
Abstract
The present study was undertaken to detect the presence of PPRV in the goats of Assam. Competitive ELISA and Sandwich ELISA are used to detect the PPR viral antibody and antigen respectively. In addition, the study also involved the assessment of specific gene targets for detection of PPRV by RT-PCR from the clinical samples. A total of 579 sera samples (68.65 % in outbreak samples and 5.29 % in random samples) collected from different parts of Assam were tested by c-ELISA, indicated overall prevalence of 27.28 in goats. The percentage prevalence of PPRV antibodies in sera samples from goats collected at the time of outbreaks were 79.26, 85.41, 58.82, 6, 29.41 and 36.36 % in Kamrup, Nalbari, Mongoldoi, Jorhat, Darrang and Barpeta respectively. However, high percent prevalence (20.83 %) was observed in district Dhubri in random samples. Among the suspected samples, high percent prevalence (85.41 %) was observed in Nalbari. The competition percentage values (ranges from 35 to 45) obtained in competitive ELISA from tested goat samples found three categories, viz. positive, doubtful and negative. Most of the serum samples (n = 158) with competition percentage less than or equal to 35 % are considered positive for the presence of PPRV antibodies, (n = 9) greater than 35 % and less than or equal to 45 % are considered doubtful and retested, and (n = 423) greater than 45 % are considered negative. The overall sensitivity, specificity, apparent prevalence and true prevalence rate was found to be 68.65, 94.70, 27.28 and 34.69 % respectively. True prevalence rate was calculated based on the sensitivity and specificity of the c-ELISA employed in the study, which has a relative specificity of 94.70 % and sensitivity of 68.65 %.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Assam is a state of Indiain the north-eastern region. Located south of the eastern between 89°5′96°1′ East Longitude and 24°3′27°58′ North Latitude Himalayas, Assam comprises the Brahmaputra Valley and the Barak river valleys along with the Karbi Anglong and the North Cachar Hills with an area of 30,285 square miles (78,438 km2). With the “Tropical Monsoon Rainforest Climate”, Assam is temperate (summer max. at 95–100 °F or 35–38 °C and winter min. at 43–46 °F or 6–8 °C) and experiences heavy rainfall and high humidity. Assam is one of the richest biodiversity zones in the world and consists of tropical rainforests, deciduous forests, riverine grassland sand numerous wetland ecosystems. Goats, one of the mainstays of subsistence farming for mainly the landless farmers and small size holders, provide a dependable source of income to most of the rural population who are below the poverty line and it contributes largely to the livelihood of the livestock-keeping households of low and medium-input farmers. As per the FAOSTAT, 2008, the population of goat in the world was approximately 861.9 million with India holding the second position with 125.7 million goats. In Assam alone, there are approximately 4,376,150 population of goats [18th Livestock Census Data (2007–2008)]. However, optimum productivity is hampered by various epidemics in goats due to infectious diseases which cause mortality rates of 50–80 % in naïve populations [4].
Peste des petites ruminant is one of the most economically significant disease. Peste-des-petits-ruminants (PPR) which literally means “Plague of small ruminants” is an acute, highly contagious, office international des Epizooties (OIE) notifiable, transboundary viral disease of sheep and goats with high morbidity and mortality which can be as high as 100 and 90 % respectively [3, 14]. It is a severe, fast spreading viral disease mainly of domestic small ruminants. The disease naturally affects mainly sheep and goats, but it is usually more severe in goats than sheep. The disease is clinically manifested with severe pyrexia, oculo-nasal discharges, necrotizing and erosive stomatitis, enteritis and pneumonia. It is a member of the morbillivirus genus of family Paramyxoviridae (sub family Paramyxovirinae) under the order Mononegavirales. PPR was first reported in the Ivory Coast, West Africa in 1942 [16] and was later from other parts of world including Arabia [3], the Middle East [20], sub-Saharan Africa, the Arabian Peninsula, the Middle East and parts of Asia [6]. In India, the first outbreak of PPR was described from Arasur village, Villupuram district in Tamil Nadu [25]. Following major outbreaks in northern India [22], the disease became endemic in the country [23]. Although, vaccination against PPR is being practiced in India and African countries, PPR still a major constraint in the productivity of small ruminants.
Till date not much work has been done on sero-surveillance of PPR in goats of Assam. Moreover, there have been few reports on detection of these viruses in goats using PCR from this part of the country. With the above views in mind, the present work was undertaken to evaluate the prevalence of PPR viral antibodies in the goat population of Assam and also to detect the presence of the viral nucleic acid by PCR.
Materials and method
Source of sample
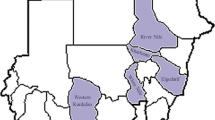
Serum samples from goats were collected randomly from different parts of Assam (Fig. 1). From each of the animals, blood samples were collected by jugular vein-puncture in vacutainer, separated the serum and transferred to small sterile screw capped plastic vials (Tarsons) labelled properly and stored at −20 °C without addition of any preservative till further use.

a Geographical location of different districts for sample collection, Green colours represents districts which showed positive for PPR. Infected goats showing typical symptoms of PPR during the time of outbreaks. b Ocular discharge, c Nasal discharge. d Diarrhoea (color figure online)
From PPR infected animals clinical samples comprising of nasal swab and whole blood were collected (Fig. 1). Whole blood samples were collected in EDTA vials (BD vacutainer®) and stored at 4 °C. From dead animals, tissue samples comprising of spleen, lymph nodes, intestine, Kidney, lungs were also collected from different parts of Assam. All the tissue materials and nasal swabs were properly preserved and stored in −20 °C till further use. Prior to analysis, the tissues samples were triturated in phosphate buffered saline (PBS) (pH 7.2) and 10 % (w/v) suspensions were prepared. The contents of the swab materials were extracted with 500 μl of PBS into eppendorf tubes.
Competitive ELISA for detection of PPR Viral antibody
In the present study c-ELISA kit (ID Screen PPR Competition, Montpellier, France) was used to detect the antibody against the nucleoprotein of pestis des petits ruminants virus in the serum samples collected from clinically affected and apparently healthy goats. The kit was used for detection of PPRV antibodies in terms of competition percentage. The microplates were precoated with purified recombinant PPR nucleoprotein (NP). To the precoated plates, samples to be tested and the controls were added after diluting in Dilution buffer provided with the kit. Plates were incubated for 45 min at 37 °C. After washing the plates three times with washing buffer, anti-NP conjugate diluted in Dilution buffer was added to the micro wells and incubated for 30 min at 21 °C. Again after washing the plates three times with washing buffer to eliminate the excess conjugate, the substrate (TMB) was added. After 15 min incubation, in the dark at 21 °C, the reaction was stopped and the results were read in the ELISA reader (TECAN) and OD was recorded at 450 nm. Samples with competition percentage ≤35 % were considered positive for the presence of PPRV antibodies, greater than 35 % and less than or equal to 45 % were considered doubtful and greater than 45 % were considered negative. Finally the sensitivity and specificity of c-ELISA was calculated using formula.
Sandwich-ELISA for detection of PPR antigen
For detection of PPR viral antigen PPR Sandwich-ELISA kit was obtained from PPR laboratory, Division of Virology, IVRI, Mukteswar. A monoclonal antibody-based sandwich ELISA (s-ELISA) kit for detecting PPRV antigenis commonly used to determine PPR clinical prevalence or diagnose PPR in India. In brief, a capture antibody (anti-rabbit polyclonal antibodies against rinderpest virus) at 1:4000 dilution (100 μl/well) in PBS was used to coat 96-well flat-bottomed ELISA plates (Maxisorp; NalgeneNunc, Germany). The plates were incubated at 37 °C for 1 h with constant shaking and then washed three times with washing buffer (0.002 mol/L PBS containing 0.05 % Tween 20). Suspensions of the clinical samples (50 μl/well) were added to the wells in duplicate. After incubation 37 °C for 1 h and washing three times with washing buffer, a PPRV-specific anti-nucleocapsid protein monoclonal antibody (1:20 dilution, 100 μl/well) was added and the plates were incubated 37 °C for 1 h. Anti-mouse horseradish peroxidase conjugates (Sigma-Aldrich, USA) diluted 1:1000 in blocking buffer (PBS containing 0.1 % Tween 20 and 0.5 % negative serum) were then added (100 μl/well). After incubating at 37 °C for 1 h, substrate solution [0.4 mg/mLo-phenylenediamine (OPD) with 4 μl of 3 % H2O2/mL of OPD] was added (100 μl/well) and the plate was incubated at 37 °C for 15 min for colour development. The reaction was stopped with 1 M H2SO4 (100 μl/well) before absorbance was read at 492 nm with an ELISA reader (LabsystemsMultiskan Plus; Thermo Fisher Scientific, USA).
RNA extraction and quantitation
The tissue samples that were preserved in chilled condition at −20 °C and used for preparing 10 % tissue suspension. The aliquots were used for viral RNA extraction. RNA isolation from tissue using Trizol® Reagent was done as per standard protocol. RNA was quantified by spectrophotometric analysis.
RT PCR for detection of PPR viral nucleic acid
Complementary DNA (cDNA) was synthesised with 2 µg of the quantified RNA using random hexamers. The mixture was heated to 70 °C for 5 min and snap chill on ice for 5 min. After spinning down, and add previously prepared master mix containing 5XRT buffer, RNAase inhibitor (Fermentas), 10 mM dNTP mix and M-MuLV RT (200 U/µl). The PCR tubes were then placed in a thermal cycler and reverse transcription was done at 42° C for 1 h followed by 35 PCR cycles. PCR amplification was carried out by a set of N gene specific forward (5′ GAT GGT CAG AAG ATC TGC A 3′) and reverse (5′ CTT GTC GTT GTA GAC CTG A 3′) primers to amplify 463 bp product. PCR was carried out in thermocycler (Flexigene Cambridge, UK) in 25 μl of total volume containing 40 ng of each primer, 200 μMdNTP mix, 1.5 mM MgCl2, 2.5 mM 10× buffer, 50–100 ng cDNA template and 1 U Taq DNA polymerase. The PCR cycling parameters were optimized as follows; initial denaturation at 94 °C for 10 min, followed by 94 °C for 30 s, 51 °C for 30 s, 72 °C for 45 s for 35 cycles and final extension at 72 °C for 10 min. The PCR products were separated by horizontal submarine agarose gel (1.5 %, free from DNAse and RNAse) electrophoresis in 1X TBE buffer at 80 V for 60 min and visualised using a gel documentation system (Gel Logic 100, KODAK).
Results
Competitive ELISA
The sera samples which were collected were tested for the presence of PPR viral antibody by c-ELISA. The c-ELISA detects PPRV antibodies in terms of competition percentage. Samples with competition percentage ≤35 % are considered positive, greater than 35 % but less than or equal to 45 % are considered doubtful and greater than 45 % are considered negative. The sensitivity and specificity of the c-ELISA employed in the study was found to have a relative specificity of 94.70 % and sensitivity of 68.65 %. The samples included 201 numbers from PPR infected animals and 378 numbers from apparently healthy animals. Out of 201 samples tested from infected animals, 138 samples showed presence of PPR viral antibody which indicated percent prevalence of 68.65 % (Fig. 2). Out of 378 sera samples collected from apparently healthy animals, only 28 number (5.29 %) of sera samples showed presence of PPR viral antibody.

a Graphical representation of the number of samples positive in cELISA. b Bar diagram representing the Sample wise positivity of PPRV by sandwich ELISA
Sandwich ELISA
A total 56 clinical samples (which includes 8 nasal swabs and 48 tissues comprising of spleen, lymph nodes, intestine, kidney, lungs) collected from a total of 35 PPR suspected goats were tested by PPR sandwich ELISA kit (Table 1). From the 35 animals suspected of PPR, 29 animals were found positive by PPR sandwich ELISA yielding an overall incidence rate of 82.85 %.
For sample wise positivity out of total 56 clinical samples tested, 37 (66.07 %) samples which include 4 nasal swabs (50 %) and 33 tissues (68.25 %) were found to be positive for PPRV (Fig. 2).
Detection of PPRV Nucleic acid by RT-PCR
Three µl of the cDNA was utilized for N gene amplification by PCR using specific primers. Five µl of the PCR amplified product was run on 1.5 % agarose gel in 1X TBE buffer at 80 V for 60 min.
Out of all the samples, 43 samples which include four blood samples and 39 clinical samples were found positive for PPRV. The remaining samples failed to produce the targeted amplification of 463 bp (Table 2).
Discussion
Information on the prevalence of PPRV antibodies in sheep, goats, cattle, buffaloes, camels, wild ruminants etc., is available from a number of countries in which the disease is reported including India [1, 7, 8, 19, 31]. The increase in the incidence of the disease was from 1995 onwards in north India [23] and in NE India from 2010 onwards. However, now the disease is enzootic in NE India in some pocktes. Majority of the reports from India indicated only the regional data from various states about the PPR seroprevalence in small ruminants and bovine [9, 26]. In India, several outbreaks go unrecorded due to underreporting/non-reporting and plenty of PPR outbreaks have occurred in the past and are now occurring regularly throughout the country. Even though India is endemic to PPR, some states especially north-eastern states (NE zone) are either free from disease or have very few reports. PPR is of increasing importance and likely to extend its geographic distribution especially in North Eastern states, as PPR outbreaks have been reported in Assam [5] and in Tripura [11, 21]. The geographical distribution of PPR in India has been widened quickly in recent times, which may be due to its economic importance, public and regulatory concerns and the availability of diagnostics [29]. The prevalence of PPRV antibodies in unvaccinated sheep and goats indicated not only subclinical or inapparent infection but also, nonlethal clinical infection or in other words, recovered infected animals, which could be of epidemiological significance.
The present study showed that PPR was widely prevalent in small ruminants in Assam. From a total of 579 serum samples, both clinical (N = 201) as well as random samples (N = 378) were tested for detection of PPR viral antibody. Based on the screening by cELISA, an overall seroprevalence of 27.28 % was found. The prevalence values reported here are in concordance with reports from India and other countries such as an overall seroprevalence of 46.01 % with a range of 42.30–52.94 % in goats were detected at different locations in the Parbhani region of Maharashtra [12]. However, the variations in seroprevalence could be due to differences in sample size, age, prevailing management practices, humidity or season, etc. [28]. Moreover, in northern India, generally goat-adapted virus circulation in the population, as most of the outbreaks from north is more severe in goats than sheep. Movement of animals from north India (where more population of goats than sheep) to the NE India are also frequently happening for the trade purpose. These could be reason for the PPR outbreaks in goats of Assam.
Higher seroprevalence was observed in suspected samples (68.65 %) than in random samples collected from apparently healthy samples (5.29 %). Greater PPR positivity in clinical samples from goats attributed to the fact that most of the suspected samples were from regions which had larger goat population [7]. Similarly, a higher mortality rate reported among infected goats than sheep in a large organized farm, which too has larger goat population. High percent prevalence (20.83 %) was also observed in border areas of Assam and Bangladesh in random samples. The presence of PPRV antibodies in goats indicate that the population was exposed to PPRV infection naturally, either directly or indirectly. It may be due to continuous surrounding of some of the NE states with PPR endemic countries like Bangladesh resulting in spread of disease in few endemic regions. Moreover it has been reported that Bangladesh PPR isolates and that of Tripura, were found similar, which may indicate cross border movement of animals between the international borders of both the countries resulting in the transmission of the virus to susceptible small ruminant population [2]. Thus, the high prevalence rate reported in the present study could be attributed to the increased animal movement from neighbouring states or countries. Twenty-nine out of the 35 animals tested were found positive by sandwich ELISA yielding an overall incidence rate of 82.85 %. Similar findings were reported an overall incidence rate of 81.25 % from Gujarat State [18]. However, 60.00 % incidence of PPR was reported in small population of sheep and goats in the same state [25]. Higher incidences of PPR with morbidity rates of 70.00, 90.00 and 100 % were also reported in India, Saudi Arabia and Ethiopia respectively [24, 27]. However, lower rates of morbidity during PPR outbreaks were also observed in different parts of India such as 25.84, 30.56 and 16 %.
The genome of PPRV contains six transcriptional units in the order of 3′ N-P/C/V-M-F–H-L 5′ with N gene getting expressed to a very high level [13]. N gene codes for an internal structural protein and also mRNAs of N gene are the most abundant transcripts of the virus, making it attractive target for development of a highly sensitive diagnostic assay. Recently, there have been a couple of attempts with promising results, targeting N gene for PCR based detection of PPRV [17]. Latest data has shown that N gene reveals improved image of PPRV epidemiology and is considered better than F gene based distribution of PPRV strains [26]. Detection of PPRV genetic material is performed by using reverse transcriptase polymerase chain reaction (RT-PCR),and it is now one of the test used most frequently in reference centers, because of its rapid, accurate and high sensitivity (FAO, 1999). In this study out of all the samples, 43 samples which include 4 blood samples and 39 clinical samples were found positive with an amplicon size of 463 bp of N gene. The remaining samples failed to produce the targeted amplification (463 bp). In another study 66.66 %similarity has been reported, where out of 18 samples, N gene based primers yielded positive results in 12 [30]. Although, RT-PCR was found to be better than virus isolation very little information was available on comparative efficiency of PPRV detection in same field samples by RT-PCR with any OIE/FAO approved field test such as sandwich-ELISA [10, 15].
In the present study, detection of PPRV was done in the same samples with both the tests i.e. sandwich-ELISA and N gene based RT-PCR. Results indicated that out of the total 56 clinical samples, PPRV could be detected in 37 samples by s-ELISA and 39 samples by N-gene based RT-PCR. Only two samples positive in RT-PCR were negative by s-ELISA. Further, comparative analysis for both the test was done, however, significant difference was not seen. In contrary to that, lower sensitivity of RT-PCR to sandwich-ELISA has been reported by George [17]. The reason for contradiction may be because of the abundance of nucleoprotein, which is targeted in both sandwich-ELISA and N-gene based RT-PCR, thus resulting in equal sensitivity in both the diagnostic tests.
References
Abraham G, Sintayehu A, Libeau G, Albina E, Roger F, Laekemariam Y, Abayneh D, Awoke KM. Antibody sero-prevalences against peste des petits ruminants (PPR) virus in camels, cattle, goats and sheep in Ethiopia. Prev Vet Med. 2005;70:51–7.
Abubakar M, Rajput ZI, Arshed MJ, Sarwar G, Ali Q. Evidence of peste des petitsruminants virus (PPRV) infection in Sindh Ibex (Capra aegagrusblythi) in Pakistan as confirmed by detection of antigen and antibody. Trop Anim Health Prod. 2011;43(4):745–7.
Abu-Elzein EME, Hassanien MM, Al-Afaleq AI, Abd-Elhadi MA, Housawi FMI. Isolation of peste des petits ruminants from goats in Saudi Arabia. Vet Rec. 1990;127(12):309–10.
Abu-Elzein EME, Housawi FMT, Bashareek Y, Gameel AA, Al-Afaleq AI, Anderson EC. Severe PPR infection in Gazelles kept under semi-free range conditions in Saudi Arabia. J Vet Microbiol B. 2004;51(2):68–71.
Anonymous. All India Co-ordinated Research Project on Animal Disease Monitoring and Surveillance (AICRP on ADMAS). Annual report 2010–2011. Guwahati: AICRP on ADMAS Collaborating Unit; 2011. p. 77–8.
Balamurugan V, Sen A, Venkatesan G, et al. Isolation and Identification of virulent peste des petits ruminants viruses from PPR outbreaks in India. Trop Anim Health Prod. 2010;42:1043–6.
Balamurugan V, Saravanan P, Sen A, Rajak KK, Venkatesan G, Krishnamoorthy P, Bhanuprakash V, Singh RK. Prevalence of peste des petits ruminants among sheep and goats in India. J Vet Sci. 2012;13(3):279–85.
Balamurugan V, Krishnamoorthy P, Raju DS, Rajak KK, Bhanuprakash V, Pandey AB, Gajendragad MR, Prabhudas K, Rahman H. Prevalence of peste-des-petits-ruminant virus antibodies in cattle, buffaloes, sheep and goats in India. Virusdisease. 2014;25(1):85–90.
Balamurugan V, Hemadri D, Gajendragad MR, Singh RK, Rahman H. Diagnosis and control of peste des petits ruminants: a comprehensive review. Virusdisease. 2014;25(1):39–56.
Brindha K, Raj GD, Ganesan PI, Thiagarajan V, Nainar AM, Nachimuthu K. Comparison of virus isolation and polymerase chain reaction for diagnosis of peste des petits ruminants. Acta Virol. 2001;45(3):169–72.
Chaudhary D, Muthuchelvan D, De A, Yadav AK, Sudhakar SB, Rajak KK, Pandey AB. Molecular characterization of PPR virus from an outbreak in Tripura reveals its origin from Bangladesh (2013). In: Asian-Pacific Congress of Virology (VIROCON 2013), Amity Institute of Virology and Immunology, Noida, UP, India from 17th to 20th Dec; p. 128.
Chavan VV, Digraskar SU, Dhonde SN, Bedarkar SN. Seromonitoring of peste des petits ruminants (PPR) in goats (Capra hircus) of Parbhani region of Maharashtra. Vet World. 2009;2:299–300.
Diallo A. Morbillivirus group: genome organisation and proteins. Vet Microbiol. 1990;23(1–4):155–63.
Diallo A, Minet C, Le Goff C, et al. The threat of peste des petits ruminants: progress in vaccine development for disease control. Vaccine. 2007;25(30):5591–7.
Forsyth MA, Barrett T. Evaluation of polymerase chain reaction for the detection and characterisation of rinderpest and peste des petitsruminants viruses for epidemiological studies. Virus Res. 1995;39(2–3):151–63.
Gargadennec L, Lalanne A. La peste des petits ruminants. Bulletin des Services Zootechniques, et des Epizooties de I’ Afrique Occidntale Francaise. 1942;5:16–21.
George AA. Comparative evaluation of different gene targets for PCR diagnosis of PPR. M.V.Sc., thesis submitted to Deemed University IVRI, Izatnagar, India; 2002.
Kerur N. Assessment of different gene targets for detection of peste des petits ruminants virus, by RT-PCR and sequence analysis of F and N gene Segments, M.V.Sc., thesis, Anand Agricultural University Anand, Gujarat; 2005.
Khan HA, Siddique M, Sajjad-ur-Rahman Abubakar M, Ashraf M. The detection of antibody against peste des petits ruminants virus in sheep, goats, cattle and buffaloes. Trop Anim Health Prod. 2008;40:521–7.
Lefevre PC, Diallo A, Schenkel S, Hussein S, Staak G. Serological evidence of peste des petits ruminants in Jordan. Vet Rec. 1991;128:110.
Majumder P. Studies on an incidence of peste des petits ruminants in the regional goat breeding farm at Debipur, Tripura. Indian J Anim Health. 1997;36:169–71.
Nanda YP, Chatterjee A, Purohit AK, Diallo A, Inui K, Sharma RN, et al. The isolation of pestedespetits ruminants virus from northern India. Vet Microbiol. 1995.
Nanda YP, Chatterjee A, Purohit AK, Diallo A, Innui K, Sharma RN, Libeau G, Thevasagayam JA, Bruning A, Kitching RP, Anderson J, Barrett T, Taylor WP. The isolation of peste des petits ruminants virus from Northern India. Vet Microbiol. 1996;51(3–4):207–16.
Roeder PL, Abraham G, Kenfe G, Barrett T. PPR in Ethiopian goats. Trop Anim Health Prod. 1994;26:69–73.
Shaila MS, Purushothaman V, Bhavasar D, Venugopal K, Venkatesan RA. Peste des petits ruminants in India. Vet Rec. 1989;125:602.
Shaila MS, Shamaki D, Forsyth MA, Diallo A, Goatley L, Kitching RP, Barrett T. Geographic distribution and epidemiology of peste des petitsruminants virus. Virus Res. 1996;43(2):149–53.
Sing VP, Cham VK, Mondhe KS. Peste des petits ruminants: an outbreak in sheep in Rajasthan. India Vet J. 1996;73:466–7.
Singh RP, Saravanan P, Sreenivasa BP, Singh RK, Bandyopadhyay SK. Prevalence and distribution of peste des petits ruminants virus infection in small ruminants in India. Rev Sci Tech. 2004;23:807–19.
Singh RK, Balamurugan V, Bhanuprakash V, Sen A, Saravanan P, Yadav MP. Possible control and eradication of peste des petits ruminants from India: technical aspects. Vet Ital. 2009;45:449–62.
Tiwari A. Prevalence of pesti des petits ruminants (PPR) virus in small ruminants of Gujarat and its characterization by RT-PCR/RFLP and SSCP profile. M.V.Sc., thesis, Anand Agricultural University, Anand, India; 2004.
Zahur AB, Ullah A, Hussain M, Irshad H, Hameed A, Jahangir M, Farooq MS. Sero-epidemiology of peste des petits ruminants (PPR) in Pakistan. Prev Vet Med. 2011;102:87–92.
Acknowledgments
The authors are thankful to the Department of Biotechnology, Government of India, for providing grant to carry out this research work. The authors are also grateful to Dr. P. Borah, Coordinator, State Biotech Hub, College of Veterinary Science, Assam Agricultural University for extending suggestion and encouragement time to time.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Devi, M., Das, S., Sharma, K. et al. Seroprevelance and molecular detection of peste des petits ruminants in goats of Assam. VirusDis. 27, 91–97 (2016). https://doi.org/10.1007/s13337-015-0291-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13337-015-0291-7