i.e. Type III-SEDDS and Sat-Type III-SEDDS, was well within the desired rheological limits, i.e. 0.1–1.0 Pas, thus predicting their good flow characteristics, and thus fulfilling the criterion for filling these developed formulations in hard gelatin capsules [76].
Drug release kinetic modelling studies
As portrayed in Fig. 8, the dissolution studies with Type III-SEDDS and Sat-Type III-SEDDS demonstrated significant improvement in drug release profile from both the formulations vis-à-vis pure drug suspension. Furthermore, the drug release kinetic model fitting into Korsmeyer-Peppas model indicated Fickian drug release mechanism from both of the developed SEDDS formulations, reporting the values of release exponent (n) of 0.331 and 0.368 for Type III-SEDDS and Sat-Type III-SEDDS, respectively. These findings are quite analogous to the observations reported previously in literature [36].

Cumulative drug release profile of Type III-SEDDS, Sat-Type III-SEDDS and pure drug. Each of the cross-bars indicate ± 1 SD
Cell line studies
Qualitative uptake studies
Figures 9(a–d) illustrate the CLSM images of Caco-2 cells incubated with C6-loaded Sat-Type III-SEDDS. Figure 9(b) reveals minuscule fluorescence inside the cells treated with dye C6 (as control), while the fluorescence intensity observed for cells treated with dye-loaded Sat-Type III-SEDDS was relatively higher (Fig. 9(d)). High fluorescence intensity indicates higher cellular uptake of Sat-Type III-SEDDS by the intestinal cells vis-à-vis plain dye. This construed a noteworthy augmentation in the uptake potential of the dye from Sat-Type III-SEDDS, ostensibly owing to the faster internalisation and enhanced permeability of the architectural constituents of formulation, i.e. lipid and emulsifying excipients through Caco-2 cells [39, 77].

The CLSM images of CaCo-2 cells indicating uptake of coumarin 6-loaded Sat-Type III-SEDDS. (a) TD image representing normal cells. (b) Cells treated with coumarin 6. (c) Normal cells. (d) Overlay of images (b) and (c) showing localization of C6-loaded Sat-Type III-SEDDS in cells
In vitro cytotoxicity study using SRB assay
In order to study the cytotoxicity potential, the SRB assay, instead of the routinely employed MTT assay, was carried out as the former is known to be more sensitive and reproducible [78]. Figure 10 (A(A) reveals a significant decline in the growth of cells with increasing concentrations of the drug, ranging between 10 and 80 μg mL−1. Significant difference in the cytotoxicity was observed for the Sat-Type III-SEDDS vis-à-vis the pure drug (p < 0.01). The positive control (i.e., adriamycin) exhibited negative percentage cell growth, as the cells not only failed to grow but started undergoing apoptosis too, ostensibly owing to its cytotoxic as well as cytocidal nature. However, SFN and Sat-Type III-SEDDS were though found to be cytostatic at concentrations > 20 μg/mL, but were cytocidal only at 80 μg/mL, resulting consequently in the positive percentage cell growth values at concentrations less than 80 μg/mL [79]. Moreover, it was found that the Sat-Type III-SEDDS inhibited proliferation of cancer cells more efficiently than pure drug. The phase-contrast micrographs of control cells and formulation-treated cells are portrayed in Fig. 10 (A(B). The cells and nuclei of control cells look to be quite normal with distinct membranes and elliptical nuclei. However, the cells treated with developed formulations exhibited membrane blebbing, along with the formation of granules within the cell, while the cells treated with SFN showed quite less number of cells undergoing cell death. Furthermore, adriamycin (i.e., positive control)-treated cells were observed to encounter cell death, evident from the lack of nucleus in the cells, distorted cell membranes and the difference between nucleus to cytoplasmic ratio (Fig. 10 (A(B)). On the basis of the morphological changes, cell growth inhibition potential of the drug, after 48 h, i.e. concentration of drug causing 50% inhibition of cell growth (GI50) and level of drug causing total inhibition of cell growth (TGI), was calculated. The values of GI50 and TGI for pure SFN and its formulations, i.e. Type III-SEDDS, Sat-Type III-SEDDS and adriamycin, are enlisted in Table 5. SFN, Type III-SEDDS and Sat-Type III-SEDDS showed GI50 in Hep G2 cells of 47 μg/mL, 18 μg/mL and < 10 μg/mL, respectively, indicating 2.6-and 4.7-fold higher efficiency of Type III-SEDDS and Sat-Type III-SEDDS vis-à-vis the pure drug. Moreover, GI50 values for Sat-Type III-SEDDS were found to be akin to the positive control, i.e. adriamycin, thus establishing high cytotoxicity potential of the developed saturated delivery system. However, TGI values indicated insignificant difference in efficacy between both the developed formulations.

(A) Graph depicting percent reduction in growth of Hep G2 cells with increase in drug concentration of Sat-Type III-SEDDS, Type III-SEDDS, pure drug and Adriamycin as positive control (n = 3). (B) Phase-contrast micrographs of cells treated with (a) Type III-SEDDS, (b) Sat-Type III-SEDDS, (c) pure drug, (d) control cells and (e) Adriamycin
In situ single-pass intestinal perfusion study
In situ SPIP studies are well-established to provide a definitive idea of the permeation (Peff and Pwall) and absorption (An and Fa) potential of a drug following its oral administration [80].
The values of Peff and Pwall are quite documented to provide an idea about uptake mechanisms of the drug across the intestine [81]. Considerable increase in these values, i.e. 9.7- and 8.9-fold in Pwall, of Sat-Type III-SEDDS and Type III-SEDDS, respectively, was noticeable with respect to those obtained with the pure drug. Likewise, the Peff values of Sat-Type III-SEDDS and Type III-SEDDS showed nearly 9.4- and 8.3-fold enhancement, correspondingly, vis-à-vis the pure SFN. Furthermore, the values of absorption number (An), an approximation of the amount of drug transversed through the intestine eventually affecting the absorption process of the drug, as well as of bioavailable fraction (Fa), an estimate of the extent of drug absorption, were also duly computed from the aforesaid SPIP studies [34, 84]. Administration of Sat-Type III-SEDDS and Type III-SEDDS resulted in significant enhancement in the magnitude of An (8.90-fold and 10.15-fold, respectively) and of Fa (10.14- and 8.92-fold, respectively) vis-à-vis those administered with pure drug. Furthermore, the results for the developed SEDDS formulations were found to be far more consistent than those obtained with pure SFN during SPIP studies, as is quite evident from the lower magnitudes of standard deviation (SD) in the former case. The percent change in calculated parameters is depicted in Fig. S6 of the Supplementary Data.
Overall, the ability of the developed SEDDS formulations to bypass the hepatic portal route was ratified in the promotion of drug absorption, possibly via lymphatic pathways and owing to their lipidic characteristics, in consonance with several earlier published reports on SEDDS [82,83,84]. In addition to this, the inherent potential of the self-emulsifying lipidic formulation to inhibit the P-glycoprotein efflux of the other architectural constituents of formulation, i.e. Labrasol and T-HP, could also increase the transcellular diffusion of drug for improving the drug absorption parameters, leading consequently to augmentation in the bioavailability of SFN [36, 83].
In vivo pharmacokinetic studies
Figure 11A portrays the mean plasma concentration-time profile at different time points in rats following single oral dose administration of pure SFN suspension, Type III-SEDDS and Sat-Type III-SEDDS. The results of pharmacokinetic studies showed significantly superior drug absorption potential of the developed self-emulsifying delivery system(s) vis-à-vis pure SFN at a dose equivalent to 200 mg/kg of SFN. Approximately, 5.43- and 8.01-fold increase in Cmax (p < 0.0001) and 3.61- and 3.98-fold in that of AUC0–72 (p < 0.0001) was observed with Type III-SEDDS and Sat-Type III-SEDDS, respectively, in comparison with the corresponding parameters obtained with the pure drug. This indicated superior drug absorption potential from the developed formulation(s). Notable improvement was also noticed in the magnitudes of first-order absorption rate constant (Ka), by 1.80- and 4.37-fold improvement in Ka, with Type III-SEDDS and Sat-Type III-SEDDS, respectively. Moreover, the values of Tmax got reduced by 25% (p < 0.05) for both Type III-SEDDS and Sat-Type III-SEDDS vis-à-vis the pure drug. However, only modest change was observed in mean residence time (MRT) and half-life (t1/2) of drug. Accordingly, all the results aforementioned unequivocally vouch remarkable improvement in the rate and extent of bioavailability of SFN with not much change in the dispositional parameters of the drug. These enhanced biopharmaceutical attributes can eventually result in augmentation of the therapeutic effect.

(A) Mean (±SD, n = 3) plasma drug level time profiles of Sat-Type III-SEDDS, Type III-SEDDS, pure drug up to 72 h. The inset portrays the % change + SD in the computed pharmacokinetic parameters for Sat-Type III-SEDDS and Type III-SEDDS. (B) Point-to-point linear level A IVIVC. (a) Sat-Type III-SEDDS, (b) pure drug and (c) represent nonlinear level A IVIVC for Type III-SEDDS
In vitro/in vivo correlation
SFN, like other BCS class II drugs, is also anticipated to follow dissolution-limited absorption and exhibit good point-to-point correlation (i.e., level A) between in vitro drug release and in vivo biopharmaceutical performance. Accordingly, a high degree of level A correlation was observed to prevail between fractions of drug dissolved (in vitro) and of drug absorbed (in vivo) at the corresponding time points. Figure 11(B (a and b)) portrays the level A IVIVC for Sat-Type III-SEDDS and pure drug, respectively. High magnitudes of R were noticed for Sat-Type III-SEDDS (i.e., R = 0.9898; p < 0.05) and for pure drug (i.e., R = 0.9970; p < 0.05) with linear model fitting observed between the two parameters, viz., percent of drug dissolved and percent of drug absorbed [36, 43]. However, Type III-SEDDS showed the prevalence of the nonlinear relationship between fractions of drug dissolved and drug absorbed as depicted in Fig. 11(B (c)). The quadratic model, therefore, showed good data fitting in this case (R = 0.9861, p < 0.05) construing curved IVIVC for this formulation. Since the fast release formulation(s), like Type III-SEDDS, are known to release the drug (in vitro) quite fast, the in vivo absorption of such formulations may not be able to keep up linearly with their corresponding rate of drug dissolution. Drug absorption, accordingly, has been found to observe nonlinear relationship with the corresponding dissolution performance, in agreement with some of the similar reports on SEDDS [85, 86]. The successful establishment of IVIVC, therefore, ratifies the robustness of the methodology for in vitro drug release evaluation, e.g., dissolution medium composition, dissolution apparatus type, stirring conditions, sampling schedule, as quite simulative of the in vivo pharmacokinetic performance of SFN. This IVIVC tool has also been found to be very useful to help in the rational prediction of the biopharmaceutical performance of the formulation following any change(s) in the scale-up, location, and/or otherwise.
Stability studies
Stability testing under controlled conditions is considered as a vital aspect in drug delivery development and their subsequent federal approval, in order to determine the safety and integrity of the delivery system during storage. The current stability studies were carried out by determining various formulation CQAs, viz., Dnm, zeta potential and percent of drug released, at the periodic intervals of 0, 30, 60 and 90 days. As the conventional Type III-SEDDS showed precipitation of drug after 15 days at all the three storage conditions, the stability studies were further carried out for Sat-Type III-SEDDS only. The results during the stability study showed that the formulation stored at 25 °C was quite stable, with only insignificant alterations in the values of Dnm and percent of drug release at 60 min (Table 6). Samples stored at 4 °C, however, showed significant increase in Dnm, along with notable reduction in percent of cumulative drug release at 1 h. This could be attributed to the fact that the drug precipitation may have started at low temperatures from the supersaturated system [87]. On the other hand, the formulations stored at high temperature (45–50 °C) showed slight colour change after 3 months of storage, along with reduction in mean values of percent of drug release as well as in their consistency.
Conclusions
In a nutshell, Type III-SEDDS formulations of SFN were systematically developed and evaluated for their physicochemical characteristics, in situ perfusion and in vivo pharmacokinetics in Wistar rats, and potential efficacy using hepatocellular carcinoma cell lines. The prepared formulations were optimised using novel paradigms of QbD and were further supersaturated, using HPMC-K25 and PVP-E5 blend as the PPI. HPMC-PVP interactions were considered to be responsible for inhibiting the growth precipitation, thus imparting prolonged stability to the developed optimised formulations, also ratified by the high magnitudes of zeta potential. The optimised formulation demonstrated Fickian diffusion-based drug release kinetic behaviour as per the Korsmeyer-Peppas model. Drug permeation, as well as absorption parameters, was found to be significantly enhanced with the Type III-SEDDS vis-à-vis the pure SFN, during SPIP studies. Furthermore, the cytotoxicity SRB assay carried out using Hep G2 cells established markedly superior efficacy of the optimised Type III-SEDDS over the pure drug. Highly significant improvement in absorptive pharmacokinetic parameters (i.e., AUC, Ka, Cmax) is a definitive testimony to the enhancement of bioavailability of SFN, when incorporated in the Sat-Type III-SEDDS formulation. This would plausibly facilitate reduction of SFN dose and the side effects associated with the therapy thereof. Establishment of high level of point-to-point relationships between in vitro drug release and in vivo pharmacokinetic performances demonstrate the superior prognostic ability the in vitro drug release evaluation methodologies in predicting the corresponding in vivo pharmacokinetic performance. Evaluation of the stability studies of SFN formulations indicated that the ambient storage conditions (i.e., at 25 °C) were quite suitable for maintaining the robustness of the self-emulsifying formulation reporting inconsequential change in the various CQAs. These results corroborate the paramount significance of these Sat-Type III-SEDDS as an alternative to improve the efficacy of similar drugs, with high doses and low aqueous solubility, along with plausible augmentation in biopharmaceutical attributes, resulting eventually in enhanced anticancer therapeutic potential.
References
Ghouri YA, Mian I, Rowe JH. Review of hepatocellular carcinoma: epidemiology, etiology, and carcinogenesis. J Carcinog. 2017;16:1–13.
Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology. 2015;61:191–9.
Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125:1780–9.
PubChem D. National center for biotechnology information. Sorafenib tosylateCID=406563,. 2019.
de Rosamel L, Blanc J-F. Emerging tyrosine kinase inhibitors for the treatment of hepatocellular carcinoma. Expert Opin Emerg Drugs. 2017;22:175–90.
Yang S, Zhang B, Gong X, Wang T, Liu Y, Zhang N. In vivo biodistribution, biocompatibility, and efficacy of sorafenib-loaded lipid-based nanosuspensions evaluated experimentally in cancer. Int J Nanomedicine. 2016;11:2329–43.
Giglio V, Viale M, Bertone V, Maric I, Vaccarone R, Vecchio G. Cyclodextrin polymers as nanocarriers for sorafenib. Investig New Drugs. 2018;36:370–9.
Wang X-q, J-m F, Y-o L, Zhao B, Jia Z-r, Zhang Q. Bioavailability and pharmacokinetics of sorafenib suspension, nanoparticles and nanomatrix for oral administration to rat. Int J Pharm. 2011;419:339–46.
Kim DH, Kim M-D, Choi C-W, Chung C-W, Ha SH, Kim CH, et al. Antitumor activity of sorafenib-incorporated nanoparticles of dextran/poly (dl-lactide-co-glycolide) block copolymer. Nanoscale Res Lett. 2012;7:91–6.
Thapa RK, Choi JY, Poudel BK, Hiep TT, Pathak S, Gupta B, et al. Multilayer-coated liquid crystalline nanoparticles for effective sorafenib delivery to hepatocellular carcinoma. ACS Appl Mater Interfaces. 2015;7:20360–8.
Truong DH, Tran TH, Ramasamy T, Choi JY, Choi HG, Yong CS, et al. Preparation and characterization of solid dispersion using a novel amphiphilic copolymer to enhance dissolution and oral bioavailability of sorafenib. Powder Technol. 2015;283:260–5.
Zhang Z, Niu B, Chen J, He X, Bao X, Zhu J, et al. The use of lipid-coated nanodiamond to improve bioavailability and efficacy of sorafenib in resisting metastasis of gastric cancer. Biomaterials. 2014;35:4565–72.
Zhang H, Zhang F-M, Yan S-J. Preparation, in vitro release, and pharmacokinetics in rabbits of lyophilized injection of sorafenib solid lipid nanoparticles. Int J Nanomedicine. 2012;7:2901–10.
Grillone A, Riva ER, Mondini A, Forte C, Calucci L, Innocenti C, et al. Active targeting of sorafenib: preparation, characterization, and in vitro testing of drug loaded magnetic solid lipid nanoparticles. Adv Healthc Mater. 2015;4:1681–90.
Bondì ML, Botto C, Amore E, Emma MR, Augello G, Craparo EF, et al. Lipid nanocarriers containing sorafenib inhibit colonies formation in human hepatocarcinoma cells. Int J Pharm. 2015;493:75–85.
Fan WH, Wang XQ. Self-microemulsifying drug delivery system used to improve the oral absorption of sorafenib. Chinese J New Drugs. 2012;12:1397–400.
Pellosi DS, Moret F, Fraix A, Marino N, Maiolino S, Gaio E, et al. Pluronic® P123/F127 mixed micelles delivering sorafenib and its combination with verteporfin in cancer cells. Int J Nanomedicine. 2016;11:4479–94.
Dokania S, Joshi AK. Self-microemulsifying drug delivery system (SMEDDS)–challenges and road ahead. Drug Deliv. 2015;22:675–90.
Elgart A, Cherniakov I, Aldouby Y, Domb AJ, Hoffman A. Improved oral bioavailability of BCS class 2 compounds by self nano-emulsifying drug delivery systems (SNEDDS): the underlying mechanisms for amiodarone and talinolol. Pharm Res. 2013;30:3029–44.
Müllertz A, Ogbonna A, Ren S, Rades T. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol. 2010;62:1622–36.
Savla R, Browne J, Plassat V, Wasan KM, Wasan EK. Review and analysis of FDA approved drugs using lipid-based formulations. Drug Dev Ind Pharm. 2017;43:1743–58.
Pouton CW, Porter CJH. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60:625–37.
Gao P, Morozowich W. Development of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs. Expert Opin Drug Deliv. 2006;3:97–110.
Rehman FU, Shah KU, Shah SU, Khan IU, Khan GM, Khan A. From nanoemulsions to self-nanoemulsions, with recent advances in self-nanoemulsifying drug delivery systems (SNEDDS). Expert Opin Drug Deliv. 2017;14:1325–40.
Singh B, Dahiya M, Saharan V, Ahuja N. Optimizing drug delivery systems using systematic “design of experiments.” Part II: retrospect and prospects. Crit Rev Ther Drug Carrier Syst. 2005;22:215–94.
Beg S, Kaur R, Khurana RK, Rana V, Sharma T, Singh B. QbD-based development of cationic self-nanoemulsifying drug delivery systems of paclitaxel with improved biopharmaceutical attributes. AAPS PharmSciTech. 2019;20:118–31.
Bandyopadhyay S, Katare OP, Singh B. Development of optimized supersaturable self-nanoemulsifying systems of ezetimibe: effect of polymers and efflux transporters. Expert Opin Drug Deliv. 2014;11:479–92.
Bharate SS, Bharate SB, Bajaj AN. Interactions and incompatibilities of pharmaceutical excipients with active pharmaceutical ingredients: a comprehensive review. J Excipients Food Chemi. 2016;1:1–24.
Bilensoy E. Cationic nanoparticles for cancer therapy. Expert Opin Drug Deliv. 2010;7:795–809.
Garg RK, Singhvi I. Optimization techniques: an overview for formulation development. Asian J Pharm Res Vol. 2015;5:217–21.
Commission IP. Indian pharmacopoeia. 2014th ed. New Delhi: GOI; 2014. p. 2775.
Khurana RK, Gaspar BL, Welsby G, Katare OP, Singh KK, Singh B. Improving the biopharmaceutical attributes of mangiferin using vitamin E-TPGS co-loaded self-assembled phosholipidic nano-mixed micellar systems. Drug Deliv Trans Res. 2018;8:617–32.
Singh B, Kaur T, Singh S. Correction of raw dissolution data for loss of drug and volume during sampling. Indian J Pharm Sci. 1997;59:196–01.
Beg S, Jena SS, Patra CN, Rizwan M, Swain S, Sruti J, et al. Development of solid self-nanoemulsifying granules (SSNEGs) of ondansetron hydrochloride with enhanced bioavailability potential. Colloids Surf B Biointerfaces. 2013;101:414–23.
Gao P, Rush BD, Pfund WP, Huang T, Bauer JM, Morozowich W, et al. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J Pharm Sci. 2003;92:2386–98.
Jain A, Kaur R, Beg S, Kushwah V, Jain S, Singh B. Novel cationic supersaturable nanomicellar systems of raloxifene hydrochloride with enhanced biopharmaceutical attributes. Drug Deliv Transl Res. 2018;8:670–92.
Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–33.
Orellana EA, Kasinski AL. Sulforhodamine B (SRB) assay in cell culture to investigate cell proliferation. https://bio-protocol.org/e1984. 2016; Acess Date: Dec 2019.
Jain AK, Thanki K, Jain S. Solidified self-nanoemulsifying formulation for oral delivery of combinatorial therapeutic regimen: part I. Formulation development, statistical optimization, and in vitro characterization. Pharm Res. 2013;31:923–45.
Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27–31.
Khurana RK, Beg S, Burrow AJ, Vashishta RK, Katare OP, Kaur S, et al. Enhancing biopharmaceutical performance of an anticancer drug by long chain PUFA based self-nanoemulsifying lipidic nanomicellar systems. Eur J Pharm Biopharm. 2017;121:42–60.
Sharma T, Khurana RK, Jain A, Katare OP, Singh B. Development of a validated liquid chromatographic method for quantification of sorafenib tosylate in the presence of stress-induced degradation products and in biological matrix employing analytical quality by design approach. Biomed Chromatogr. 2018;32:e4169.
Bandyopadhyay S, Beg S, Katare OP, Sharma G, Singh B. QbD-oriented development of self-nanoemulsifying drug delivery systems (SNEDDS) of valsartan with improved biopharmaceutical performance. Curr Drug Deliv. 2015;12:544–63.
WHO. Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. In: preparations Ecosfp, editor. 2018.
Bandyopadhyay S, Katare OP, Singh B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf B Biointerfaces. 2012;100:50–61.
Beg S, Sandhu PS, Batra RS, Khurana RK, Singh B. QbD-based systematic development of novel optimized solid self-nanoemulsifying drug delivery systems (SNEDDS) of lovastatin with enhanced biopharmaceutical performance. Drug Deliv. 2014:1–20.
Swenson ES, Milisen WB, Curatolo W. Intestinal permeability enhancement: efficacy, acute local toxicity, and reversibility. Pharm Res. 1994;11:1132–42.
Wadhwa J, Asthana A, Shilakari G, Chopra AK, Singh R. Development and evaluation of nanoemulsifying preconcentrate of curcumin for colon delivery. Sci World J. 2015;2015:1–13.
Nazzal S, Smalyukh II, Lavrentovich OD, Khan MA. Preparation and in vitro characterization of a eutectic based semisolid self-nanoemulsified drug delivery system (SNEDDS) of ubiquinone: mechanism and progress of emulsion formation. Int J Pharm. 2002;235:247–65.
Avachat AM, Patel VG. Self nanoemulsifying drug delivery system of stabilized ellagic acid-phospholipid complex with improved dissolution and permeability. Saudi Pharm J. 2015;23:276–89.
Bansal T, Akhtar N, Jaggi M, Khar RK, Talegaonkar S. Novel formulation approaches for optimising delivery of anticancer drugs based on P-glycoprotein modulation. Drug Discov Today. 2009;14:1067–74.
Cui SX, Nie SF, Li L, Wang CG, Pan WS, Sun JP. Preparation and evaluation of self-microemulsifying drug delivery system containing vinpocetine. Drug Dev Ind Pharm. 2009;35:603–11.
Azeem A, Rizwan M, Ahmad FJ, Iqbal Z, Khar RK, Aqil M, et al. Nanoemulsion components screening and selection: a technical note. AAPS PharmSciTech. 2009;10:69–76.
Singh B, Kumar R, Ahuja N. Optimizing drug delivery systems using systematic “design of experiments.” Part I: fundamental aspects. Crit Rev Ther Drug Carrier Syst. 2005;22:27–105.
Singh B, Bhatowa R, Tripathi CB, Kapil R. Developing micro-/nanoparticulate drug delivery systems using “design of experiments”. Int J Pharm Invest. 2011;1:75–87.
El-Malah Y, Nazzal S, Khanfar NM. D-optimal mixture design: optimization of ternary matrix blends for controlled zero-order drug release from oral dosage forms. Drug Dev Ind Pharm. 2006;32:1207–18.
Eide I, Johnsen HG. Mixture design and multivariate analysis in mixture research. Environ Health Perspect. 1998;106:1373–6.
Singh B, Garg B, Chaturvedi SC, Arora S, Mandsaurwale R, Kapil R, et al. Formulation development of gastroretentive tablets of lamivudine using the floating-bioadhesive potential of optimized polymer blends. J Pharm Pharmacol. 2012;64:654–69.
Gupta B, Poudel BK, Pathak S, Tak JW, Lee HH, Jeong J-H, et al. Effects of formulation variables on the particle size and drug encapsulation of imatinib-loaded solid lipid nanoparticles. AAPS PharmSciTech. 2016;17:652–62.
Abdel-Salam FS, Elkheshen SA, Mahmoud AA, Ammar HO. Diflucortolone valerate loaded solid lipid nanoparticles as a semisolid topical delivery system. Bull Fac Pharm Cairo Univ. 2016;54:1–7.
Chen ZQ, Liu Y, Zhao JH, Wang L, Feng NP. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int J Nanomedicine. 2012;7:1115–25.
Wei Y, Ye X, Shang X, Peng X, Bao Q, Liu M, et al. Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Coll Surf A: Physicochem Eng Aspects. 2012;396:22–8.
Brouwers J, Brewster ME, Augustijns P. Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci. 2009;98:2549–72.
Douroumis D, Fahr A. Stable carbamazepine colloidal systems using the cosolvent technique. Eur J Pharm Sci. 2007;30:367–74.
Gao P, Akrami A, Alvarez F, Hu J, Li L, Ma C, et al. Characterization and optimization of AMG 517 supersaturatable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorption. J Pharm Sci. 2009;98:516–28.
Nan Z, Lijun G, Tao W, Dongqin Q. Evaluation of carbamazepine (CBZ) supersaturatable self-microemulsifying (S-SMEDDS) formulation in-vitro and in-vivo. Iran J Pharm Res. 2012;11:257–64.
Singh SK, Verma PR, B Razdan. Development and characterization of a lovastatin-loaded self-microemulsifying drug delivery system. 1;15(5):. Pharm Dev Technol. 2010;469–483.
Tripathi CB, Beg S, Kaur R, Shukla G, Bandopadhyay S, Singh B. Systematic development of optimized SNEDDS of artemether with improved biopharmaceutical and antimalarial potential. Drug Deliv. 2016;23:3209–23.
Baoding Hualian Chemical Co. L. Hydroxypropyl methylcellulose (HPMC). http://www.hualianhg.com/en/product. Acess Date: Dec 2019.
nanoComposix. Polyvinylpyrrolidone (PVP) Surface. https://nanocomposix.eu/pages/polyvinylpyrrolidone-pvp-surface. Access Date: Dec 2019.
Gao P, Guyton ME, Huang T, Bauer JM, Stefanski KJ, Lu Q. Enhanced oral bioavailability of a poorly water soluble drug PNU-91325 by supersaturatable formulations. Drug Dev Ind Pharm. 2004;30:221–9.
Thomas N, Holm R, Garmer M, Karlsson JJ, Mullertz A, Rades T. Supersaturated self-nanoemulsifying drug delivery systems (Super-SNEDDS) enhance the bioavailability of the poorly water-soluble drug simvastatin in dogs. AAPS J. 2012;15:219–27.
Warren DB, Benameur H, Porter CJ, Pouton CW. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: a mechanistic basis for utility. J Drug Target. 2010;18:704–31.
Raghavan SL, Trividic A, Davis AF, Hadgraft J. Effect of cellulose polymers on supersaturation and in vitro membrane transport of hydrocortisone acetate. Int J Pharm. 2000;193:231–7.
Thomas N, Holm R, Mullertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J Control Release. 2012;160:25–32.
Marques RC, Cole E, Kruep D, Gray V, Murachanian D, Brown WE, et al. Liquid-filled gelatin capsules. Pharmacopeial Forum. 2009;35:1029–41.
Singh B, Singh R, Bandyopadhyay S, Kapil R, Garg B. Optimized nanoemulsifying systems with enhanced bioavailability of carvedilol. Colloids Surf B Biointerfaces. 2013;101:465–74.
Keepers YP, Pizao PE, Peters GJ, van Ark-Otte J, Winograd B, Pinedo HM. Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur J Cancer Clin Oncol. 1991;27:897–900.
Dhingra N, Kar A, Sharma R. Inhibition of aromatase and cell proliferation of breast cancer and human placenta choriocarcinoma by Prunus persica extracts. IJPS. 2018;80:903–10.
Singh B, Bandopadhyay S, Kapil R, Singh R, Katare OP. Self-emulsifying drug delivery systems (SEDDS): formulation development, characterization, and applications. Crit Rev Ther Drug Carrier Syst. 2009;26:427–521.
Johnson DA, Amidon GL. Determination of intrinsic membrane transport parameters from perfused intestine experiments: a boundary layer approach to estimating the aqueous and unbiased membrane permeabilities. J Theor Biol. 1988;131:93–106.
Chen Y, Li G, Wu X, Chen Z, Hang J, Qin B, et al. Self-microemulsifying drug delivery system (SMEDDS) of vinpocetine: formulation development and in vivo assessment. Biol Pharm Bull. 2008;31:118–25.
Lin YM, Wu JY, Chen YC, Su YD, ., Ke WT, Ho HO, Sheu MT In situ formation of nanocrystals from a self-microemulsifying drug delivery system to enhance oral bioavailability of fenofibrate. Int J Nanomedicine 2011;6:2445–2457.
Liu Y, Zhang P, Feng N, Zhang X, Wu S, Zhao J. Optimization and in situ intestinal absorption of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2009;365:136–42.
Polli J. IVIVR versus IVIVC. 2000.
Bigora S, Piscitelli D, Dowell J, Butler J, Farrell C, Devane J, et al. Use of nonlinear mixed effects modelling in the development of in vitro-in vivo correlations. Adv Exp Med Biol. 1997;423:207–15.
Library C. Saturated solutions and solubility. https://chem.libretexts.org/Bookshelves/General_Chemistry. Acess date Dec 2019.
Acknowledgements
The authors gratefully appreciate the generosity of M/s Stat-Ease Inc., Minneapolis, USA, for providing a a perpetual licence of Design Expert 11.0 version to the corresponding author (B.S.) and its multiple single-user software licences to his research team ex-gratis as a mark of recognition for his exemplary contribution in the QbD-oriented drug delivery research work.
Funding
One of the authors (T.S.) acknowledges the Depatment of Science and Technology (DST), New Delhi, India, for providing financial grants to carry out the present research work as a Senior Research Fellow (IF:150273), under DST-INSPIRE fellowship scheme.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sharma, T., Jain, A., Kaur, R. et al. Supersaturated LFCS type III self-emulsifying delivery systems of sorafenib tosylate with improved biopharmaceutical performance: QbD-enabled development and evaluation. Drug Deliv. and Transl. Res. 10, 839–861 (2020). https://doi.org/10.1007/s13346-020-00772-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-020-00772-x