
Abstract
Transdermal drug delivery is a useful route of administration that avoids first-pass metabolism and more invasive delivery options. However, many drugs require enhancers to enable sufficient drug absorption to reach therapeutic effect. Alpha-tocopheryl phosphate (TP) and di-alpha-tocopheryl phosphate (T2P) are two phosphorylated forms of vitamin E which form tocopheryl phosphate mixture (TPM) when combined, and have been proposed to enhance the dermal and transdermal delivery of actives of interest. Here, we report the physicochemical characteristics and morphological properties of TPM formulations, including particle size, deformability and morphology, and its ability to facilitate the transport of carnosine, vitamin D3, CoEnzyme Q10 and caffeine into, and across, the skin. Results demonstrate that TPM self-assembles to form vesicular structures in hydroethanolic solutions ranging in mean size from 101 to 162 nM depending on the amount of TPM and ethanol present in the formulation. The ratio of TP to T2P in TPM formulations altered vesicle size and elasticity, with vesicles high in TP found to be more deformable than those rich in T2P. TPM produced a significant (p < 0.05) 2.4–3.4-fold increase in the absorption of carnosine, vitamin D3, CoEnzyme Q10 and caffeine into, or through, the skin. The TPM delivery platform was able to deliver a diverse range of actives with differing size and solubility profiles and therefore has significant potential to expand the number and types of drugs available for topical application and transdermal delivery.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transdermal drug delivery (TDD) systems facilitate the dermal or topical route for drug administration, both for local and systemic drug delivery [1]. The transdermal route offers numerous advantages compared with oral drug administration, including avoidance of first-pass effect, ease of use, better patient compliance, continuous drug delivery and decreased side effects [2–6]. However, compounds must exhibit the correct physicochemical properties to enable their effective delivery across the skin [3, 4, 7]. Transdermal delivery is limited by the barrier function of the stratum corneum, which limits successful drug candidates to those that are small in size, of appropriate lipophilicity and of high potency [2, 6, 7]. Encapsulation of the drugs in lipid-based vesicular structures (liposomes) is one approach to overcoming this limitation. Liposomes are artificial vesicles that enclose an aqueous volume within one or more phospholipid bilayers [1, 6]. When these lipids are exposed to an aqueous environment, the hydrophobic chains associate to minimise contact with the aqueous environment, resulting in the formation of closed bilayers, encapsulating the drug passively inside [6]. It is hypothesised that liposomes interact with the lipid bilayers of the stratum corneum, enabling penetration past epidermal barriers and release of the enclosed drug [3, 4, 6].
While numerous studies have reported that ‘first-generation’ liposomal vesicles were unable to deeply penetrate the skin [1], intensive research has led to the development of a new class of highly deformable, elastic (or ultraflexible) liposomes. These ‘second-generation’ liposomes are formed by (i) incorporating surfactant molecules which act as ‘edge activators’ (typically ethanol) to the bilayers, destabilising the lipid bilayers of the vesicles and increasing deformability, or (ii) by mixing with certain hydrophilic solutes of high mobility [1, 4, 8–10]. While some studies have reported that these deformable liposomes improve the penetration of a range of molecules, enabling drug delivery at therapeutic concentrations [11–16], reports on their efficiency have been inconsistent to date, with several studies reporting negative or inconclusive results [17–22]. The differences across these studies can, at least in part, be explained by the fact that vesicles with different lipid compositions and physicochemical characteristics were used in different studies [4]. Nevertheless, the postulated interaction of liposomes with lipid structures in the stratum corneum appears to depend greatly on the composition of the liposomes and as such, identifying lipids that optimally facilitate such interactions is one route to improving transdermal drug delivery for drugs that require a liposomal carrier system.
Vitamin E (alpha-tocopherol, tocopherol) is the most common lipid-soluble antioxidant in humans and a key biological component of all membranes [23]. Direct topical application of vitamin E provides a number of benefits, including protection against ultraviolet (UV)-induced oxidative stress, skin photocarcinogenesis and erythema [24–26]. Vitamin E has also been shown to enhance human skin penetration and transdermal absorption [27]. It is hypothesised that vitamin E acts as a penetration enhancer by interacting with the lipid bilayer region of the stratum corneum and altering membrane permeability [27]. Unlike other well-known enhancers, vitamin E is generally thought to be non-irritating and possesses antioxidant and emollient properties [27], hence its inclusion in an assortment of cosmetic and dermatological products.
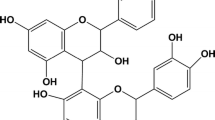
Tocopheryl phosphate mixture (TPM), also referred to as mixed tocopheryl phosphates (MTP), is a new lipidic material made up of two phosphorylated forms of vitamin E which has demonstrated enhanced transdermal or topical delivery of a range of active molecules in the laboratory [28]. TPM is formulated from a mixture of two phosphorylated forms of the most biologically active vitamin E isomer alpha-tocopherol [29]: alpha-tocopheryl phosphate (TP), a phosphoric acid ester of alpha-tocopherol esterified at the hydroxyl group of tocopherol [30], and di-tocopheryl phosphate ester (T2P), obtained by esterification of two tocopherol moieties with one phosphate group (Fig. 1) [13]. Both TP and T2P molecules consist of a chroman head (with two rings: one phenolic and one heterocyclic) and a phytyl tail [13], and are able to self-assemble into vesicular structures in aqueous environments. In this way, TPM differs from liposomes which assemble from phospholipids and/or surfactants.

Chemical structures of the phosphorylated tocopherols, tocopheryl phosphate (TP) and di-tocopheryl phosphate ester (T2P) that make up tocopheryl phosphate mixture (TPM)
Here, we investigate the physicochemical characteristics and morphological properties of the TPM delivery system and evaluate its ability to facilitate the transport of a variety of molecules with differing size and solubility profiles into and across the skin.
Materials and methods
Chemicals
Alpha-tocopheryl phosphate and di-alpha-tocopheryl phosphate were obtained from Phosphagenics Ltd. (Melbourne, VIC, Australia). Methanol, 1-propanol (high-performance liquid chromatography (HPLC) grade) and formic acid were purchased from Merck Millipore (Bayswater, VIC, Australia). Ethanol was purchased from Ajax Finechem (Sydney, NSW, Australia). Acetonitrile was purchased from Honeywell Burdick & Jackson (Muskegon, MI, USA). 1,4-Benzoquinone, cholecalciferol (vitamin D) and L-carnosine were purchased from Sigma-Aldrich (St. Louis, MO, USA). L-carnosine (b-alanine 3-3H) was purchased from American Radiolabelled Chemicals Inc. (St. Louis, MO, USA). CoQ10 was purchased from Kaneka (Osaka, Honshu, Japan). Solvable™ and Ultima Gold™ were purchased from Perkin Elmer (Melbourne, VIC, Australia). Carbopol 934 was purchased from Lubrizol (Cleveland, OH, USA). Caffeine was purchased from Sigma Chemical Company (St. Louis, MO, USA). All other reagents and solvents were of analytical grade and were purchased from Sigma Chemical Company (St. Louis, MO, USA), unless otherwise specified. Milli-Q water was produced from a Milli-Q® system (Merck Millipore, Bayswater, VIC, Australia).
Preparation of TP/T2P carriers
The mixture of TP/T2P (TPM®, Phosphagenics, Ltd.) has been described previously by Munteanu et al. (2004) [13]. The TPM vesicular systems investigated here were composed of 0.5–4 % w/w TPM, 5–30 % w/w ethanol, active as described and water to 100 % w/w. TPM and actives were dissolved in ethanol with heating at 50 °C in a water bath. Milli-Q water was heated to 50 °C in a separate vessel. The TPM/drug/ethanol solution was added slowly to the warm water whilst being stirred by a magnetic stirring bar at 600 rpm. The formulation was cooled to room temperature while mixing over approximately 20 min. All formulation steps were carried out in air-tight vessels with minimal head space to prevent ethanol evaporation. The liquid formulations were assessed for particle size, deformability, encapsulation efficiency and dermal/transdermal absorption using in vitro diffusion (Franz cell) studies. For topical application in vivo, formulations were thickened by the addition of 1 % w/w Carbopol 934 and NaOH (final pH 4.5) to create a semi-solid gel. For the preparation of caffeine and carnosine formulations, the caffeine and carnosine were added to the water phase prior to mixing with the TPM/ethanol phase and the formulation continued as described above.
Particle size distribution
The mean diameter and particle size distribution of vesicles were determined using dynamic light scattering (DLS) technique with a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK). This system used a 4 mW helium/neon laser at 633 nm wavelength and the samples were measured in backscatter mode at a detection angle of 173°. Results are presented as an average diameter of the vesicles in suspension (Z-average mean) with the polydispersity index (PDI). PDI values <0.15 are considered monodisperse and suitable for presentation of data as Z-average; in cases where the PDI >0.15, caution is required, as the reported mean particle size is not likely to reflect that of the majority of particles. Sample viscosity and refractive index were calculated by the Zetasizer software using known values for ethanol and water. All measurements were run in duplicate at 22 °C and the mean presented.
Assessments of nanoparticle deformability
TPM nanoparticle suspensions (3 mL) were manually extruded through a Millex-W 0.1-μm syringe-driven filter (Millipore, Bedford, MA) using a 3-mL syringe (Terumo, NSW, Australia). The filtrate was collected and the amount of TP and T2P quantified using high-performance liquid chromatography (HPLC) using the method described in Gianello et al. [29]. TP and T2P had retention times of 22.9 and 37.4 min, respectively, using this method.
Cryogenic transmission electron microscopy
A laboratory-built humidity-controlled vitrification system was used to prepare the samples for cryo-TEM. Humidity was kept close to 80 % for all experiments, and ambient temperature was 22 °C. Copper grids (200-mesh) coated with perforated carbon film (Lacey carbon film, ProSciTech, Queensland, Australia) were glow discharged in nitrogen to render them hydrophilic. Aliquots (4 μL) of the sample were pipetted onto each grid prior to plunging. After 30-s adsorption time, the grid was blotted manually using Whatman 541 filter paper for approximately 2 s. Blotting time was optimized for each sample. The grid was then plunged into liquid ethane cooled by liquid nitrogen. Frozen grids were stored in liquid nitrogen until required. The samples were examined using a Gatan 626 cryoholder (Gatan, Pleasanton, CA, USA) and a Tecnai 12 transmission electron microscope (FEI, Eindhoven, The Netherlands) at an operating voltage of 120 kV. The sample holder was operated at T = −175.5 ± 1 °C. At all times, low dose procedures were followed, using an electron dose of 8–10 electrons/Å2 for all imaging. Images were recorded using a FEI Eagle 4kx4k CCD camera at magnifications ranging from ×15,000 to ×50,000.
Skin permeation studies
Selection of actives and formulations
TPM formulations with actives of differing properties were studied for skin permeation in vitro. A range of actives with different sizes and chemistries, such as small (vitamin D3) and larger (CoEnzyme Q10 (CoQ10)) lipophilic molecules, small hydrophilic drugs (caffeine) and di-peptides (carnosine), were included. The active permeation profile of CoQ10 formulated with TPM also tested in vivo. The composition of the formulations is provided in Table 1. These formulations were determined after preliminary formulation work identified compositions with acceptable physical stability and appropriate preliminary permeation profiles. The TPM used in all formulations contained TP and T2P at a 2:1 ratio.
Encapsulation efficiency
The formulations described in Table 1 were assessed for drug encapsulation within the TPM particles using two independent methods: centrifugation and filtration. Both methods were used in parallel to account for the differing solubility profile of the various drugs in the formulations. TPM formulations were made as described above in conjunction with the appropriate control formulations lacking TPM. An additional control formulation was used for each of the lipophilic ingredients, which was 0.5 % w/w CoQ10 and 0.05 % w/w vitamin D dissolved in ethanol.
Centrifugation studies
Samples (500 mg) of each formulation were weighed out into 2-mL Eppendorf tubes and centrifuged at 14,000 rpm for 40 min to pellet the TPM particles. The supernatant was removed and made up to 10 mL in the following solvents: caffeine 90 % IPA in water, CoQ10 and vitamin D in ethanol, carnosine in 75 % acetonitrile in water. The pellet was resuspended in 100 μL of diethyl ether and left standing for 20 min at room temperature to dissolve, before being diluted to 10 mL with appropriate buffer (caffeine 90 % IPA in water, CoQ10and vitamin D in ethanol, carnosine in 75 % acetonitrile in water). All samples were passed through an Acrodisc syringe filter (0.45 μM filter; Pall Corporation, Ann Arbor MI, USA) prior to analysis. Drug detected in the supernatant was considered to represent unencapsulated material, whilst drug detected in the pellet was considered encapsulated/associated with the TPM particles. Results were compared against the control formulations lacking TPM and expressed as a percentage of the starting material.
Filtration studies
Formulations (6 mL) were passed through an Acrodisc syringe filter (0.45 μM) and the filtrates assayed for drug content. The filtrate was collected and 500 mg of filtrate diluted in appropriate buffer (caffeine 90 % IPA in water, CoQ10 and vitamin D in ethanol, carnosine in 75 % acetonitrile in water) for HPLC analysis as described below.
In vitro permeation experiments
The in vitro permeation profiles of TPM formulations were obtained using static vertical diffusion Franz cells and full thickness rat skin. Rat skin was harvested with ethical approval by the Monash Animal Research Platform (Clayton, VIC, Australia) under approval number MARP/2011/076. Rats were euthanized by asphyxiation using CO2 gas, after which time the abdominal area was carefully shaved and excised. All underlying fat and connective tissue were removed. Skin was frozen flat between sheets of aluminium foil and stored at −20 °C until the morning of experimentation, when the skin was thawed at room temperature and assembled in 12 mL vertical Franz diffusion cells (Permegear, Hellertown, PA, USA). The receptor solution in the Franz cells was PBS (12 mL) and the exposed skin area had a surface area of 1.77 cm2. The skin was removed from the Franz cells after 4 h and the unabsorbed active remaining on the surface carefully washed off. The absorbed active was then extracted from the skin overnight using appropriate solvents and quantitated using HPLC (described below). For experiments examining percutaneous absorption, infinite dosing conditions were used (200 μL). The receiver solution was sampled (500 μL) over a 9-h period and the active content quantified. Fresh PBS was used to replace the sampled aliquots in the receiver solution.
In vivo active permeation analysis
The active permeation profile of TPM formulations was also tested in vivo with CoQ10 as the model compound. All animal experiments were approved by the School of Biomedical Sciences Animal Ethics Committee based at Monash University (Clayton, VIC, Australia) under approval number BAM/B/2005/18. Male Sprague-Dawley rats (n = 6) were anaesthetised and prepared for experiments by shaving an area ∼5 × 4 cm from the upper back [35]. TPM formulations and control formulations were applied topically to the skin the day after shaving. The following day, rats were euthanized and blood collected via venipuncture. Blood was centrifuged to separate the plasma which was frozen immediately and stored at −20 °C until analysis. The dorsal skin at the application site was washed gently to remove unabsorbed material, before being extracted overnight in 1-propanol as described below for HPLC analysis.
Analytical methods and validation
CoQ10
For in vitro and in vivo dermal absorption samples, skin was placed in a 50-mL Falcon tube containing 1-propanol (5 mL) and left overnight. The following morning, samples were sonicated and then vortexed for 1 h before being passed through an Acrodisc syringe filter (0.45-μm filter) and the filtrate collected in vials for HPLC analysis. For experiments examining encapsulation efficiency, samples were prepared as described in Sections 2.6.2.1. and 2.6.2.2. CoQ10 was quantitated using an isocratic reversed-phase HPLC method on a Waters Alliance Separation Module 2790 liquid chromatography system equipped with a PDA detector. Separations were carried out using a Waters Sunfire™, C-18 column. CoQ10 was detected at 275 nm with a mobile phase of ethanol/methanol (80:20) at a flow rate of 1 mL/min.
Vitamin D
For in vitro diffusion samples, skin samples were soaked in 5 mL methanol overnight at 4–8 °C. The following morning, the samples were sonicated and then vortexed. An aliquot (600 μl) was filtered using a 0.2-μM GHP 96-well filter plate into a 1-mL round bottom 96-well plate. For experiments examining encapsulation efficiency, samples were prepared as described in Sections 2.6.2.1. and 2.6.2.2. Vitamin D was quantitated using an isocratic reversed-phase HPLC method on a Waters Alliance Separation Module 2790 liquid chromatography system equipped with a PDA detector. Separations were carried out using a Waters Symmetry, C-18 column (3.5 μm, 4.6 × 150 mm). Vitamin D was detected at 280 nm with a mobile phase of methanol/acetonitrile (50:50) at a flow rate of 1 mL/min.
Caffeine
For in vitro diffusion sample, skin samples were soaked in methanol (5 mL) overnight at 4–8 °C. The following morning, the samples were sonicated and then vortexed. A 1-mL aliquot of the extract solution was transferred to an Eppendorf tube and water (1 mL) added. The samples were mixed well before being centrifuged at 14,000 rpm for 20 min at 5 °C. Supernatants were transferred to HPLC vials for analysis. Caffeine was quantitated using a reversed-phase HPLC method on a Waters Alliance Separation Module 2795 liquid chromatography system equipped with a PDA detector. Separations were carried out using a symmetry, C-18 column. Caffeine was detected at 280 nm with a gradient of mobile phase A (0.1 % formic acid in water) and mobile phase B (0.1 % formic acid in methanol) at a flow rate of 1.5 mL/min. Samples of receiver solutions were centrifuged at 14,000 rpm for 20 min at 5 °C before being analysed by HPLC as described above.
For experiments examining encapsulation efficiency, samples were prepared as described in Sections 2.6.2.1. and 2.6.2.2. Caffeine was quantified using the HPLC method described above but using detection at 250 nm and a flow rate of 1 mL/min.
Carnosine
For in vitro diffusion studies, skin samples were digested overnight at 50 °C in 2 mL of Solvable™. Peroxide (200 μL) was added to the samples and incubated at 50 °C for 1 h. Samples were cooled to room temperature before 500 μL of sample was mixed with 500 μL water in a scintillation vial. Ultima Gold™ (6 mL) was added to the sample and mixed. Samples were left for 2 h before measurement in a Tri-Carb Liquid Scintillation Counter (Perkin Elmer, VIC, Australia).
For experiments examining encapsulation efficiency, samples were prepared as described in Sections 2.6.2.1. and 2.6.2.2. Carnosine was quantitated on a Waters Alliance Separation Module 2795 liquid chromatography system equipped with a PDA detector. The separation was carried out on an Atlantis HILIC silica column (2.1 × 50 mM, 3 μM) at 40 °C. Carnosine was detected at 214 nM with a gradient of mobile phase A containing 0.65 mM ammonium acetate, pH 5.5 in water/acetonitrile (25:75) and mobile Phase B containing 4.55 mM ammonium acetate, pH 5.5 in water/acetonitrile (70:30) at a flow rate of 0.8 mL/min.
CoQ10 quantitation in rat plasma
Plasma samples were thawed at room temperature for 30 min before being vortexed briefly and centrifuged at 14,000 rpm for 10 min. A 150-μL aliquot was transferred to a 2-mL Eppendorf tube and 20 μL of 1,4-benzoquinone (5 mg/mL in 1-propanol) was added. The sample was vortexed for 10 s before being allowed to stand for 10 min. 1-Propanol (730 μl) was added to the sample and before being vortexed for 30 s. The samples were then centrifuged for 10 min at 14,000 rpm at 10 °C. Supernatant (500 μL) was transferred to vials for HPLC analysis of CoQ10 content using the conditions described above.
Statistical analysis
Results from in vitro and in vivo absorption experiments were compared using Student’s t test (two-tailed) using the software PRISM (Graph Pad). A value of p < 0.05 was considered statistically significant.
Results
TPM forms particles in water with composition-dependent size distribution and morphology
Increasing the concentration of TPM in the formulation, while keeping the ethanol concentration constant at 30 % w/w, induced a steady increase in both the mean size and polydispersity index (Fig. 2) of the TPM particles. At TPM concentrations between 0.5 and 3 % w/w, relatively monodisperse (PDI <0.15) vesicle populations ranging in mean size from 115 to 162 nm were evident. Upon increasing the TPM concentration to 4 % w/w, the distribution became more polydisperse (PDI = 0.22), with an increase in mean particle size to 218 nm.

Effect of increasing TPM concentration on mean particle size and polydispersity index, at constant ethanol concentration. PDI polydispersity index, TPM tocopheryl phosphate mixture
Decreasing the ethanol concentration, while holding the TPM concentration at 1 % w/w, also induced a small but consistent increase in mean particle size from 137 to 165 nm, although the PDI remained low (Fig. 3).

Effect of reduced ethanol concentration on mean particle size and polydispersity, while maintaining TPM content at 1 % w/w. PDI polydispersity index, TPM tocopheryl phosphate mixture
The effect of TPM composition itself was also investigated. Formulations containing different ratios of TP and T2P (Fig. 1) were prepared to examine the effect on particle size distribution. Total TPM content (TP + T2P) was maintained at 1 % w/w and ethanol at 30 % w/w. Formulations prepared with a higher TP content (1:0 to 7:3, Fig. 4 ) were typically smaller (101 to 104 nm) than those with higher T2P content (128 to 116 nm), with the variation among all ratios being relatively minor (Fig. 4). In contrast, there was a strong dependence on TPM composition for the polydispersity index of the formulations. The homogenous formulations comprising only TP or T2P were considerably polydisperse, with recorded PDIs of 0.417 and 0.416, respectively, while heterogeneous mixtures had PDI <0.15. The high PDI of the TP- and T2P-only formulations makes interpretation of mean particle size values difficult, a point raised later in the discussion of electron microscopy results.

Effect of TP/T2P ratio on the mean particle size and polydispersity index of dispersions containing 1 % w/w total TP + T2P and 30 % w/w ethanol. PDI polydispersity index, TP tocopheryl phosphate, T2P di-tocopheryl phosphate ester
The dispersion of TP alone, T2P alone or the TPM mixture, all at 1 % w/w total lipid and 30 % w/w ethanol, displayed a systematic change in structures formed in dispersion when imaged by cryo-TEM. Formulations composed of 100 % TP were predominantly smaller, unilamellar vesicles; however, a small proportion of tubular micelle structures were also present in the sample (Fig. 5a). Formulations composed of 100 % T2P, however, were in the form of emulsion particles, with no bilayer vesicles present in the micrographs (Fig. 5b). Particles prepared using TPM mixture containing 0.66 % TP and 0.33 % T2P were dense but with bilayer structures consistent with multilamellar vesicles (Fig. 5c).

Dependence of particle morphology on lipid composition, with increasing T2P content, evident in representative cryo-TEM images for a TP, b T2P, c TPM (2:1 TP/T2P). Total phosphorylated tocopherol was 1 % w/w, ethanol 30 % w/w. TP tocopheryl phosphate, T2P di-tocopheryl phosphate ester, TPM tocopheryl phosphate mixture
Dependence of particle deformability on composition
The TPM formulations were assessed for extrudability through a 0.1-μm filter, as a standardised measure of deformability. The formulations contained 1 % w/w TPM in 30 % w/w ethanol, and were once again composed of various ratios of TP to T2P. The total amount of phosphorylated tocopherol passing through the filter was quantified and expressed as a percentage of the starting material. Particles high in TP content were able to pass through the filter easily, with little resistance to the syringe and a high percentage of starting material detected in the filtrate (Fig. 6). As the amount of T2P was increased in the formulation, the force required to pass the particles through the filter increased significantly, which was reflected in the reduced amounts of phosphorylated tocopherol detected in the filtrate. In the absence of TP (0:100 TP/T2P), no particles composed of T2P could be passed through the filter for detection in the filtrate. This occurred despite the similar size ranges presented for all particles in Fig. 4 and the constant concentration of ethanol.

Dependence of particle deformability on the formulation ratio of TP and T2P, at constant total phosphorylated tocopherol (1 % w/w) and ethanol (30 % w/w). TP tocopheryl phosphate, T2P di-tocopheryl phosphate ester, TPM tocopheryl phosphate mixture
Drug solubility influences encapsulation efficiency
In order to quantify the amount of drug encapsulated within TPM particles, two approaches were used in tandem: filtration and centrifugation.
The lipophilic ingredients, CoQ10 and vitamin D, are readily soluble in ethanol. The addition of TPM was enough to dissolve the lipophilic drugs, suggesting that CoQ10 and vitamin D were solubilised by, and therefore encapsulated within, TPM particles. Both caffeine and carnosine were readily soluble in the hydroethanolic solvents with, or without, TPM.
The extent to which TPM encapsulated and solubilised drugs was examined by passing the candidate formulations through 0.45-μM microfilters. As seen in Table 2, when CoQ10 or vitamin D was dissolved in ethanol, these solutions were able to pass through the filter without any loss of material. However, they had poorer solubility in the hydroalcoholic solvents used in these formulations (10–30 % ethanol in water; Table 1) and formed lipid droplets in suspension rather than dissolving completely. Consequently, 96.1 % of CoQ10 and 99.6 % of vitamin D were removed by the 0.45-μM filter from the ‘No TPM’ controls. Filtration was therefore appropriate for determining whether CoQ10 or vitamin D was dissolved in solution, or remained as larger, insoluble oil droplets.
TPM enabled the passage of 91.3 % of CoQ10 and 94.9 % vitamin D (Table 2) through the 0.45-μM filter. TPM must therefore be responsible for solubilising the lipophilic drugs, with the logical conclusion that the drugs are entrapped within the particles. Comparison with the control formulations suggested that the entrapment efficiency is likely to be ∼90–95 % for CoQ10 and vitamin D.
Caffeine and carnosine were readily soluble in the hydroalcoholic dispersive medium of the formulations and were therefore able to pass through the 0.45-μM filter in the presence, or absence, of TPM without significant loss (Table 2). Filtration was therefore unable to provide any information regarding the encapsulation efficiency of caffeine or carnosine.
Centrifugation was used to pellet the TPM particles, separating them from the dispersive medium in order to quantify the amount of encapsulated drug. Non-entrapped material remained in the supernatant and was collected and analysed after centrifugation. CoQ10 and vitamin D3 were found in the TPM pellet (85.2 and 53.1 % respectively; Table 2), in line with the data obtained from filtration. In the absence of TPM, the majority of CoQ10 (66.5 %) and vitamin D3 (30.1 %) was present in the pellet. This was expected given the poor solubility of the molecule in the hydroalcoholic solvents used here. Over 90 % of the CoQ10 and 98.0 % of the vitamin D remained in the supernatant when dissolved in ethanol.
The majority of the caffeine and carnosine remained in the supernatant. Only 6.5 % of the caffeine and 8.9 % of the carnosine were detected with the TPM pellet (Table 2). Importantly, the supernatants of the negative TPM controls contained 99.7 % of the caffeine and 100 % of the carnosine, demonstrating that the dissolved molecules would not ordinarily pellet under the centrifugation conditions used here. The caffeine and carnosine detected in the TPM pellet were therefore considered to be encapsulated or associated with the TPM particles.
In summary, the results of the filtration assay demonstrated that TPM was responsible for solubilising greater than 90 % of CoQ10 and vitamin D3. The high rate of entrapment was driven by the lipophilicity of the molecules and their relatively poor solubility in the dispersive medium. In contrast, caffeine and carnosine had good solubility in the dispersive medium and were therefore not driven to be encapsulated within the TPM particles at high efficiency.
In vitro active permeation studies
The dermal absorption of the four molecules, each formulated with TPM, was compared to control formulations (of an identical composition and active concentration to each test formulation, except without TPM) using static vertical diffusion Franz cells. Although the absolute concentrations were significantly different, in each case there was a 2.4–3.4-fold increase in absorption across the four compounds compared to that of the control (Fig. 7).

Relative in vitro dermal absorption of carnosine, CoQ10, caffeine and vitamin D3 following topical application of formulations with and without TPM. For each compound, relative delivery has been normalised to 100 % using the amount of drug delivered by the ‘−TPM’ control formulation. The relative increase in dermal absorption produced by TPM (carnosine, 3.41-fold increase; CoQ10, 2.95-fold; caffeine, 2.47-fold; vitamin D, 2.35-fold) was statistically significant for each drug (p < 0.05). Data are presented as the mean of n = 6. Bars represent SEM. CoQ 10 CoEnzyme Q10, TPM tocopheryl phosphate mixture
The mean amount of caffeine detected in skin extracts with TPM present was 314.6 ± 41.1 μg compared to 127.2 ± 46.3 μg for the control. In the case of CoQ10, the mean concentration detected in the skin with TPM present was 1464.0 ± 178.7 ng versus 496.7 ± 24.3 ng for the control, while for vitamin D3, the amounts extracted from the skin were 7.5 ± 0.5 μg and 3.2 ± 0.8 μg in the presence and absence of TPM, respectively. For L-carnosine, the mean CPM detected in skin extracts with TPM present was 2892.0 ± 454.6.4 compared to 848.7 ± 83.4 for the control. The increased absorption brought about by TPM for each compound was statistically significant (Student’s t test; p < 0.05).
In addition to the increased dermal absorption, TPM was also examined for its ability to promote transdermal delivery. Percutaneous absorption of caffeine was examined in Franz cells under infinite dosing conditions over a 9-h period (Fig. 8). At the conclusion of the experiment, the caffeine formulation containing TPM delivered 2.6 times more caffeine to the receiver solution (8.48 ± 0.54 μg/mL) when compared to the control formulation (3.30 ± 1.97 μg/mL). The rate of delivery (flux) was also faster, with calculated mean flux rates of 8.05 ± 0.40 and 2.85 ± 1.58 μg/cm2·h in the presence and absence of TPM, respectively. These differences were statistically significant (Student’s t test; p < 0.05). TPM was therefore able to promote both increased dermal and transdermal delivery in vitro.

Cumulative percutaneous absorption of caffeine in vitro. TPM significantly (p < 0.05) increased the amount (2.6 times) and rate (2.9 times) of caffeine delivered across the skin. Flux calculations used the linear portion of the graphs (3–6 h). Data are presented as the mean of n = 3. Bars represent standard deviation (SD). TPM tocopheryl phosphate mixture
In vivo permeation studies
Application of CoQ10 formulated with TPM to the dorsal region of rats produced a significant increase in the amount of CoQ10 present within plasma and skin. Twenty-four hours after the application of the TPM/CoQ formulation, mean CoQ10 levels detected in skin were 6.14 ± 0.98 μg/g of tissue (Fig. 9a). This was significantly higher (p < 0.05) than the endogenous amounts of CoQ10 detected in both the untreated sample (0.24 ± 0.04 μg/g) and TPM control (without CoQ10) sample (0.32 ± 0.02 μg/g). In the absence of TPM, the CoQ Control formulation delivered approximately 12 times less CoQ10 into the skin (0.74 ± 0.20 μg/g) than the TPM/CoQ formulation (p < 0.05).

Amounts of CoQ10 detected in skin (a) and in plasma (b) following topical application of TPM/CoQ10. TPM significantly (p < 0.05) increased the amount of CoQ10 delivered into the skin and plasma after topical application. Data are presented as the mean of n = 6. Error bars represent standard deviation (SD). CoQ 10 CoEnzyme Q10, TPM tocopheryl phosphate mixture
Mean CoQ10 levels in plasma (Fig. 9b) were also increased (p < 0.05) after topical application of TPM/CoQ (59.33 ± 16.48 ng/mL) relative to the endogenous CoQ10 levels in the untreated (27.67 ± 4.97 ng/mL) and TPM control (33.67 ± 7.06 ng/mL) samples, as well as plasma concentrations produced by the CoQ control formulation (35 ± 6.45 ng/mL). No apparent increase in plasma CoQ10 concentration relative to endogenous levels was evident after the topical application of the CoQ10 control formulation demonstrating that TPM is required to facilitate the transdermal delivery of CoQ10.
Discussion
TPM is a patented commercialised lipid excipient being investigated for a wide range of drug delivery applications. The data presented in this manuscript demonstrates that the excipient forms particles in hydroethanolic solutions suitable for topical application, that the particles are deformable depending on the composition and that they consequently enhance the dermal and transdermal delivery of actives of varying physicochemical properties. This set of physicochemical and delivery attributes is reminiscent of ethanolic phospholipid systems (so-called ‘ethosomes’ or ‘transfersomes’) but does not contain any glycero-phospholipids. First- and second-generation liposomal vesicles, which rely on bilayer ingredients and component-selection methods for TDD, have generated mixed reports of efficacy, and the potential for local irritation at the site of application has also been suggested [11–22]. TPM is a vesicular formulation derived from tocopherol that has demonstrated efficacy in the absorption of a wide variety of structurally different actives across the skin. The morphology, size and fluidity of the nanocarriers are largely dependent on the ratio of the constituent molecules.
Results demonstrated that TPM formed vesicles with internal bilayers in ethanolic solutions, although the internal morphology appeared more complex than for typical multilamellar vesicles exhibiting concentric bilayers. The composition of TPM formulations was shown to affect particle size distribution and morphology, with increasing concentrations of TPM relative to ethanol inducing a steady increase in both mean size and polydispersity index. Formulations containing the different ratios of TP and T2P also formed different structures in dispersion. Particles prepared using TPM mixture (TP and T2P) formed vesicles with multiple bilayers present, while TP alone formed predominantly unilamellar vesicles and T2P alone formed unstructured emulsion particles. This is likely due to the fact that TP is expected to be much more hydrophilic than T2P, but also amphiphilic, with the charged phosphate head group and lipid phytyl tail leading to the simpler unilamellar structures and tubular micelle formation observed in the cryo-TEM images. Although the particle size for the TP dispersion was seemingly much greater when measured using DLS (Fig. 4), the polydispersity index was very high, meaning that it is possible that the true dominant particle population was much smaller in size than the 100 nm (Fig. 4). The larger tubular micelles evident in the cryo-TEM images of TP samples are possibly responsible for increasing both the recorded particle size and polydispersity index. Conversely, T2P is hydrophobic, and the phosphate group is positioned between the two hydrophobic moieties, possibly preventing self-assembly when present alone. When mixed together, however, the combination of packing tendencies allows bilayer formation to occur, with structured particles resulting that were of more uniform size than for either parent compound alone.
The TPM particles were able to encapsulate lipophilic drugs at high efficiency. This was unsurprising, as the TPM bilayers of the particles represent the only lipophilic domains in the formulation available to dissolve CoQ10 or vitamin D. The hydrophilic caffeine and carnosine were encapsulated at much lower efficiency, likely through passive entrapment during particle formation. For topical formulations, poor aqueous solubility of the active compounds can limit the concentration of drug within the vehicle or force the inclusion of co-solvents or oils that are adverse or impair dermal absorption. TPM encapsulation may allow increased concentrations of lipophilic ingredients to be formulated in order to maximise therapeutic potential. Independent of any role in dermal absorption, TPM’s performance as a solubilising excipient may benefit a range of other dosage forms that struggle with ingredients having poor water solubility. This is especially true of injectable dosage forms that often resort to high concentrations of lipids, surfactants or organic solvents in order to support drug solubility [31].
Data also demonstrated that TPM particle deformability (elasticity) was dependent on the formulation composition. The ratio of TP to T2P was shown to directly affect particle deformability in TPM carriers, a key characteristic for dermal/transdermal delivery as it allows the possibility of penetration through channels of the stratum corneum [32]. T2P was found to enhance particle rigidity while TP increased particle deformability (Fig. 6). Previously, particle deformability has been reported to be dependent on the presence of ethanol for liposomal carrier systems [8].
These deformable particles were also shown to enhance the dermal and transdermal delivery of a range of actives of different sizes and chemistries across the skin, including small (vitamin D3) and larger (CoQ10) lipophilic molecules, small hydrophilic drugs (caffeine) and di-peptides (carnosine). In each case, 2.4–3.4-fold increases in absorption across the four compounds were observed compared to the control (Fig. 7). Since the formulations for each active and corresponding control were identical except for the addition of TPM, the additional absorption must be attributed to the TPM acting as a penetration enhancer. These data demonstrate the suitability of TPM for topical application and suggest that it may be applicable to a broad range of candidate drugs.
TPM may enhance transdermal delivery of drugs via two classical mechanisms of action, namely by acting as a drug carrier system and by disturbing the packing of the lipids in the stratum corneum. Both mechanisms of action rely upon the lipid structure of TPM and the way in which it interacts with lipids and membranes in the stratum corneum to effectively deliver the drug. In the drug carrier system hypothesis, TP and T2P assemble into vesicles in the presence of low-to-mid concentrations of solvent. These vesicles, which are able to entrap the drug, are highly deformable, and a growing body of literature has shown that deformable vesicles are able to improve transdermal delivery [1, 3, 33]. This is often attributed to their deformability and consequent ability to penetrate between the corneocytes in the stratum corneum: delivery is increased using deformable vesicles compared to standard liposomes that are more rigid [34]. Despite relatively poor encapsulation efficiency, the TPM mediated increase in absorption of caffeine and carnosine was similar to that seen for CoQ10 and vitamin D, both of which demonstrated high rates of encapsulation. This suggested that increased absorption was not predominantly a result of TPM particles transporting encapsulated cargo into the skin. Instead, the TPM particle may directly alter the structure of the stratum corneum to allow increased absorption of unentrapped material. This phenomenon has previously been identified for other elastic particles [33], although the relative absorption of the unentrapped material was not as great as results presented here. It may be that TPM particles produce greater ultrastructural changes to the strata corneum than other elastic vesicular systems. While the mechanism of elastic vesicle–skin interaction has not yet been clearly defined, the deformable nature of TPM vesicles is attributed to the TP structure. Unlike standard membrane forming phospholipids such as phosphatidylcholine, TPs form very flexible membranes. This property is thought to be a result of the methyl side chains on the carbon tail and the chroman head group, which prevents tight packing of the molecules.
The second hypothesised mechanism of action for these systems is the disruption of lipid packing in the stratum corneum. This typically results in reduced barrier quality and increased absorption and is the mechanism by which many established penetration enhancers work [35, 36]. The poor lipid packing properties of TPM, as evidenced by particle deformability, is likely to affect the packing of lipids in other systems. Laboratory experiments have demonstrated that TPM is able to systematically modulate bilayer lipid packing upon the addition of TP to phospholipids, altering the expected liposomal structures and producing long flexible sheets of membrane and other structures. It is therefore hypothesised that TPM is able to disturb the packing of lipid components of the stratum corneum in a similar way. As such, penetration enhancement may be possible in the absence of particles, with the TPM acting more like a conventional penetration enhancer directly on the stratum corneum [37]. A direct penetration enhancing effect may also help explain the increased absorption reported for caffeine and carnosine, despite relatively poor encapsulation efficiency. This may be useful for dosage forms that do not contain water and are therefore unable to maintain TPM in a vesicular system, such as adhesive transdermal delivery matrix patch.
Of further interest is the biological activity of the tocopheryl phosphates themselves, which have been shown to have enhanced vitamin E activity in terms of cardioprotective properties [30], protection from ultraviolet irradiation [38] and protection from atherosclerosis and inflammation [39, 40], and have also demonstrated attractive anti-erythema properties for the skin [41]. Unlike nanoparticle delivery systems composed of biologically inert phospholipids or surfactants, it may be that TPM itself has attractive properties beyond simply drug delivery, namely the ability to reduce irritation of adverse effects brought about by the delivery of irritating or sensitizing drug molecules. This will be examined in the future work.
Conclusions
TPM forms particles on dispersion in the approximate size range of 100–250 nm. The formulation components are important in dictating the particle size and deformability, while the TP/T2P ratio can alter the internal particle morphology. TPM encapsulates poorly soluble molecules at high efficiency. The addition of TPM to formulations of a number of different actives results in enhanced uptake into, and through, rodent skin in vitro. In an in vivo rat model, the uptake into skin was also correlated with enhanced plasma levels of CoQ10. The findings indicate the potential application of TPM as a new penetration enhancing excipient in transdermal drug formulations.
References
Akhtar N. Vesicles: a recently developed novel carrier for enhanced topical drug delivery. Curr Drug Deliv. 2014;11(1):87–97.
Prausnitz MR, Langer R. Transdermal drug delivery. Nat Biotechnol. 2008;26(11):1261–8. doi:10.1038/nbt.1504.
Jain A, Jain P, Kurmi J, Jain D, Jain R, Chandel S, et al. Novel strategies for effective transdermal drug delivery: a review. Crit Rev Ther Drug Carrier Syst. 2014;31(3):219–72.
Honeywell-Nguyen PL, Bouwstra JA. Vesicles as a tool for transdermal and dermal delivery. Drug Discov Today Technol 2005;2(1):67–74. doi:10.1016/j.ddtec.2005.05.003.
Paudel KS, Milewski M, Swadley CL, Brogden NK, Ghosh P, Stinchcomb AL. Challenges and opportunities in dermal/transdermal delivery. Ther Deliv. 2010;1(1):109–31.
Alexander A, Dwivedi S, Ajazuddin Giri TK, Saraf S, Saraf S et al. Approaches for breaking the barriers of drug permeation through transdermal drug delivery. J Control Release 2012;164(1):26–40. doi:10.1016/j.jconrel.2012.09.017.
Kligman AM. Skin permeability: dermatologic aspects of transdermal drug delivery. Am Heart J. 1984;108(1):200–6.
Touitou E, Dayan N, Bergelson L, Godin B, Eliaz M. Ethosomes - novel vesicular carriers for enhanced delivery: characterization and skin penetration properties. Journal of controlled release: official journal of the Controlled Release Society. 2000;65(3):403–18.
Benson HA. Transfersomes for transdermal drug delivery. Expert Opin Drug Deliv. 2006;3(6):727–37. doi:10.1517/17425247.3.6.727.
Benson HA. Elastic liposomes for topical and transdermal drug delivery. Methods Mol Biol. 2010;605:77–86. doi:10.1007/978-1-60327-360-2_4.
Elsayed MM, Abdallah OY, Naggar VF, Khalafallah NM. Deformable liposomes and ethosomes: mechanism of enhanced skin delivery. Int J Pharm. 2006;322(1–2):60–6. doi:10.1016/j.ijpharm.2006.05.027.
Elsayed MM, Abdallah OY, Naggar VF, Khalafallah NM. Deformable liposomes and ethosomes as carriers for skin delivery of ketotifen. Die Pharm. 2007;62(2):133–7.
Munteanu A, Zingg JM, Ogru E, Libinaki R, Gianello R, West S, et al. Modulation of cell proliferation and gene expression by alpha-tocopheryl phosphates: relevance to atherosclerosis and inflammation. Biochem Biophys Res Commun. 2004;318(1):311–6. doi:10.1016/j.bbrc.2004.04.028.
Song YK, Kim CK. Topical delivery of low-molecular-weight heparin with surface-charged flexible liposomes. Biomaterials. 2006;27(2):271–80. doi:10.1016/j.biomaterials.2005.05.097.
Srisuk P, Thongnopnua P, Raktanonchai U, Kanokpanont S. Physico-chemical characteristics of methotrexate-entrapped oleic acid-containing deformable liposomes for in vitro transepidermal delivery targeting psoriasis treatment. Int J Pharm. 2012;427(2):426–34. doi:10.1016/j.ijpharm.2012.01.045.
Trotta M, Peira E, Carlotti ME, Gallarate M. Deformable liposomes for dermal administration of methotrexate. Int J Pharm. 2004;270(1–2):119–25.
Cevc G, Gebauer D, Stieber J, Schatzlein A, Blume G. Ultraflexible vesicles, transfersomes, have an extremely low pore penetration resistance and transport therapeutic amounts of insulin across the intact mammalian skin. Biochim Biophys Acta. 1998;1368(2):201–15.
El Maghraby GM, Williams AC, Barry BW. Skin delivery of oestradiol from deformable and traditional liposomes: mechanistic studies. J Pharm Pharmacol. 1999;51(10):1123–34.
El Maghraby GM, Williams AC, Barry BW. Skin delivery of oestradiol from lipid vesicles: importance of liposome structure. Int J Pharm. 2000;204(1–2):159–69.
Gillet A, Compere P, Lecomte F, Hubert P, Ducat E, Evrard B, et al. Liposome surface charge influence on skin penetration behaviour. Int J Pharm. 2011;411(1–2):223–31. doi:10.1016/j.ijpharm.2011.03.049.
Manosroi A, Khanrin P, Lohcharoenkal W, Werner RG, Gotz F, Manosroi W, et al. Transdermal absorption enhancement through rat skin of gallidermin loaded in niosomes. Int J Pharm. 2010;392(1–2):304–10. doi:10.1016/j.ijpharm.2010.03.064.
Sinico C, Manconi M, Peppi M, Lai F, Valenti D, Fadda AM. Liposomes as carriers for dermal delivery of tretinoin: in vitro evaluation of drug permeation and vesicle-skin interaction. J Control Release: Off J Control Release Soc. 2005;103(1):123–36. doi:10.1016/j.jconrel.2004.11.020.
Traber MG, Sies H. Vitamin E in humans: demand and delivery. Annu Rev Nutr. 1996;16:321–47. doi:10.1146/annurev.nu.16.070196.001541.
Gensler HL, Magdaleno M. Topical vitamin E inhibition of immunosuppression and tumorigenesis induced by ultraviolet irradiation. Nutr Cancer. 1991;15(2):97–106. doi:10.1080/01635589109514117.
Lopez-Torres M, Thiele JJ, Shindo Y, Han D, Packer L. Topical application of alpha-tocopherol modulates the antioxidant network and diminishes ultraviolet-induced oxidative damage in murine skin. Br J Dermatol. 1998;138(2):207–15.
Trevithick JR, Xiong H, Lee S, Shum DT, Sanford SE, Karlik SJ, et al. Topical tocopherol acetate reduces post-UVB, sunburn-associated erythema, edema, and skin sensitivity in hairless mice. Arch Biochem Biophys. 1992;296(2):575–82.
Trivedi JS, Krill SL, Fort JJ. Vitamin E as a human skin penetration enhancer. Eur J Pharmaceut Sci. 1995;3:241–3.
Gavin P, Griffey A, Gianello R, Kennedy N, Keah HH, Cottrell J, et al. Transdermal delivery of various molecules in vivo using alpha-tocopheryl phosphate. Drug delivery. Technology. 2008;9(9):34–41.
Gianello R, Libinaki R, Azzi A, Gavin PD, Negis Y, Zingg JM, et al. Alpha-tocopheryl phosphate: a novel, natural form of vitamin E. Free Radic Biol Med. 2005;39(7):970–6. doi:10.1016/j.freeradbiomed.2005.05.016.
Mukherjee S, Lekli I, Das M, Azzi A, Das DK. Cardioprotection with alpha-tocopheryl phosphate: amelioration of myocardial ischemia reperfusion injury is linked with its ability to generate a survival signal through Akt activation. Biochim Biophys Acta. 2008;1782(9):498–503. doi:10.1016/j.bbadis.2008.05.002.
Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21(2):201–30.
Honeywell-Nguyen PL, Frederik PM, Bomans PH, Junginger HE, Bouwstra JA. Transdermal delivery of pergolide from surfactant-based elastic and rigid vesicles: characterization and in vitro transport studies. Pharm Res. 2002;19(7):991–7.
Verma DD, Verma S, Blume G, Fahr A. Liposomes increase skin penetration of entrapped and non-entrapped hydrophilic substances into human skin: a skin penetration and confocal laser scanning microscopy study. Eur J Pharm Biopharm: Off J Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV. 2003;55(3):271–7.
Subongkot T, Pamornpathomkul B, Rojanarata T, Opanasopit P, Ngawhirunpat T. Investigation of the mechanism of enhanced skin penetration by ultradeformable liposomes. Int J Nanomedicine. 2014;9:3539–50. doi:10.2147/IJN.S65287.
Moghadam SH, Saliaj E, Wettig SD, Dong C, Ivanova MV, Huzil JT, et al. Effect of chemical permeation enhancers on stratum corneum barrier lipid organizational structure and interferon alpha permeability. Mol Pharm. 2013;10(6):2248–60. doi:10.1021/mp300441c.
Aungst BJ. Absorption enhancers: applications and advances. AAPS J. 2012;14(1):10–8. doi:10.1208/s12248-011-9307-4.
Lopes LB, Garcia MT, Bentley MV. Chemical penetration enhancers. Ther Deliv. 2015;6(9):1053–61. doi:10.4155/tde.15.61.
Nakayama S, Katoh EM, Tsuzuki T, Kobayashi S. Protective effect of alpha-tocopherol-6-O-phosphate against ultraviolet B-induced damage in cultured mouse skin. J Invest Dermatol. 2003;121(2):406–11. doi:10.1046/j.1523-1747.2003.12351.x.
Ogru E, Libinaki R, Gianello R, West S, Munteanu A, Zingg JM, et al. Modulation of cell proliferation and gene expression by alpha-tocopheryl phosphates: relevance to atherosclerosis and inflammation. Ann N Y Acad Sci. 2004;1031:405–11. doi:10.1196/annals.1331.058.
Libinaki R, Tesanovic S, Heal A, Nikolovski B, Vinh A, Widdop RE, et al. Effect of tocopheryl phosphate on key biomarkers of inflammation: implication in the reduction of atherosclerosis progression in a hypercholesterolaemic rabbit model. Clin Exp Pharmacol Physiol. 2010;37(5–6):587–92. doi:10.1111/j.1440-1681.2010.05356.x.
Rerek ME, Mills OH, Wood R, Verdicchio R, West S. Disodium Lauriminodipropionate tocopheryl phosphates: a potent new anti-inflammatory. Cosmet Toiletries. 2003;118(7):63–8.
Acknowledgments
We thank Dr. Nicholas Kennedy, Mr. Giacinto Gaetano and Dr. Billie Nikolovski for the conduct of in vitro permeation studies and Ms. Gisela Ramirez and Mr. Mathew Parsons for the conduct of HPLC analysis. Lynne Waddington (CSIRO, Australia) is also thanked for the provision of cryo-TEM services.
Writing assistance was provided by Gemma Williams, Biosector 2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Paul Gavin, Mahmoud El-Tamimy and Hooi Hong Keah are employees of Phosphagenics Limited, a company commercialising TPM for use in transdermal drug delivery.
Ben J. Boyd declares that he has no conflict of interest.
All institutional and national guidelines for the care and use of laboratory animals were followed.
Rights and permissions
About this article

Cite this article
Gavin, P.D., El-Tamimy, M., Keah, H.H. et al. Tocopheryl phosphate mixture (TPM) as a novel lipid-based transdermal drug delivery carrier: formulation and evaluation. Drug Deliv. and Transl. Res. 7, 53–65 (2017). https://doi.org/10.1007/s13346-016-0331-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-016-0331-x