
Abstract
Phytic acid (PA) has momentous chemotherapeutic potential. Due to the chelate formation and rapid elimination, it is not popular in cancer treatment. The present work was inquested to develop a surface-modified nanoformulation of PA which prevents its speedy elimination and maximizes chemotherapeutic action. Chloroauric acid was reduced with pectin to produce pectin-gold nanoparticles (PGNPs). PGNPs were incubated with PA and jacalin for drug loading and surface modifications, respectively, to form PA-loaded jacalin-pectin-gold nanoparticles (PA-J-PGNPs). Formulation(s) were assessed for various pharmaceutical/pharmacological parameters. To validate the efficacy against colon carcinogenesis, formulation(s) were assessed in 1,2-dimethylhydrazine (DMH)-treated Wistar rats. DMH treatment distorted colonic architecture, oxidative, and hemodynamic parameters, which were favorably restored by PA-J-PGNP administration. To further confirm our deliberations, formulation(s) were also examined against DMH-altered metabolic changes and expression of markers pertaining to cellular proliferation, which was reinstated by PA-J-PGNPs. Our findings establish PA formulation(s) as a promising approach for suppression of colon carcinogenesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Colon cancer is escalating its threat due to high mortality rate. It is made known as the third most common cancer found in men and women, worldwide. Presently, it constitutes approximately 10% of the cancer burden. Colorectal cancer is likely to expand by 60% and > 2.2 million new cases have been predicted by 2030 [1].
To facilitate cancer prevention, efforts are being made amongst which, the use of naturally occurring constituents as chemopreventive and chemotherapeutic agents has seen a rise, due to their safety, availability, and general acceptance [2]. Phytic acid (PA) is one such naturally occurring carbohydrate, generally found in legumes, nuts, fruit, etc. Various researches proved that intake of PA containing legumes is linked with the lessening of incidences of colon cancers [3, 4]. However, this bioactive carbohydrate is largely underutilized due to its shortcoming of fast chelation and elimination from the body, within an hour of oral administration [5].
To eliminate its limitation and to maximize the chemotherapeutic activity, a surface-modified nanoformulation of PA may be developed. Gold nanoparticles have drawn attention due to their distinctive properties which make them valuable for application as controlled drug delivery agents, contrast agents, and biosensors for cancer detection and treatment [6,7,8]. Active targeting of a nanoparticulate system may be accomplished through surface functionalization of the nanoparticles with antibodies, proteins, peptides, and aptamers. Consequently, conjugation of biomolecules to the gold core may contribute to its additional interaction and advanced potential [9,10,11].
Atypical glycosylation is found in all types of human as well as experimental cancers. The associated glycosyl epitopes form antigens that can be exploited for binding/targeting [12]. Lectins are proteins of non-immune origin and have the capacity to spot aberrant glycosylation. Thus, lectins have been used by researchers to target glycosylation linked with metastasis [13, 14]. Jacalin is a lectin obtained from the seed of jackfruit [15]. This lectin has a superior capability of identifying tumor-linked antigenic disaccharides [16, 17] which get overexpressed in almost 85% of human and experimental carcinomas including colon and breast [18].
The innovative/pioneering strategy utilized in this paper is the loading of PA and jacalin on pectin-encrusted gold nanoparticles. Efforts were made to ensure enhancing residence time of PA and targeting the formulation in required site to achieve superior anticancer efficacy. Moreover, various in vivo evaluations have been carried out to examine the ability of the nanoconjugated system against 1,2-dimethylhydrazine (DMH)-induced neoplastic damage in colonic mucosa of Wistar rats.
Experimental section
Materials
Chloroauric acid was purchased from Loba Chemie, Mumbai. PA and jacalin were purchased from Sigma Co. (St. Louis, MO, USA). Pectin and DMH were purchased from Acros Organics (Thermo Fisher Scientific, NJ, USA). Deoxyribonucleotide triphosphate (dNTPs), RT-PCR kit, Taq DNA polymerase, and PCR primers were procured from Bangalore Genei, India. All other chemicals were procured from local commercial sources and were of analytical grade.
Preparation and characterization of PA-loaded jacalin-pectin-gold nanoparticles
A green method of reduction of chloroauric acid using pectin was utilized for production of pectin-gold nanoparticles (PGNPs). Pectin solution (0.5% w/v) was prepared in distilled water. To 9 ml of pectin solution, 1 ml of chloroauric acid solution (3 mM) was added. Concentration of chloroauric acid was optimized (data not shown). The mixture was allowed to react in a water bath at 55 °C till reddish purple color was obtained. The UV–visible spectroscopy (LT-2910, Labtronics, India) was used for examining the synthesis of PGNPs. The wavelength range for analysis was set from 400 to 600 nm. To this PGNP suspension, PA (1 mg/ml) and jacalin (for surface-modified batch) 1 mg/ml solution were added in 1:1 ratio and gently stirred at 4 °C for 24 h, protected from light. Prepared suspension was centrifuged (REMI CPR-24, Mumbai, India) at a temperature of 4 °C and 20,000 rpm for 30 min in order to separate supernatant (containing free PA and jacalin) and pellets of PA-loaded jacalin-pectin-gold nanoparticles (PA-J-PGNPs). The separated supernatant was taken to estimate free PA (at 520 nm) and jacalin (at 280 nm) by UV-Vis spectroscopy and percentage loading was calculated by using the Eq. 1 mentioned below:
The pellets were collected, washed, and redispersed with deionized water and stored for further characterization. The particle size and polydispersity index (PDI) of PA-J-PGNPs was evaluated through the standard method by using a particle size analyzer (Nanoplus, Zeta/nano particle analyzer, Japan). Fourier transform infrared (FTIR) spectroscopy was used to examine the chemical nature/identification of chemical groups involved in the reaction of pectin, chloroauric acid, jacalin, PA, and PA-J-PGNPs. The FTIR spectra of samples were recorded spectrometrically at room temperature by Nicolet 6700, Thermo Scientific, USA. Morphology and shape of prepared PGNPs and PA-J-PGNPs were observed via transmission electron microscopy (TEM) (H-7500, Hitachi Made, Japan) through the established procedure, at an accelerating voltage of 100 kV and magnification of ×30,000. The in vitro drug release studies of PA-PGNPs and PA-J-PGNPs were carried out with the pre-treated dialysis membrane (HiMedia Laboratories, Mumbai, India) of 12,000–14,000 kDa molecular weight cutoff, by previously reported methods [1,2,3].
In vivo studies
The study was approved/conducted as per guidelines of Institutional Animal Ethics Committee (approval no. SDCOP&VS/AH/CPCSEA/01/0028). All animals were handled as per norms of institutional animal ethics and care was taken that all guidelines be followed with a humane approach. Albino rats of Wistar strain, weighing 100–120 g, were used for the study. Animals were housed in polypropylene cages under well-ventilated, standard conditions of temperature (22 ± 2 °C) and a photoperiodic cycle of 12 h dark/light with access to the commercial pellet diet and water ad libitum. Animals were acclimatized for a week before the commencement of the study. Animals were divided into five groups containing eight animals each. DMH (20 mg/kg/week, subcutaneous) was administered, and all other treatment details are mentioned in Table 1, given for 6 weeks. At the end of the sixth week, animals were sacrificed with cervical dislocation. Before sacrifice, animals were examined for their electrocardiogram (ECG) and heart rate variability (HRV) paradigms and blood was collected through retro-orbital plexus. The blood serum, colon tissue, and colon content were collected and stored for further analysis.
Hemodynamic changes
Animals were anesthetized by a combination injection (intramuscular) of ketamine hydrochloride (100 mg/kg) and diazepam (5 mg/kg) and mounted on a wax tray. ECG signals were recorded by placing platinum hook electrodes on the skin of dorsal and ventral thorax. Electrodes were attached with channel power lab (ML-826) and bio-amplifier (ML-136) to change analogue signals to digital ones (Lab Chart Pro-8, AD Instruments, Australia). The recorded ECG signals were saved and analyzed offline. HRV analysis was made over different segments of ECG signals. Raw signals were scrutinized manually to make sure that all the R waves are perceived correctly and HR was computed by plotting the R wave number/time. Time and frequency domains of HRV were also determined offline.
Estimation of weight variation, pH, total acidity, and aberrant crypt foci count
Weight variation among the animals of each group was determined by using the following formula (Eq. 2). The collected colon content was evaluated for colonic pH with a pen-type digital pH meter (Hanna Instrument HI 98107) and total acidity calculated by the previously described procedure [4, 5]. Aberrant crypt foci (ACF) analysis was performed by an earlier reported method [6], under a light microscope with the magnification of ×40.
Antioxidant markers
The distal part of colon tissue (10% w/v) was homogenized with 0.15 M KCl and centrifuged at 4 °C and at an rpm of 10,000. The supernatant was analyzed for several antioxidant markers including protein carbonyl, thiobarbituric acid reactive substances (TBARS), glutathione (GSH), superoxide dismutase (SOD), and catalase by following the established procedure of our laboratory [7].
Morphological evaluation by scanning electron microscopy
The colon tissue was examined for its morphology by scanning electron microscopy (SEM). Tissue samples were fixed for 6 h with 2.5% glutaraldehyde at 4 °C and rinsed with 0.1 M phosphate buffer with three changes, each for 15 min at 4 °C. Osmium tetroxide (1% w/v) was used for fixation, at 4 °C for 2 h, and samples washed with 0.1 M phosphate buffer for three changes, each for 15 min at 4 °C. Tissue specimens were dehydrated through increasing acetone concentrations, viz. 30, 50, 70, 90, 95, and 100% v/v, for 30 min each at 4 °C. Air-dried samples were mounted on an aluminum stub and observed under SEM JEOL (JSM-6490LV, Japan) at ×500 magnification.
Histopathology
The samples were fixed overnight with paraformaldehyde and further treated overnight with 70% v/v isopropanol. Afterwards, specimens were exposed to isopropanol of variable concentrations (70, 90, and 100% v/v) and dehydrated with 100% v/v xylene. The prepared tissue specimens were embedded with paraffin wax to prepare blocks, sectioned with a microtome, and stained with hematoxylin and eosin. Stained sections were observed under a digital biological microscope (N120, BR Biochem Life Sciences, New Delhi, India) at ×40 magnification.
Western blotting
Total protein was extracted from the colon tissue by freeze-thaw method and quantified by Bradford protein assay. Tissue lysate was prepared with 20 mM Tris buffer (pH 7.5). Prepared tissue lysate (equivalent to 50 μg proteins) was resolved on 10% SDS-PAGE and transferred onto methanol-soaked PVDF membrane (Millipore Co., USA), using transfer buffer. Consequently, the membrane was blocked with blocking solution for 3 h and probed overnight with primary antibodies (dilution 1:1000) against PI3K (no. 4257), Akt (no. 4691) (Cell Signaling Technology, USA), and COX-2 (ALX-210-711-1) (Alexis Biochemicals, USA). Primary antibody incubation was followed by horseradish peroxidase-conjugated secondary antibody (Bangalore Genie) incubation. Antibody binding signals were visualized by using Chemiluminescence HRP detection system (Millipore) over Versa Doc (Bio-Rad) and simultaneously band strength was computed by Syngene gene tool. PVDF membranes were stripped and reprobed using β-actin antibody (4970) (Cell Signaling Technology Inc., USA) [8].
Reverse transcription polymerase chain reaction
Reverse transcription polymerase chain reaction (RT-PCR) (Applied Biosystems, Verity, USA) was used for scrutinizing colon tissue samples for their mRNA content. Total RNA content was extracted with TRIzol reagent (Invitrogen) according to manufacturer’s directives. DNA contaminations were removed by using DNaseI (Ambion Co.). c-DNA synthesis was made by using c-DNA preparation kit and taking RNA equivalent volume to 2 ng. c-DNA was used for quantification of particular mRNA through primers (MWG Bio Tech, Germany) mentioned in Table 2. The reaction mixture (20 μl) was prepared by using 100 ng of c-DNA, MgCl2 (1.5 mM), dNTPs (1.5 mM), 10 pM each primer, and 1 unit Ampli Taq DNA polymerase enzyme. The product was amplified via thermal cycles (denaturation at 94 °C for 5 min, 95 °C for 60 s, annealed at 72 °C for 60 s) ×35, and final extension was for 4 min at 72 °C. Agarose gel (1.5%) containing ethidium bromide was used to resolve/visualize prepared PCR products. Quantification was made utilizing gene tool Syngene software. β-actin was used as internal control [9].
1H NMR spectroscopic analysis of serum samples
Detailed procedures of serum sample preparation, NMR measurements, spectral assignment, and multivariate statistical analysis have been mentioned in the supplementary section.
Results
Preparation and in vitro characterization of PA-J-PGNPs
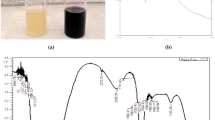
PA-J-PGNPs were prepared through reduction of chloroauric acid using pectin, loaded with PA, and surface-modified with jacalin. Biosynthesis of PGNPs showed color change from pale yellow to reddish purple. The absorption maxima range was evident between 520 and 530 nm, through UV spectroscopy. Percentage loading of PA and jacalin onto prepared PA-J-PGNPs was found to be 76.1 ± 2.69 and 68.3 ± 5.9%, respectively. PA-J-PGNPs showed particle size of 283 ± 4.94 nm and PDI value of 0.32 ± 0.08. FTIR spectroscopy of pectin, chloroauric acid, PA, jacalin, and PA-J-PGNPs has been shown in Fig. 1. All excipients exhibited their characteristic peaks. In the case of pectin, the peak observed at 2935.8 and 1746.8 cm−1 corresponds to C–H and C=O bond (indicating –COCH3 groups) vibrations [10]. For chloroauric acid, peaks at 1631.4 and 3389.8 cm−1 correspond to the C–O and –OH stretches, respectively [11]. The peaks near 1654.4 cm−1 show the main vibrational bands of amide groups of jacalin [12] and the bands near 1378.8 cm−1 show vibrational mode of the COOH and C–O groups of aspartic acid, glutamic acid-like amino acids, present in jacalin. The peak at 3430.3 cm−1 in case of PA relates to OH stretching [13]. PA-J-PGNPs showed the shift in peaks of hydroxyl and carbonyl and also displayed all the characteristic peaks of PA and jacalin. In vitro drug release studies of PA-PGNPs as well as PA-J-PGNPs displayed similar pattern of drug release from the formulations. Results showed that release was significantly low in acidic pH. However, it increased after changing the pH of media to 6.8 and was found to be even higher in media containing cecal content (pH 7.4) (Fig. 2a). Morphological evaluation of PGNPs through TEM revealed spherical shape, while PA-J-PGNPs displayed a corona surrounding the surface (Fig. 2b).

FTIR spectrums of pectin, chloroauric acid, jacalin, PA, and PA-J-PGNPs

a Percent of PA released as a function of time (h) from PA-PGNPs and PA-J-PGNPs. b Morphological evaluation of PGNPs (inset) and PA-J-PGNPs through TEM analysis
Hemodynamic changes
The PA, PA-PGNPs, and PA-J-PGNPs affected various parameters of ECG when administered to albino Wistar rats (Fig. 3). Heart rate, P duration, and the QRS complex were found higher in case of toxic control which was normalized after PA-J-PGNP treatment. The lower frequency (LF) was reduced in toxic control, whereas it increased significantly in the treatment groups. An analogous pattern was found in higher frequency variables (HF) (Tables 3 and 4).

Representative ECG recordings of normal control (NC), toxic control (TC), standard (PA), and PA-PGNP- and PA-J-PGNP-treated animals
Weight variation, pH, total acidity, and ACF count
The toxic group showed weight loss, decrease in pH along with an increase in total acidity, and the formation of aberrant crypts in comparison to control. Concomitant treatment with PA-PGNPs and PA-J-PGNPs favorably regulated the weight and pH in the treated animals. The total acidity along with ACF was also regulated near to normal after PA-J-PGNP treatment (Table 5).
Antioxidant markers
DMH application significantly increased protein carbonyl and malondialdehyde (MDA) levels when compared with the control group. PA, PA-PGNPs, and PA-J-PGNPs significantly (< 0.001) lowered the level of both protein carbonyl as well as MDA. The downregulated GSH, SOD, and catalase in the toxic control group were found to be significantly increased in PA-PGNPs and PA-J-PGNPs treated groups (Table 6).
Morphological and histopathological evaluation
Toxic control animals showed the highest ACF count, abrupt crypts and loss of goblet cells, crypts, and distorted lamina propria. Treatment with PA, PA-PGNPs, and PA-J-PGNPs tried to restore normal structure of colonic mucosa. Animals treated with PA-PGNPs showed maximum restoration towards normal (Fig. 4).

a−e Methylene blue-stained colon tissues. a Normal control, b toxic control, c standard, d PA-PGNPs and e PA-J-PGNPs,  denotes aberrant crypts. f–j SEM (×500): f normal control, g toxic control, h standard, i PA-PGNPs and J- PA-J-PGNPs,
denotes aberrant crypts. f–j SEM (×500): f normal control, g toxic control, h standard, i PA-PGNPs and J- PA-J-PGNPs,  denotes aberrant crypts/ neoplastic lesions. k–o Histopathological evaluation: k normal control, l toxic control, m standard, n PA-PGNPs and O- PA-J-PGNPs,
denotes aberrant crypts/ neoplastic lesions. k–o Histopathological evaluation: k normal control, l toxic control, m standard, n PA-PGNPs and O- PA-J-PGNPs,  denotes aberration in colonic mucosa
denotes aberration in colonic mucosa
COX-2
Evaluation of altered gene expressions
Change in expressions of PI3K, Akt, and COX-2 genes was examined through western blot and RT-PCR for protein and mRNA levels, respectively. Normal control was considered as the baseline (with no up/ down regulation) and all comparisons were made with respect to this. The expressions of PI3K, Akt, and COX-2 proteins, as well as mRNA, increased markedly after DMH administration in the toxic control group. Treatment with PA, PA-PGNPs, and PA-J-PGNPs helped to restore the expressions of aforesaid genes. PA-J-PGNPs revealed thoroughgoing restoration of protein and mRNA levels (Fig. 5).

Effect of PA, PA-PGNPs, and PA-J-PGNPs on DMH-induced dysregulation of PI3K, Akt, and COX-2. Quantitative analysis at protein level (a and c) and at mRNA level (b and d). †p < 0.05, ††p < 0.01, †††p < 0.001 (with respect to normal control) and *p < 0.05, **p < 0.01, ***p < 0.001 (with respect to toxic control)
Serum metabolic profiling using 1H NMR spectroscopy
The representative 1D 1H CPMG NMR spectra of rat serum samples obtained from different groups with the assigned resonances of relevant metabolites are shown in Fig. 6. The major metabolites in the spectra were identified according to data reported in the literature and the Human Metabolome Database. The NMR spectra showed signals from various metabolites including lipids/lipoproteins, polyunsaturated fatty acids (PUFAs), and amino acids (e.g., alanine, valine, lysine, leucine, isoleucine, phenylalanine, histidine, tyrosine, glutamine, and glutamate). Other identified metabolites were glucose, choline, creatine, creatinine, pyruvate, acetate, citrate, ethanol, methanol, and lactate. Multivariate data analysis was carried out to find the DMH-induced metabolic alterations and further to reveal the effect of prepared formulations on these alterations (Fig. 7).

Stack plot of representative 1D 800-MHz 1H CPMG NMR spectra (δ 0.7–4.6 and δ 5.2–8.6) of rat serum of toxic control (TC), PA-J-PGNPs (TII), PA-PGNPs (TI), PA (Std), and normal control (NC). The region of δ 5.2–8.6 (b) is magnified eight times compared with the corresponding region of δ 0.7–4.6 (a) for the purpose of clarity. LDL/VLDL, low/very-low-density lipoproteins; PUFA, polyunsaturated fatty acids; Ile, isoleucine; Leu, leucine; Val, valine; Pyr, pyruvate; Ch, choline; GPC, glycerophosphocholine. Glucose resonances have been indicated using symbol asterisk “*”; NAG, N-acetyl glycoprotein; OAG, O-acetyl glycoprotein; DMA, dimethylamine

Representative box-cum-whisker plots showing quantitative variations of relative signal integrals for serum metabolites. Boxes denote interquartile ranges, horizontal lines inside the box denote the median, and bottom and top boundaries of boxes are 25th and 75th percentiles, respectively. Lower and upper whiskers are 5th and 95th percentiles, respectively. toxic control (TC), Std (PA), T1 (PA-PGNPs), T2 (PA-J-PGNPs), and normal control (NC)
Metabolic changes
Unsupervised Principal Component Analysis (PCA) score plot was constructed for an initial overview of the data sets which exhibited a clear trend of clustering in separate groups and no outlier sample was detected. To obtain a satisfactory classification and select metabolite markers, pair-wise Partial Least Squares Discriminant Analysis (PLS-DA) analysis was further performed on NMR data matrices. The combined PLS-DA score plot for all groups (Fig. 8) and pair-wise PLS-DA score plots (supplementary Fig. S1(A)) demonstrated that the DMH-treated rats are well separated from normal control (NC) group with a significantly higher quality of fit and predictability (R2 = 0.98, Q2 = 0.94), suggesting significant metabolic changes in DMH-treated rats as compared to NC. The metabolites responsible for the discrimination of the two groups are shown in the loading plot (supplementary Fig. S1(B)) in the first latent variable of PLS-DA. The loading plots were color-coded according to the absolute value of correlation coefficients (|r|), where a hot-colored signal (red) indicated a more significant contribution to class separation than a cold-colored one (blue). The significantly distinguishing metabolites were determined according to values of variable importance on projection (VIP) score, > 1 and p < 0.05. A number of metabolites differed in the DMH-treated group when compared with the normal control group. It was found that DMH-treated rats had higher levels of lipids, VLVL/LDL lipoprotein, lactate, polyunsaturated fatty acids, and pyruvate in their sera, whereas they had decreased levels of glucose and several amino acids when compared with the normal control group (Fig. 7).

a PLS-DA score plot derived from 1D CPMG spectra of toxic control, PA, PA-PGNPs, PA-J-PGNPs, and normal control groups. b Variable importance in projection (VIP) plot demonstrating the metabolites responsible for the separation of various groups in the corresponding score plot shown in (a)
The PLS-DA score plot shown in Fig. 8 depicts a clear differentiation between DMH-treated rats and the group of rats receiving treatment with PA and formulation(s). Further, it was found that some of the metabolic alterations which were observed in DMH-treated group got ameliorated after PA-PGNP and PA-J-PGNP treatment. Figure 7 shows representative box-cum-whisker plots of the normalized integral area of a few metabolites which were modulated due to the action of PA-PGNP and PA-J-PGNP treatment.
Discussions
PA-J-PGNPs were produced through a green method by the one-pot reduction of chloroauric acid using pectin as reducing/capping agent and jacalin for surface modification. The use of pectin as a reducing or capping agent was motivated by virtue of it being a natural substance. Further, pectin is nontoxic and has the presence of carboxylate units which facilitate activation through jacalin. Jacalin has the ability to recognize and target tumor-associated disaccharides which are usually expressed highly in almost all tumor cells. It bears functional groups like hydroxyl and amine which facilitated pectin activation [12, 14, 15]. In this study, nanoparticles were selected because the drug was to be attached on to the pectin with gold as a core, jacalin, and PA as the corona. Pectin facilitated interaction between the drug and gold core. For this purpose, small nanoparticles provide a much larger surface area, whereas if bigger particles were utilized as the core, they may provide a higher volume but lower surface area and hence gold nanoparticles proved to be a better choice. The color change from pale yellow to reddish purple confirmed the formation of PGNPs. The surface plasmon resonance (SPR) of Au+ is responsible for the color change. When chloroauric acid is added to the pectin solution, –OH group of pectin would be bind to gold ions, probably through electron-rich oxygen atoms of –OH groups, as is expected to show interaction in an alkaline environment with electropositive metal cations. This would lead to the reduction of Au+ ion to elemental gold. The possible attachment mechanism of pectin with Au+ ions, jacalin, and PA is also represented in the graphical abstract. Additionally, pectin may also be responsible for the prevention of aggregation through the layer formed on the gold nanoparticles, thereby acting as a reducing as well as capping agent [12, 14, 16]. The UV–Vis spectra of formulation exhibited absorption maxima at 520–530 nm which indicated the formation of GNPs [16]. The prepared metal particles were found to be homogenous with PDI values ≤ 0.5. FTIR analysis of PA-J-PGNPs showed a shift in peaks of amines (1655.9, 1515.0, and 1203.2 cm−1) and hydroxyl (3417.5 cm−1) of jacalin and PA, respectively, which may be attributed to their involvement in complex formation. Furthermore, the spectra of PA-J-PGNPs displayed all the characteristic peaks of PA and jacalin indicating corona of PA and jacalin. The absence of the peak of gold indicated complete coverage by this corona. Lesser drug release at acidic pH and higher in alkaline pH may be attributed to the fact that pectin contains carboxyl groups in its structure which ionize in neutral to alkaline pH. Further, pectin degrades in the colonic microflora [17, 18]. Due to the ionization/enzymatic degradation of pectin, the corona containing jacalin and PA would breakdown thereby releasing the drug. Morphological analysis of PA-J-PGNPs through TEM showed a corona surrounding the particulate surface which may be attributed to the formation of a layer of jacalin and PA around the PGNPs.
Depreciated ECG and HRV are related to cancer patients. Higher HRV is related to the low-level tumor markers and lower HRV is associated with autonomic dysfunction in advanced cases of cancer. An analogous pattern of reduced HRV was reflected in the DMH-treated animals, in time as well as frequency domain. Treatment with PA, PA-PGNPs, and PA-J-PGNPs revealed significant restoration of time and frequency domain paradigms towards normal values (Table 4) which may be ascribed to the action of PA.
Oxygen radicals augment all stages of the multistep process of cancer progression [19]. There is a proven link between colon cancer progression and serious oxidative stress. Earlier studies have reported increased levels of lipid peroxide and protein carbonyl and reduced SOD, catalase, and GSH in clinical stage IV of colon cancer [20]. The present study also confirmed the decrease in SOD, catalase, and GSH levels in colon tissue of DMH-treated animals which may be attributed to the induction of colon cancer [21, 22]. PA-J-PGNPs showed maximum restoration (Table 6) of these biochemical markers towards normal validating the maximized efficacy of PA in the form of PA-J-PGNPs.
Cell deterioration and improper proliferation lead to abnormalities in the architecture of crypts known as aberrant crypts, ultimately causing invasive cancers [23, 24]. The maximum restoration of ACF count by PA-J-PGNPs (Fig. 4) may be attributed to the targeted delivery into the affected cells due to the presence of jacalin. The histopathological analysis of normal colon tissue portrays three layers including mucosa, submucosa, and muscularis. The intact architecture of colonic mucosa was seen in the normal control group describing all the three layers, in contrast to the toxic group animals which showed abrupt/impaired arrangement of cells (Fig. 4l). The PA-J-PGNP-treated groups showed maximum similarity with the normal control group which could again be a call out of targeting the required site because of jacalin.
Previous studies have reported the involvement of PA in the inhibition of PI3K/Akt [25, 26] and β-catenin and COX-2 [27] pathways. Consequently, authors considered it significant to analyze the efficacy of PA formulation(s) against DMH-induced dysregulation of PI3K, Akt, and COX-2. PI3K is a lipid kinase and a second messenger which translocates Akt to the plasma membrane, where it phosphorylates to activate PDK1 and PDK2. Alterations in the PI3K-Akt signaling pathway have already been reported frequently in human cancers. The developments, as well as the progression of cancers, are the result of interruptions in the equilibrium of cell proliferation and cell apoptosis. Eventually, PI3K-Akt signaling is linked with both of these events and imparts a vital role in tumor growth as well as tumor treatment [26, 28]. Application of DMH uncovered significant upregulation of PI3K and Akt at protein and mRNA levels, and treatment with PA-PGNPs and PA-J-PGNPs conveyed favorable restoration. To further boost up the deliberations, the authors examined the expression of COX-2. COX-2 is a well-documented inflammatory marker involved in tumor progression [29]. DMH application incited a significant upregulation of the COX-2 protein and mRNA expression, in toxic control. Concomitant application of the PA-PGNPs and PA-J-PGNPs showed significant restoration (Fig. 5) which validate the inhibitory action of PA for aforesaid enzymes.
1H NMR-based metabolic profiling was performed to explore the biochemical changes associated with colon cancer and to further see how these changes get modulated in the rats receiving the treatment with PA formulation(s). Increased levels of lipoproteins and PUFAs in DMH-treated animals imply rapid cellular regeneration and the high cancer cell lipid demand for cell membrane biosynthesis [30]. The lower concentration of choline and its derivatives can be correlated with the rapid cellular regeneration in tumors. Serum creatine which was reduced significantly in DMH-treated rats can be associated with the increase in energy consumption due to rapid cell division. Increased levels of amino acids in DMH rats suggest an increased catabolism due to the rapid cell proliferation [31, 32]. Further, a decreased level of glucose indicates the increase in glucose metabolism through glycolysis to generate ATP instead of oxidative phosphorylation which enhances glucose uptake in tumor cells to meet the energy requirement of quick proliferation. A higher glycolytic rate in tumor cells also results in the generation of a huge quantity of lactate from pyruvate and thus supports our findings of increased serum levels of lactate in toxic control rats. The observed decrease in the levels of glucose and an increase in lactate levels are consistent with previous results reported in studies dealing with human rectal cancer [33]. Dimethylamine, the product of choline metabolism, was also found to be altered in the DMH-treated rats indicating the disturbance of choline metabolism. Further, increased serum levels of N- and O-acetyl glycoproteins reflect a nonspecific inflammatory response to DMH-treated rats. The disturbed metabolic pathways discussed above lead us to infer the acceleration of cell proliferation in the process of tumor formation, hyperplasia in colonic mucosa, and an increase in inflammation in DMH-treated rats. Nearly most metabolic changes in DMH-treated animals were retuned back towards normal after PA formulation(s) treatment, suggesting that the formulation(s) has the potential to normalize the DMH-altered metabolic changes.
Conclusions
Conclusively, PA-J-PGNPs displayed a significant effect against progression of colon carcinogenesis via positively modulating the physiological markers, oxidative stress, inflammatory markers, and hemodynamic changes. This effect was attributed to the targeting property of nanoparticles to the desired site due to surface modification by jacalin. Detailed in vitro study at the molecular level has been performed to perceive the effect and exact localization/targeting potential of PA-J-PGNPs in human colorectal carcinoma cell lines and is being communicated as a separate study. Simple PA-PGNPs also revealed the analogous pattern of results but exhibited lesser effect, which may be due to the fact that pectin carried the intact nanoparticle to the colon and drug released in the vicinity of the desired site but could not be targeted as it did not contain surface functionalization with the targeting agent. PA (taken as standard) showed minimal restoration of abovementioned changes which can be justified due to the fact that free drug gets exposed and metabolizes to form chelates (property of PA) [9] and get absorbed and eliminated within an hour of administration. All in all, in vivo results confirmed that PA formulation(s) could constitute a promising tool for the anticipation of DMH-induced neoplastic damage, dysregulation of proliferation markers, and metabolic changes. Our previous study and current findings underlined the potential role of PA formulations in the suppression of carcinogenesis and open new vistas of research towards exploring its effect against various carcinomas.
References
Singh S, Kotla NG, Tomar S, Maddiboyina B, Webster TJ, Sharma D, et al. A nanomedicine-promising approach to provide an appropriate colon-targeted drug delivery system for 5-fluorouracil. Int J Nanomedicine. 2015;10:7175.
Liu J, Zhang L, Jia Y, Hu W, Zhang J, Jiang H. 558301. Preparation and evaluation of pectin-based colon-specific pulsatile capsule in vitro and in vivo. Arch Pharm Res. 2012;35(11):1927–34.
Izadi Z, Divsalar A, Saboury AA, Sawyer L. β-lactoglobulin–pectin nanoparticle-based oral drug delivery system for potential treatment of colon cancer. Chem Biol Drug Des. 2016;88(2):209–16.
Shukla K, Raj P, Kumar A, Kumar M, Kaithwas G. Effect of monotherapy and combination therapy of pantoprazole and aprepitant in gastric esophageal reflux disease in albino rats. Sci World J. 2014;2014:1–7.
Khinchi P, Saha S, Saraf SA, Kaithwas G. Combination therapy of gamma-aminobutyric acid derivative promotes proton pump inhibitor based healing of reflux esophagitis in animal model. Pharmacol Rep. 2014;66(1):165–8.
Bird RP. Observation and quantification of aberrant crypts in the murine colon treated with a colon carcinogen: preliminary findings. Cancer Lett. 1987;37(2):147–51.
Kumar S, Singh M, Rawat JK, Gautam S, Saraf SA, Kaithwas G. Effect of rutin against gastric esophageal reflux in experimental animals. Toxicol Mech Methods. 2014;24(9):666–71.
Singh M, Kanoujia J, Parashar P, Arya M, Tripathi CB, Sinha V, et al. Augmented bioavailability of felodipine through an α-linolenic acid-based microemulsion. Drug Deliv Transl Res .2017;1–22.
Arya M, Tiwari P, Tripathi CB, Parashar P, Singh M, Sinha P, et al. Colloidal vesicular system of phytic acid to counteract DMBA induced dysregulation of markers pertaining to cellular proliferation/differentiation and inflammation of epidermal layer in mouse model. Mol Pharm. 2017;14(3):928–39.
Guolin H, Jeffrey S, Kai Z, Xiaolan H. Application of ionic liquids in the microwave-assisted extraction of pectin from lemon peels. J Anal Methods Chem. 2012;2012:1–8.
Koperuncholan M. Bioreduction of chloroauric acid (HAuCl4) for the synthesis of gold nanoparticles (GNPs): a special empathies of pharmacological activity. Int J Phytopharm. 2015;5(4):72–80.
Marangoni VS, Paino IM, Zucolotto V. Synthesis and characterization of jacalin-gold nanoparticles conjugates as specific markers for cancer cells. Colloids Surf B Biointerfaces. 2013;112:380–6.
Daneluti ALM, Matos JR. Study of thermal behavior of phytic acid. Braz J Pharm Sci. 2013;49(2):275–83.
Verma J, Kanoujia J, Parashar P, Tripathi CB, Saraf SA. Wound healing applications of sericin/chitosan-capped silver nanoparticles incorporated hydrogel. Drug Deliv Transl Res. 2017;7(1):77–88.
Madhusudhan A, Reddy GB, Venkatesham M, Veerabhadram G, Kumar DA, Natarajan S, et al. Efficient pH dependent drug delivery to target cancer cells by gold nanoparticles capped with carboxymethyl chitosan. Int J Mol Sci. 2014;15(5):8216–34.
Suganya KU, Govindaraju K, Kumar VG, Karthick V, Parthasarathy K. Pectin mediated gold nanoparticles induces apoptosis in mammary adenocarcinoma cell lines. Int J Biol Macromol. 2016;93:1030–40.
Liu L, Fishman ML, Kost J, Hicks KB. Pectin-based systems for colon-specific drug delivery via oral route. Biomaterials. 2003;24(19):3333–43.
Dongowski G, Lorenz A, Anger H. Degradation of pectins with different degrees of esterification by Bacteroides thetaiotaomicron isolated from human gut flora. Appl Environ Microbiol. 2000;66(4):1321–7.
Skrzydlewska E, Stankiewicz A, Sulkowska M, Sulkowski S, Kasacka I. Antioxidant status and lipid peroxidation in colorectal cancer. J Toxicol Environ Health A. 2001;64(3):213–22.
Skrzydlewska E, Sulkowski S, Koda M, Zalewski B, Kanczuga-Koda L, Sulkowska M. Lipid peroxidation and antioxidant status in colorectal cancer. World J Gastroenterol. 2005;11(3):403–6.
Raj P, Singh M, Rawat JK, Gautam S, Saraf SA, Kaithwas G. Effect of enteral administration of α-linolenic acid and linoleic acid against methotrexate induced intestinal toxicity in albino rats. RSC Adv. 2014;4(104):60397–403.
Kaithwas G, Majumdar DK. In vitro antioxidant and in vivo antidiabetic, antihyperlipidemic activity of linseed oil against streptozotocin-induced toxicity in albino rats. Eur J Lipid Sci Technol. 2012;114(11):1237–45.
Khare S, Chaudhary K, Bissonnette M, Carroll R. Aberrant crypt foci in colon cancer epidemiology. Methods Mol Biol. 2009;472:373–86.
Wargovich MJ, Brown VR, Morris J. Aberrant crypt foci: the case for inclusion as a biomarker for colon cancer. Cancers (Basel). 2010;2(3):1705–16.
Vucenik I, Stains J. Cancer preventive and therapeutic properties of PA: efficacy and mechanisms. Period Biol. 2010;112(4):451–8.
Liu G, Song Y, Cui L, Wen Z, Lu X. Phytic acid suppresses growth and induces apoptosis in HT-29 colorectal cancer cells in culture: PI3K/Akt pathway as a potential target. Int J Clin Exp Pathol. 2015;8(2):1402–10.
Shafie NH, Mohd Esa N, Ithnin H, Md Akim A, Saad N, Pandurangan AK. Preventive phytic acid extracted from rice bran inhibits colorectal cancer through involvement of Wnt/β-catenin and COX-2 pathways. Biomed Res Int. 2013;2013:1–10.
Osaki M, Ma O, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9(6):667–76.
Edelman MJ, Hodgson L, Wang X, Kratzke RA, Vokes EE. Cyclooxygenase-2 (COX-2) as a predictive marker for the use of COX-2 inhibitors in advanced non-small-cell lung cancer. J Clin Oncol. 2012;30(16):2019–20.
Chan ECY, Koh PK, Mal M, Cheah PY, Eu KW, Backshall A, et al. Metabolic profiling of human colorectal cancer using high-resolution magic angle spinning nuclear magnetic resonance (HR-MAS NMR) spectroscopy and gas chromatography mass spectrometry (GC/MS). J Proteome Res. 2008;8(1):352–61.
Leichtle AB, Nuoffer J-M, Ceglarek U, Kase J, Conrad T, Witzigmann H, et al. Serum amino acid profiles and their alterations in colorectal cancer. Metabolomics. 2012;8(4):643–53.
Koh TJ, Dockray GJ, Varro A, Cahill RJ, Dangler CA, Fox JG, et al. Overexpression of glycine-extended gastrin in transgenic mice results in increased colonic proliferation. J Clin Invest. 1999;103(8):1119–26.
Wang L, Chen J, Chen L, Deng P, Xiang P, Li M, et al. 1 H-NMR based metabonomic profiling of human esophageal cancer tissue. Mol Cancer. 2013;12(1):25.
Acknowledgments
The authors are grateful to Dr. Dinesh Kumar and Mr. Amit Kumar, CBMR, SGPGIMS Campus Lucknow for their help in NMR data acquisition. The University Grants Commission, Government of India granted a senior research fellowship to MA (F1-17.1/2014-15/RGNF-2014-15-SC-UTT-70422) and PP (F1-17.1/2014-15/RGNF-2014-15-SC-UTT-63524), Department of Science and Technology (DST), Government of India, DST INSPIRE Faculty Award (Ref. No. DST/Inspire Faculty Award 2014/LSBM-120) to AG, and ICMR (Ref. no.74/1/2017-Pers (EMS)) to KPG and SAS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 260 kb)
Rights and permissions
About this article

Cite this article
Arya, M., Singh, P., Tripathi, C.B. et al. Pectin-encrusted gold nanocomposites containing phytic acid and jacalin: 1,2-dimethylhydrazine-induced colon carcinogenesis in Wistar rats, PI3K/Akt, COX-2, and serum metabolomics as potential targets. Drug Deliv. and Transl. Res. 9, 53–65 (2019). https://doi.org/10.1007/s13346-018-00605-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-018-00605-y