
Abstract
The occurrence of efflux mechanisms via Permeability-glycoprotein (P-gp) recognized as an important physiological process impedes drug entry or transport across membranes into tissues. In some instances, either low oral bioavailability or lack of brain penetration has been attributed to P-gp mediated efflux activity. Therefore, the objective of development of P-gp inhibitors was to facilitate the attainment of higher drug exposures in tissues. Many third-generation P-gp inhibitors such as elacridar, tariquidar, zosuquidar, etc. have entered clinical development to fulfil the promise. The body of evidence from in vitro and in vivo preclinical and clinical data reviewed in this paper provides the basis for an effective blockade of P-gp efflux mechanism by elacridar. However, clinical translation of the promise has been elusive not just for elacridar but also for other P-gp inhibitors in this class. The review provides introspection and perspectives on the lack of clinical translation of this class of drugs and a broad framework of strategies and considerations in the potential application of elacridar and other P-gp inhibitors in oncology therapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
P-glycoprotein inhibitors (P-gp) are gaining momentum in the present-day drug therapy because these provide an opportunity for tissue-specific accumulation of drug by overcoming the natural or acquired efflux mechanism(s) of the local environment. |
Many third-generation P-gp inhibitors such as tariquidar, elacridar, zosuquidar, laniquidar, ONT-093, etc. have advanced to clinical development as a combination therapy in different therapeutic areas most notably in the cancer area. |
While strong evidence reported from in vitro and in vivo preclinical and clinical data reviewed in this paper provide the mechanistic basis for an effective blockade of P-gp efflux by elacridar, the clinical translation of the promise has been elusive not just for elacridar but also for other third-generation P-gp inhibitors. |
A number of critical questions and key development strategies need to be considered to understand and overcome the observed dilemma of lack of clinical translatability of this class of P-gp inhibitors. |
1 Introduction
During the past 2 decades, researchers have identified many membrane transporters specifically related to drug pharmacokinetics and disposition [1]. These transporters play a crucial role in the absorption, distribution and elimination of drugs and their metabolite(s). Initially, the role of the transporters was established for drug excretion and subsequently identified in the drug absorption process [2]. The solute carriers (SLC) and ATP-binding cassette (ABC) transporters are considered as the primary classes of drug transport proteins that are widely distributed in different tissues in the body and exhibit broad substrate specificity [3]. These transporters modulate the access of the drugs into the cells and thus control the subsequent pharmacological and toxicological effects. They are classified as influx and efflux transporters based on their direction of substrate translocation. ABC transporters are considered as primary efflux transporters that use energy generated from adenosine triphosphate (ATP) hydrolysis to transport the substrate from intracellular to extracellular locale, mostly against a concentration gradient [2]. Amongst the ABC transporters, P-glycoprotein (P-gp) plays a significant role in modulating the drug uptake and access to different tissues. P-gp, a glycosylated membrane protein consists of 1280 amino acids with 12 hydrophobic, helical transmembrane segments, two intracellular ATP binding sites and a molecular weight of 170 Da. P-gp was first identified in cancer cells and later found in normal tissues such as the apical membrane of intestinal epithelial cells, the biliary cannalicular membrane of liver, the luminal membrane of proximal tubular epithelial cells in kidney and the luminal membrane of the endothelial cells forming the blood–brain barrier (BBB), blood-cerebro spinal fluid barrier (BCSFB), and blood-testis barrier [4,5,6,7,8]. P-gp located across the BBB limits the access of the drugs/xenobiotics into the central nervous system (CNS). Intestinal P-gp effluxes some of the drugs administered orally, thus reducing the bioavailability [9].
Several approaches were followed to overcome the issue of P-gp-mediated reduced oral bioavailability of important drugs that included developing suitable dosage form using nanotechnology, use of alternative route of drug administration and co-administration of P-gp inhibitors [10].
P-gp inhibitors are classified into three generations based on their specificity and affinity [2]. The first-generation inhibitors include several commonly used drugs such as verapamil, cyclosporin A, reserpine, quinidine, yohimbine and tamoxifen [2]. However, the major drawback of this class of P-gp inhibitors is the dose-related toxicity to achieve desirable P-gp inhibition. Second generation P-gp inhibitors, namely non-immunosuppressive analogues of cyclosporin A, PSC 833; D-isomer of verapamil, dexverapamil; and others such as biricodar (VX-710), GF120918 and MS-209 were devoid of the pharmacological activity and exhibited higher affinity as compared to the first-generation inhibitors. Another major limitation of second-generation P-gp inhibitors is their non-specific interaction with cytochrome P450 (CYP) enzymes [11]. However, third-generation inhibitors are found to be more specific and are regarded to display a better toxicity profile. Various third-generation inhibitors those are under development include tariquidar, elacridar, zosuquidar, laniquidar and ONT-093. The third-generation inhibitors do not interact with CYP enzymes; however, some interaction has been observed with other transporters outside of both P-gp and breast cancer resistance protein (BCRP) [11, 12]. These P-gp inhibitors show their activity by (a) blocking drug binding site either competitively, non-competitive or allosterically and (b) interfering with the ATP hydrolysis and (c) by altering integrity of cell membrane lipids [13, 14].
Amongst the third-generation P-gp inhibitors, elacridar has been extensively investigated. The chemical name of elacridar (also known as GF120918) is (N-[4-[2-(3,4-Dihydro-6,7-dimethoxy-2(1H)-isoquinolinyl)ethyl]phenyl]-9,10-dihydro-5-methoxy-9-oxo-4-acridinecarboxamide). It is a white coloured powder with a molecular weight of 563.64 and exhibits a solubility of 2 mg/ml in dimethyl sulfoxide (DMSO) [11]. Elacridar is a potent and specific non-competitive inhibitor of P-gp [11]. Elacridar acts by inhibiting the ATP hydrolysis by modulating the ATPase activity [15].
Along with elacridar, two other P-gp inhibitors, namely tariquidar and zosuquidar, are currently under clinical development [11]. Tariquidar binds specifically and noncompetitively to the P-gp and has exhibited dose linearity in systemic exposure in healthy male subjects. Clinical pharmacokinetic study in healthy subjects suggest that the maximum plasma concentration (C max) was 2.3 μM, area under the curve (AUC0–48) was 12.6 μM h, clearance was 0.19 l/h/kg, volume of distribution was 246 l/m2 and the terminal elimination half-life was 26 h, at a dose of 2 mg/kg [16].
Zosuquidar is characterised by high potency and low toxicity and has entered the clinical trial in combination with vinorelbine and doxorubicin for various types of advanced malignancies [17, 18]. Zosuquidar has been reported to be the most specific of the third-generation inhibitors of ABCB1 with little measurable effect on ABCG2 or ABCC1 transporters, or CYP in vitro [17]. Pharmacokinetic study in cancer patients suggested that the clearance of zosuquidar is independent of the dose. The half-life (t 1/2) following intravenous administration of 640 mg/m2/day was found to be 17 h, suggesting slow elimination [18]. However, the major reported side-effect is neurotoxicity and potential drug–drug interactions with vinorelbine and doxorubicin [19]. P-gp inhibitors are gaining significant importance in drug development because of the promise of avoiding efflux mechanism to ensure higher attainment of drug exposures in the desired regions of the body [20,21,22]. Table 1 summarizes the clinical development status of third-generation P-gp inhibitors.
Increasing efforts are also underway to develop novel P-gp inhibitors that exhibit higher specificity and affinity along with minimal toxicity to overcome the issues of multi-drug resistance and compromised drug bioavailability.
2 Scope
As elacridar has shown promise as a P-gp inhibitor, this review compilation was instituted to understand its pharmacokinetic aspects. The focus of this review is towards compilation of nonclinical and clinical pharmacokinetics of elacridar and critically probe the drug development considerations of this class of P-gp inhibitors using elacridar as the probe. The literature review was done using Pubmed® search (NCBI 2016), SCIFINDER® and Google Scholar databases with specific key words such as P-gp inhibitor, elacridar, pre-clinical, clinical, pharmacokinetics, absorption, distribution, metabolism, excretion, bioavailability, disposition, drug–drug interaction, transporters, enzymes, animal and human to collect the related full-length articles and abstracts.
This review is organized to provide the following: (a) an overarching compilation of the in vitro P-gp inhibitory potential exhibited by elacridar applicable to various therapeutic areas; (b) a tabular summary of the status of P-gp inhibitors in drug development; and (c) summarize the reported preclinical and clinical pharmacokinetic studies of elacridar (Tables 2, 3). The individual tabular pharmacokinetic summary was designed to succinctly capture study designs, objectives and evaluable pharmacokinetic parameters with key remarks. Additionally, a discussion section provides perspectives; strategies and considerations in the applicability of elacridar and/or this class of drugs for potential use in oncology therapies where P-gp inhibition may show benefits.
3 In Vitro P-gp Inhibition Activity
3.1 Anticancer Therapy
O’Neill et al. observed that elacridar increased the cellular uptake of docetaxel in resistant DU-145 R (moderate P-gp expression) and 22RV1 R (high P-gp expression) prostate cancer cell lines, but it was not the case with respect to the resistant PC-3 cell lines that were devoid of P-gp expression. Thus, it may be inferred that multiple mechanisms contribute towards docetaxel resistance including P-gp efflux [23].
Elacridar increased the response of hepatoblastoma cell line (i.e., HepT1) to facilitate the treatment with doxorubicin. The IC50 for doxorubicin + elacridar was 1.7 times lower as compared to native doxorubicin [24]. Another study suggested that the cellular uptake of doxorubicin from a formulated doxorubicin + elacridar (polymer-lipid hybrid nanoparticles) as well as non-formulated native doxorubicin + elacridar was more than 1.5 times higher as compared to the cellular uptake of native doxorubicin alone [25].
Marchetti et al. described the impact of elacridar on the cellular uptake of topotecan and gimetecan in different breast cancer cell lines. The IC50 of topotecan + elacridar in T8 and MDCKII-BCRP1 cell lines were 32 and 57 times lower as compared to native topotecan. Similarly, gimetecan + elacridar exhibited 3 and 6 times lower IC50 in T8 and MDCKII-BCRP1 cell lines as compared to native gimetecan [26].
The studies in hepatocellular carcinoma cell lines showed that elacridar increased the cellular uptake of irinotecan and its metabolite SN-38 in KYN-2 (expressing BCRP, CYP3A4/5 and UGT1A1) and KYN-1 cell lines (expressing BCRP only), thus suggesting that BCRP is one of the chemo-sensitivity determinants of irinotecan in hepatocellular carcinoma cell lines and its inhibition might be critical for cells expressing abundant BCRP [27].
The intestinal absorptive and secretory transport of irinotecan was investigated using Caco-2 cell monolayers and engineered Madin-Darby canine kidney (MDCK) II cells overexpressing P-gp, cannalicular multi-specific organic anion transporter (cMOAT) and MRP1 [28]. Elacridar (IC50—0.38 ± 0.06 µM) significantly decreased the secretory efflux of irinotecan [28].
Elacridar increased the sensitivity of ixabepilone, a novel microtubule targeting agent in MDCK and MDCK-MDRI cell lines. The IC50 of elacridar + ixabepilone was 90 times lower as compared to native ixabepilone [29]. Xia et al. observed that elacridar increased the apical-to-basolateral (A-to-B) transport and decreased the basolateral-to-apical (B-to-A) transport of methotrexate in Caco-2 cells [30].
Mitoxantrone uptake was 4.6 times higher in the presence of elacridar (5 µM/L) when evaluated in the human choriocarcinoma cell line BeWo, an in vitro model of the human trophoblast [31]. Tallkvist et al. observed that elacridar reduced secretion and increased accumulation of mitoxantrone in both undifferentiated and differentiated mammary epithelial HC11 cells [32]. The effect of elacridar on the sensitivity of human IGROV-1 ovarian cancer cell line and its cisplatin resistant variant IGROVCDDP towards paclitaxel, docetaxel and epirubicin was observed. The results showed that IC50 of elacridar + paclitaxel, elacridar + docetaxel and elacridar + epirubicin were 1.54, 61 and 2.6 times lower as compared to native paclitaxel, docetaxel and epirubicin in IGROV-1 cell lines. However, for IGROVCDDP cell lines, the IC50 of elacridar + paclitaxel, elacridar + docetaxel and elacridar + epirubicin were 404, 102,812 and 129 times lower as compared to native paclitaxel, docetaxel and epirubicin [33]. O’Conner et al. observed the potentiating effect of elacridar when bortezomib and elacridar combination was evaluated in several different P-gp-resistant cancer cell lines, including DLKP-A (lung cancer) NCI-Adr/res (ovarian cancer) and RPMI-Dox40 (MM) cells. The findings of the study showed that bortezomib is a substrate of P-gp, and this resistance was greatly reduced when P-gp efflux was inhibited by elacridar [34]. Elacridar increased the intracellular accumulation of CGP74588, a pharmacologically active metabolite of imatinib by 5 times in rat C6 glioma cells [35]. Sato et al. observed that the sunitinib resistance could be reversed by the co-treatment with elacridar in renal carcinoma cell lines 786-O; however, similar outcome was not observed in ACHN and Caki-1 cell lines [36].
3.2 Antiretroviral Area
Neumanova et al. observed that elacridar increased the apical-to-basolateral (A-to-B) transport and decreased the B-to-A transport of abacavir in MDCK-II cell lines [37]. A significant decrease in the B-to-A transport was observed in MDCK–MDR1 for zidovudine and MDCK–MDR1 and Caco-2 for lamivudine [38].
3.3 Miscellaneous
The findings from in vitro trans-well assays suggested that elacridar completely inhibited the efflux of EPZ-6438, an inhibitor of Enhancer of Zeste Homolog 2 (EZH2) and implicated in multiple gliomas and increased the brain penetration [39]. Elacridar increased the intracellular accumulation of kaempferol by 15, 2.11 and 1.5 times at concentrations of 5, 10 and 15 µM, respectively, in MDCK-II cell lines thus indicating that kaempferol is a substrate for P-gp. However, it was important to note that no dose-linear relationship was observed with respect to efflux ratio [40]. Sugano et al. described that elacridar (0.5 µM/L) reversed the resistance of non-small cell lung cancer cell line EBC-1R towards PHA-665752, a MET inhibitor by inhibiting the cancer-stem cells like property that leads to activation of ABCB1 [41]. Elacridar (5.0, 1.0 and 0.2 µM) decreased the efflux and increased the absorption of Ochratoxin A (OTA), a major secondary metabolite formed by various fungal species of the genus Penicillum and Aspergillus in Caco-2 cell model [42].
Elacridar decreased the efflux of danofloxacin in a dose-dependent manner (0.04–5 µM/L). A 1.5 times decrease in the efflux was observed at a dose of 5 µM/L as compared to native danofloxacin [43]. Miller et al. described that elacridar decreased the ATPase concentration and efflux of darifenacin in MDCK-MDRI cell lines [44]. Elacridar also reduced the basolateral to apical permeability of digoxin in MDCK-MDR1, Caco-2 and CPT-B2 cell lines [45]. The resistance of Leishmaniasis cell line DNM-R150 towards miltefosine was reversed by elacridar (1 µM). A 30% inhibition in the growth and 50% reversal were observed in the presence of elacridar [46]. An investigation in sandwich-cultured human hepatocytes revealed that co-incubation of tolvaptan with elacridar (10 µM) reduced DM-4107 (metabolite of tolvaptan) accumulation by 23.0% relative to control, with no effect on the accumulation of tolvaptan, thus suggesting that elacridar might have facilitated the cannalicular transport and modulated CYP enzymes in the human hepatocytes [47].
4 Ex-vivo P-gp Inhibition Activity
The role of placental P-gp with respect to maternal to fetal transfer was evaluated for L-α-acetylmethadol (a congener of methadone that has been used for treatment of the adult opiate addict) and paclitaxel using human placental lobule. The results suggested that the fetal rate transfer, maternal clearance and clearance index for L-α-acetylmethadol were 1.23, 1.16 and 1.26 times higher, respectively, in the presence of elacridar as compared to the control group [48]. Similarly, elacridar increased the fetal rate transfer, maternal clearance and clearance index by 2.0, 2.0 and 1.75 times, respectively, for paclitaxel as compared to the control group [48]. Elacridar increased the absorptive permeability of digoxin by 3.3 times when evaluated using the rat intestinal brush-border vesicles [49].
5 Pharmacokinetic Properties of Elacridar
5.1 Preclinical Pharmacokinetics
5.1.1 Absorption
Ward and Azzarano described the pharmacokinetic profile of elacridar in mouse, rat, dog and monkey. The studies were conducted at 3 and 30 mg/kg for all the species, whereas additional experiment was conducted in mice and rats at 300 mg/kg. Linear dose–concentration relationship could be established across the tested dose levels [50]. Sane et al. observed that the absolute bioavailability of elacridar in mice was 0.22 for oral administration and 0.01 for intra-peritoneal administration. Low aqueous solubility and high lipophilicity of elacridar are considered responsible for poor oral absorption [51].
5.1.2 Distribution
Plasma protein binding was found to be 98.5, 99.0, 99.8, 99.9 and 99.9 for mouse, rat, dog, monkey and human, respectively [50]. Biodistribution study in mice showed that the brain-to-plasma partition coefficient of elacridar in the wild-type mice was 0.82, as compared with 3.5 in MDR1a/b(−/−) mice indicating that P-gp limits the brain distribution of elacridar [52]. The brain-to-plasma partition coefficient after intravenous, intra-peritoneal and oral dosing was 0.82, 0.43 and 4.31, respectively [51].
5.1.3 Metabolism
Metabolism study using human P450 enzymes such as CYP1A2, 2C9, 2C19, 2D6 AND 3A4 showed that elacridar is not a potent inhibitor of CYP enzymes. The IC50 value ranged from 10.5 to 49.9 µM across all the tested enzymes [50].
5.1.4 Elimination
Elacridar was observed to be eliminated with a modest half-life of 3–6 h in all the preclinical species and appeared to be dose independent [50]. The mean hepatic extraction at 3 mg/kg dose for rat, dog and monkey were found to be 57.0, 24.1 and 76.2, respectively, whereas at 30 mg/kg dose was 40.5, 23.4 and 76.4, respectively [50].
5.2 Clinical Pharmacokinetics
With respect to the clinical pharmacokinetic data, no reports have been published showing the pharmacokinetic profile of elacridar alone. However, Kuppens et al. reported the pharmacokinetic profile for elacridar (dose ranging from 100 to 500 mg) upon co-administration with topotecan. No linear increase in the systemic exposure (either C max or AUC) for elacridar was observed with 5-times escalation in the dose and the time to reach maximum plasma concentration (T max) ranged from 4 to 6 h across the all the tested doses [53]. The observations drawn from the plasma concentration versus time profiles of elacridar suggested a long half-life value for elacridar although it was not reported [53]. As elacridar was dosed along with topotecan, it may be difficult to predict the actual pharmacokinetic behaviour of elacridar because the effect of topotecan on the pharmacokinetic profile of elacridar cannot be ignored. Elacridar potentially inhibited BCRP to a greater extent than compared to P-gp that has been evident from the biodistribution study in mice. Therefore, brain-to-plasma partition coefficient of elacridar in the wild-type mice, which was only 0.82, was dramatically enhanced to 3.5 in MDR1a/b(−/−) mice, 6.6 in BCRP1(−/−) mice and 15 in MDR1a/b(−/−)BCRP1(−/−) mice, indicating that both P-gp and BCRP limit the brain distribution of elacridar [52].
6 Preclinical Studies
6.1 Anticancer
Tang et al. observed that elacridar increased the brain accumulation of carbazitaxel by 9.6-fold in wild-type mice and the observed concentrations were similar to that observed for ABCB1a/1b;ABCG2−/− mice treated with or without elacridar. However, no significant enhancement in the plasma concentration of carbazitaxel was observed in the wild-type animals, which may be due to the higher selective brain uptake of carbazitaxel without altering systemic levels [54]. The brain-to-liver and brain-to-kidney concentrations were 10- to 14-fold higher in the presence of elacridar and were equivalent to that observed in both ABCB1-deficient strains. The findings of this study inferred that intravenous elacridar co-administration could completely inhibit the activity of mouse ABCB1 in the BBB, leading to highly increased cabazitaxel concentrations in the brain and, therefore, may translate into better efficacy [54]. Elacridar significantly increased the plasma concentrations of SN-38 (metabolite of topotecan) in wild-type and ABCC4, ABCB1 and ABCG2 knock-out mice suggesting that plasma level of SN-38 is mainly due to the inhibition of ABCG2-mediated elimination because ABCC4 + ABCB1 + ABCG2 knockout animals showed 2 times higher plasma concentration of SN-38 as compared to ABCC4 + ABCB1 knockout animals [55]. With respect to brain concentrations, the results suggested that only one of the aforementioned three genes is sufficient to maintain the similar brain concentration equivalent to wild-type animal’s brain concentration of SN-38 [55]. Elacridar administration had no effect on the plasma concentration of gimatecan in wild-type animals which may be perhaps due to the sub-efficacious dose of elacridar to inhibit ABCB1a/b;ABCG2 significantly. However, a 1.5 and 3 times higher plasma concentration was observed for gimatecan in ABCB1a/b;ABCG2 knock-out animals at 1 and 4 h, respectively. A marginal increase in the brain concentration of gimatecan was observed in the wild-type and knock-out animals dosed with elacridar [55]. Elacridar increased the oral bioavailability of a campothecin analogue AR-67. Rats dosed with elacridar and AR-67 lactone orally resulted in 5.5- and 11-fold increase in the plasma lactone and carboxylate concentrations, respectively, by decreasing the clearance [56]. Similarly, animals dosed with AR-67 carboxylate resulted in 4.2- and 5.2-fold increase in the plasma lactone and caroboxylate concentrations, respectively [56]. Another study by de Vries et al. showed that elacridar increased the brain concentrations of topotecan in both wild-type, MDR1a/b(−/−) and BCRP1(−/−) knock-out mice but was not significant in MDR1a/b(−/−);BCRP1(−/−) knock-out mice suggesting that both MDR1a/b and BCRP1 are responsible for limiting the brain access of topotecan [57]. Elacridar increased the plasma concentrations of topotecan by six- and ninefold in P-gp deficient and wild-type mice by increasing the intestinal uptake and decreasing the clearance and hepatobiliary excretion [57]. The relative fetal penetration of topotecan was twofold higher in P-gp deficient mice as compared to vehicle control animals, suggesting a function for BCRP in the maternal–fetal barrier of the placenta [58]. The results of positron emission tomography (PET) study in mice using [11C]-topotecan showed a twofold increase in the brain concentration of elacridar as compared to the group treated only with [11C]-topotecan [59].
Kemper et al. described that elacridar increased the brain concentration of docetaxel by 59% in P-gp knock-out mice [60]. Elacridar increased the plasma concentration of paclitaxel by 3.8 times in wild-type mice [61]. Kemper et al. observed that elacridar increased brain concentration of paclitaxel by fivefold in P-gp knockout animals as compared to wild-type control animals [62]. Oral co-administration of elacridar increased the bioavailability of paclitaxel and docetaxel by 10.7- and 4-fold in CYP3A4-humanised mice. Although the brain concentration of both paclitaxel and docetaxel increased in the presence of elacridar, the brain-to-plasma ratio remained unaffected [63].
Minocha et al. observed that elacridar increased the brain distribution of pazopanib by 2.1 times in FVB wild-type mice without affecting the plasma concentration. This may be due to the preferential uptake of pazopanib into the brain tissues and similar to what was observed for erlotinib and canertinib. No significant difference in the clearance and half-life of pazopanib was observed in the presence of elacridar. However, elacridar increased the volume of distribution of pazopanib by 1.4 times as compared to native pazopanib [64]. Elacridar increased the brain-to-plasma ratio of cobimetinib by 11, 6 and 7 times in MDR1a/b(−/−) and MDR1a/1b/BCRP1(−/−) knock-out and wild-type mice [65]. Tang et al. described that elacridar increased the plasma concentration and brain-to-plasma ratio of crizotinib by 2.2- and 12-fold in wild-type mice, respectively, compared with the vehicle-treated group and the concentrations were equivalent to that obtained from ABCB1a/1b;ABCG2(–/–) mice [66]. The brain levels of dasatinib increased by fivefold in wild-type mice in the presence of elacridar [67]. Another study by Lagas et al. showed a 2-time increase in the plasma concentration of dasatinib in wild-type mice in the presence of elacridar, but no change was observed in ABCB1a/1b;ABCG2−/− mice [68]. In the same study, the brain levels of dasatinib increased by 11- and 1.6-times in elacridar-treated wild-type and knock-out mice, respectively, as compared to the vehicle-treated counterpart [68]. Thus, the brain-to-plasma ratio of dasatinib increased by 2.3and 1.3-times in elacridar-treated wild-type and knock-out mice as compared to the vehicle-treated counterpart [68]. However, elacridar was not found effective in increasing the brain concentration of dasatinib in platelet-derived growth factor-B (PDGF-B)-driven brainstem glioma model in mice [69]. Co-treatment with elacridar increased the concentration of erlotinib in the tumour core by fourfold and by 14-fold in the regions around the brain as observed in a study on U87 rat xenograft model [70]. Radiolabelled pharmacokinetic study of [11C]-erlotinib in the presence of elacridar showed a 5.3-time increase in the brain concentration in wild-type mice as compared to vehicle-treated group and the observed level was equivalent to that of ABCB1a/b(−/−)ABCG2(−/−) mice [71]. Elacridar increased the brain-to-plasma ratio of gefitinib by fourfold in wild-type mice [72]. Kawamura et al. described that the brain-to-blood ratio of [11C]gefitinib increased by 4 and 11times following intravenous injection at 5 and 50 mg/kg dose levels, respectively [73]. Elacridar increased the blood concentration of imatinib in a dose-dependent manner where 1.8-fold increase was observed at a dose of 3 mg/kg and 4.4-fold at 30 mg/kg. However, the pattern was not same with respect to the brain-to-blood ratio [74]. The brain-to-blood ratio of animals treated with elacridar + imatinib showed ninefold higher value as compared to the animals treated with imatinib only [74]. Elacridar also increased the blood and brain concentration of the radioactive metabolites of imatinib by 1.7- and 2.8-fold, respectively, in Mdr1a/1b(−/−) mice [74]. Breedveld et al. demonstrated that elacridar increased the brain penetration of imatinib (following intravenous administration) by 4.2-fold and also reduced the clearance by 1.7-fold in wild-type mice [75]. Bihorel et al. described that elacridar increased the brain uptake of imatinib in wild-type (4.1-fold) and MDR1a/1b(−/−) mice (1.2-fold) [76]. Co-administration of elacridar increased the (AUC0-inf) oral and (AUC0-inf) IV by 3.3- and 2.0-fold, respectively, in wild-type and 2.7- and 1.3-fold in MDR1a/1b/BCRP1−/− mice. The percentage increase in oral bioavailability of imatinib when elacridar was co-administered was 105 and 102% in wild-type and MDR1a/1b/BCRP1−/− mice, respectively [77]. Elacridar also increased the brain concentration of sunitinib by 10 times in wild-type mice; however, no significant change in the plasma concentration was observed which may be due to the preferential uptake of sunitinib in the brain relative to increase plasma concentrations [78]. Tang et al. observed that elacridar increased the plasma con centration of N-desethyl sunitinib (active metabolite of sunitinib) by 1.4 times in wild-type mice. N-desethyl sunitinib was not detectable in the brain of wild-type mice in the absence of elacridar; however, 10 ng/g concentration of the metabolite was observed in the presence of elacridar [79]. Oberoi et al. described that brain-to-plasma ratio for sunitinib after co-administration of elacridar in wild-type mice was ∼12 compared with ∼17.3 in MDR1a/b(−/−)BCRP1(−/−) mice [80]. Elacridar was found to increase the brain concentration of palbociclib by 22 times in wild-type mice as compared to the group treated with only palbociclib; however, no significant change in plasma concentration was observed [81]. Although no significant enhancement in the plasma concentration of sorafenib was observed in the presence of elacridar, the brain concentration was 7 times higher in wild-type mice, which may be due to the preferential uptake of sorafenib in the brain [82]. Elacridar increased the brain-to-plasma ratio of sorafenib by 8 times in wild-type mice [83]. Minocha et al. observed that elacridar increased the brain-to-plasma ratio of vandetanib up to fivefold in FVB wild-type mice [84]. A 3- to 5-fold increase in the brain concentration of vemurafenib was observed upon co-treatment with elacridar [85].
6.2 Anti-retroviral
Edwards et al. observed 8 times increase in the brain-to-blood ratio of amprenavir in the presence of elacridar in rats [86]. Similarly, a 100-fold increase in the brain-to-plasma ratio of nelfinavir was observed in the presence of elacridar in rats [87]. Elacridar-treated wild-type mice showed a significant increase in atazanavir Cbrain/Cplasma (12.3-fold) and Ctestes/Cplasma (13.5-fold) ratios compared to those in vehicle-treated counterparts [88]. Huisman et al. observed a 4.4-fold increase in the plasma concentration and tenfold increase in the brain concentration of saquinavir upon co-treatment with elacridar [89]. The oral bioavailability of ritonavir was increased by 2.7 times upon co-administration of elacridar in rats [90].
6.3 Miscellaneous
Elacridar increased the plasma concentration of convallatoxin (cardiovascular agent) by 1.5 times and by 2 times in the brain of in rats [91]. Zhang et al. observed that elacridar increased the brain-to-plasma ratio of EZH2 inhibitor that is used for the treatment of gliomas by 12-fold without affecting the plasma concentration [39]. The brain distribution of GSK2126458, phosphoinositide 3-kinase inhibitor, was enhanced by sevenfold in wild-type mice [92]. Elacridar increased the systemic exposure of GV196771, an N-methyl-D-aspartate receptor antagonist, by more than tenfold in wild-type mice [93]. The brain-to-plasma ratio of YQA-14, a novel dopamine D3 receptor antagonist, increased by more than 75-fold with co-administration of GF120918 in mice [94].
A twofold decrease in the biliary clearance was observed for acetaminophen sulfate in rats upon co-administration of elacridar [95]. Lee et al. observed a 2-times increase in the brain uptake of radiolabelled dehydroepiandrosterone in wild-type mice [96]. In the presence of elacridar, the ratio of brain to plasma ratio in mouse increased 2-, 4- and 38-fold, respectively, for talinolol, digoxin and quinidine, whereas in rat, a 70-fold increase was observed for quinidine [97]. Pre-treatment of mice infected with Cryptococcus neoformans with elacridar significantly increased the cerebral concentration of elacridar [98]. Elacridar increased the brain distribution of loperamide by 3.5-fold in rats [99]. Barraud de Lagerie et al. observed that elacridar increased the brain concentration of (+) and (−)-mefloquine by 2.5 and 1.5 times in mice without affecting the plasma concentration. The efflux clearance from the brain decreased for both enantiomers, with a larger decrease for (+)-mefloquine [100]. Elacridar did not alter the plasma levels of quinidine and verapamil in rats. However, there was a twofold increase in the plasma concentration of digoxin in the presence of elacridar. The brain and CSF levels of quinidine, verapamil and digoxin increased 21–26- and 6–11-fold, respectively [101]. Elacridar increased the brain tissue:serum concentration ratio of morphine by approximately threefold in rats [102]. The half-life of unbound morphine in brain extracellular fluid was approximately threefold longer in elacridar-treated rats compared with normal counterparts [102]. The fraction unbound of morphine in whole blood was not altered significantly in the presence of elacridar as compared with controls [102]. The CNS uptake of riluzole was increased by 3 times in mice, in the presence ofelacridar [103]. Elacridar increased the brain distribution of verapamil in rats by 11-fold [104].
7 Clinical Studies
Sparreboom et al. described that elacridar increased the systemic exposure of doxorubicinol (major metabolite of doxorubicin) following doxorubicin administration by 1.5 times at a dose level of 200 mg b.i.d (elacridar) and by more than 2 times at 400 mg b.i.d (elacridar) dose in cancer patients [105]. Planting et al. observed no linear increase in the response for doxorubicin with the escalation of elacridar dose from 50 to 100 mg b.i.d: however, linear increase in the plasma concentration was observed from 100 to 400 mg b.i.d in a clinical study in 46 cancer patients. Varying doses of doxorubicin (50, 60, 75 mg/m2) had no impact on the pharmacokinetic profile of elacridar. Significant interpatient variability was observed in the pharmacokinetics of elacridar. The AUC of doxorubicin was only marginally influenced by elacridar and only at the highest dose level; however, elacridar significantly increased the systemic exposure of doxorubicinol [46]. Elacridar (Dose: 1000 mg) increased the oral bioavailability of paclitaxel by 5 times upon co-administration in cancer patients (N = 6) and was found to be well tolerated. AUC ratio for the metabolites 6α-hydroxypaclitaxel and 3′p-hydroxypaclitaxel was 1.1 (0.40/0.36) after oral drug administration with elacridar, whereas this ratio was 3.5 (1.69/0.48) when paclitaxel was combined with cyclosporine A which might be due to higher P-gp inhibition potential of paclitaxel at the tested dose as compared to elacridar [106]. The oral bioavailability of topotecan was found to be more than 102% higher in the presence of elacridar (Dose: 100 mg) administered concomitantly in 39 cancer patients. Two dose-limiting toxicities were seen at the 2.5 mg topotecan dose level [53].
8 Discussion
The choice of elacridar for this review compilation stemmed from the fact that elacridar has been extensively studied amongst all other reported P-gp inhibitors which are in development. More importantly, the observations and conjectures drawn using elacridar as an example can be extrapolated to the whole class despite differences in the pharmacokinetic properties which may be compensated by appropriate dose sizes.
Plethora of in vitro and in vivo data reviewed in this paper provides the basis for an effective blockade of P-gp efflux mechanism by elacridar. The beneficial effect of such a blockade if successfully translated in clinical practice would revolutionize many therapeutic areas most notably in the oncology area based on the huge body of collective evidence for various chemotherapeutic drugs Fig. 1.

Chemical structure of elacridar
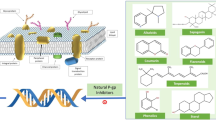
An important introspection worth considering is why drugs of this class including elacridar with so much promise and potential have not reached the stage of market approval yet. In this context, elacridar along with other P-gp inhibitor drugs in this class provides a distinct advantage as compared to its predecessors in not having cytochrome P450-related inhibition liability. However, despite such a differential feature in its disposition, it appeared that there was still lacuna in the complete understanding of key requirements when elacridar and/or other P-gp inhibitors are given in a combination therapy [107]. To address this important question, it is necessary to closely examine the ADME characteristics of elacridar along with other drugs of this class (Table 4); unfortunately, the pharmacokinetic reports for the listed drugs are scanty. It appeared that elimination half-life for several P-gp inhibitors including elacridar were relatively long (>18 h); however, it would be uncertain as to what this would mean at the tissue level of P-gp inhibitors. Therefore, would once-a-day dosing for P-gp inhibitors be justified is an important question that needs to be addressed. As displayed in Fig. 2, the key ADME points of interaction by elacridar which may be representative of the entire class would likely incorporate multiple non-specific interactions of the efflux mechanisms from the site of absorption to various distribution sites including brain, liver, kidney, etc. Also, the excretory mechanisms such as biliary and/or renal may also be affected by elacridar. Interestingly, there may be altered entero-hepatic recycling phenomenon because of elacridar on the indigenous transporters both at enterocytes and biliary efflux transporters. To underscore the above views, Srinivas has suggested that there was a need to understand the possible interplay of various mechanisms of a P-gp inhibitor drug when co-administered with cytotoxic drugs using tariquidar as an example [22]. For instance, if the P-gp inhibitor has an inhibitory role in the biliary excretion of the cytotoxic drug, the co-administration would result in an increased systemic exposure of the cytotoxic drug. However, if the same P-gp inhibitor simultaneously decreases efflux within tumour cells, it may promote greater accumulation of the cytotoxic drug within the tumour environment. Therefore, the net effect on systemic exposure may be neutral although the excretory pathway of the cytotoxic drug was impacted by the P-gp inhibitor.

Representation of interaction liability of elacridar during absorption, distribution, metabolism and excretion (ADME) process. P-gp permeability-glycoprotein, BCRP breast cancer resistance protein, CYP cytochrome P450
There are a number of developmental considerations that need to be strategized for ensuring that optimal delivery of elacridar and/or other members of this class occur in the patient population. First, do elacridar and/or other P-gp inhibitors of this class reach the purported site of action at the threshold concentration to elicit the efflux inhibitory response? Because in both clinical studies elacridar was orally administered, the issue of absorption and oral bioavailability is not an important consideration. One important question would be whether increasing oral doses of elacridar have an impact on its own rate and extent of absorption and, therefore, the ability of elacridar to reach the purported site of action would be expected to be highly variable. Similar issues would also prevail with other P-gp inhibitors in this class such as tariquidar, zosuquidar, laniquidar, etc. Besides debating on the lack of effectiveness of oral P-gp inhibitors, the lack of convincing clinical data on the effectiveness of P-gp inhibitors even after giving intravenous doses does raise a concern. Second, few other questions such as (a) adequacy of intravenous dose size of the P-gp inhibitor drug, (b) choice of the right regimen/schedule of the P-gp inhibitor (whether it should closely mimic the chosen cytotoxic drug), (c) distribution characteristics and elimination kinetics of the chosen P-gp inhibitor in relation to the cytotoxic drug need to be probed to possibly to ascertain areas of concern for remedial measures, if any. Third, what are the inadvertent consequences of lack of specificity of elacridar and/or P-gp inhibitor drugs? Because of extensive tissue distribution of such P-gp inhibitor drugs (i.e., the distribution volume of tariquidar is >100 times the total blood volume), it may be possible that it may promote accumulation of cytotoxic drug(s) in another area (i.e., brain, kidney, intestine, etc.) leading to undesired adverse reactions or side effects. However, from a benefit:risk analysis, the greater good of tumour shrinkage should outweigh such episodes of adverse events and/or safety issues if they not considered life-threatening or fatal. Fourth, because of non-specific nature of P-gp inhibition, it should be generally expected that elacridar and/or other P-gp inhibitors belonging to this class, should also impede the excretion mechanisms of the co-administered cytotoxic drugs (Fig. 2). Therefore, either reduced clearance or biliary excretion of cytotoxic drugs should only promote increased systemic circulation of cytotoxic drugs. While in theory, a more sustained concentration of cytotoxic drug is to be expected in circulation in presence of elacridar and/or other P-gp inhibitor drugs, and this in turn should provide a reservoir effect to promote increased uptake of cytotoxic drugs into the tumour tissues where P-gp efflux mechanisms would be expected to be turned off. But the only caveat would be how to measure the transfer of cytotoxic drug from the systemic circulation to tumour tissues whether it is primary tumour or metastasized tumour sites. However, with the availability of current day modelling tools including physiology based pharmacokinetic modelling it may be possible to try to answer this question. Fifthly, it may be equally important to match the pharmacokinetics of the cytotoxic drug (distribution, excretory mechanisms and half-life) with the chosen pharmacokinetic attribute of the P-gp inhibitor to ensure that an overlapping synergy exists to maximize the therapeutic effectiveness. Therefore, one important realization should be that not all cytotoxic drugs may be targeted with the same dose size of elacridar and/or other P-gp inhibitors and there would be a clear need for the optimization of the P-gp inhibitor dose based on cytotoxic drug(s) and perhaps, the type of tumours being targeted.
Similar scenario of interactions of small molecule tyrosine kinase inhibitors used in cancer therapy with P-gp inhibitors is likely to manifest during absorption, distribution and excretion of such tyrosine kinase inhibitors [108]. However, because several tyrosine kinase inhibitors may also affect efflux or uptake transporters, the net consequences of co-administration of tyrosine kinase inhibitors with P-gp inhibitors is difficult to predict. Also, since cancer treatment is very much a combination therapy that may include cytotoxic drug(s) along with tyrosine kinase inhibitors, the use of P-gp inhibitor need to be made with caution to avoid dangerous drug–drug interaction potential which may be difficult to predict a priori using in vitro tools and/or in silico models.
P-gp inhibition and the relevance to antidepressant therapy have been a topic of great interest because of the opportunity to deliver antidepressant drugs effectively to brain overcoming the P-gp based efflux mechanism. A review published several years ago has provided a critical assessment on issues pertaining to evolution of the field of antidepressant therapy with P-gp inhibitors [109]. While P-pg inhibitors may provide an opportunity to increase the brain penetration of antidepressants for treating refractory patients, the translatability in the clinic is yet to be established in an unambiguous manner [109].
From all the above viewpoints, it is important that both study design (pre-treatment with P-gp inhibitor versus simultaneous dosing) and dose size (due to differences in excretory mechanisms for P-gp inhibitors relative to cytotoxic drugs) be given careful consideration. To underscore the above point, biodistribution of the labelled tariquidar suggested relatively higher uptake of the drug in liver, spleen and kidneys in humans and, therefore, the importance of excretory pathways such as hepatobiliary and renal to rapidly remove P-gp inhibitor drug such as tariquidar from circulation should be factored in both study design and dosing decisions [110].
9 Conclusions
Innovative approaches to counter efflux mechanisms via P-gp inhibitors have been tried using third-generation P-gp inhibitors such as elacridar, tariquidar, zosuquidar, laniquidar, etc. While the promise demonstrated by scores of in vitro and preclinical in vivo data of elacridar and other drugs in the class was encouraging, unfortunately the translatability in a desired clinical outcome is yet to be established in oncology trials where P-gp inhibitors are given in combination with cytotoxic drugs. There are number of critical questions that need to be considered while attempting to understand and solve this problem. Key developmental considerations would encompass the following: (a) whether P-gp inhibitors be pre-treated as opposed to given simultaneously with the coadministered drug; (b) as to how should one choose the dose size and schedule; (c) as to what are the overlapping ADME mechanisms that need to be considered to assess the drug interaction potential; (d) whether plasma concentration (i.e., threshold level) of P-gp inhibitor drug be used as a surrogate from a dosing strategy perspective. It appears that a thorough understanding of the interplay of various mechanisms is a key for the successful development of P-gp inhibitor drugs in assessing the potential risks versus therapeutic benefits in combination therapies. In summary, the clinical translation into a desirable outcome has still been elusive for P-gp inhibitor drugs not only in cancer therapeutics but also in other areas; however, the promise of avoidance of efflux mechanisms in desired regions would revolutionize current treatment options in multiple therapeutic areas.
References
Russel FGM (2010) Transporters: Importance in drug absorption, distribution, and removal. In: Pang K, Sandy, Rodrigues, A. David, Peter, Raimund M (eds) In Enzyme- and transporter-based drug–drug interactions. Springer: New York
Varma MV, Ashokraj Y, Dey CS, Panchagnula R. P-glycoprotein inhibitors and their screening: a perspective from bioavailability enhancement. Pharmacol Res. 2003;48:347–59. doi:10.1016/S1043-6618(03)00158-0.
Fredriksson R, Nordstrom KJ, Stephansson O, Hagglund MG, Schioth HB. The solute carrier (SLC) complement of the human genome: phylogenetic classification reveals four major families. FEBS Lett. 2008;582:3811–6. doi:10.1016/j.febslet.2008.10.016.
Mukhopadhyay T, Batsakis JG, Kuo MT. Expression of the mdr (P-glycoprotein) gene in Chinese hamster digestive tracts. J Natl Cancer Inst. 1998;80:269–75. doi:10.1093/jnci/80.4.269.
Demeule M, Labelle M, Regina A, Berthelet F, Beliveau R. Isolation of endothelial cells from brain, lung, and kidney: expression of the multidrug resistance P-glycoprotein isoforms. Biochem Biophys Res Commun. 2001;281:827–34. doi:10.1006/bbrc.2001.4312.
Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, Bertino JR. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci. 1989;86:695–8.
Fromm MF. P-glycoprotein: a defence mechanism limiting oral bioavailability and CNS accumulation of drugs. Int J Clin Pharmacol Ther. 2000;38:69–74.
Wijnholds J, deLange EC, Scheffer GL, van den Berg DJ, Mol CA, van der Valk M, Schinkel AH, Scheper RJ, Breimer DD, Borst P. Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. J Clin Invest. 2000;105:279–85. doi:10.1172/JCI8267.
Westphal K, Weinbrenner A, Giessmann T, Stuhr M, Franke G, Zschiesche M, Oertel R, Terhaag B, Kroemer HK, Siegmund W. Oral bioavailability of digoxin is enhanced by talinolol: evidence for involvement of intestinal P-glycoprotein. Clin Pharmacol Ther. 2000;68:6–12. doi:10.1067/mcp.2000.107579.
Patil S, Dash RP, Anandjiwala S, Nivsarkar M. Simultaneous quantification of berberine and lysergol by HPLC-UV: evidence that lysergol enhances the oral bioavailability of berberine in rats. Biomed Chromatogr. 2012;26:1170–5. doi:10.1002/bmc.2674.
Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting P-glycoprotein. Cancer Control. 2003;10:159–65.
König J, Müller F, Fromm MF. Transporters and drug–drug interactions: Important determinants of drug disposition and effects. Pharmacol Rev. 2013;65:944–66. doi:10.1124/pr.113.007518.
Shapiro AB, Ling V. Effect of quercetin on Hoechst 33342 transport by purified and reconstituted P-glycoprotein. Biochem Pharmacol. 1997;53:587–96. doi:10.1016/S0006-2952(96)00826-X.
Drori S, Eytan GD, Assaraf YG. Potentiation of anticancer drug cytotoxicity by multidrug-resistance chemosensitizers involves alterations in membrane fluidity leading to increased membrane permeability. Eur J Biochem. 1995;228:1020–9. doi:10.1111/j.1432-1033.1995.1020m.x.
Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447–59. doi:10.1586/14737140.7.4.447.
Stewart A, Steiner J, Mellows G, Laguda B, Norris D, Bevan P. Phase I trial of XR9576 in healthy volunteers demonstrates modulation of P-glycoprotein in CD56 + lymphocytes after oral and intravenous administration. Clin Cancer Res. 2000;6:4186–91.
Lê LH, Moore MJ, Siu LL, Oza AM, MacLean M, Fisher B, Chaudhary A, de Alwis DP, Slapak C, Seymour L. Phase I study of the multidrug resistance inhibitor zosuquidar administered in combination with vinorelbine in patients with advanced solid tumours. Cancer Chemother Pharmacol. 2005;56:154–60. doi:10.1007/s00280-004-0942-7.
Sandler A, Gordon M, De Alwis DP, Pouliquen I, Green L, Marder P, Chaudhary A, Fife K, Battiato L, Sweeney C, Jordan C, Burgess M, Slapak CA. A Phase I trial of a potent P-glycoprotein inhibitor, zosuquidar trihydrochloride (LY335979), administered intravenously in combination with doxorubicin in patients with advanced malignancy. Clin Cancer Res. 2004;210:3265–72. doi:10.1158/1078-0432.CCR-03-0644.
Falasca M, Linton KJ. Investigational ABC transporter inhibitors. Investigational ABC transporter inhibitors. Expert Opin Investig Drugs. 2012;21:657–66. doi:10.1517/13543784.2012.679339.
Colabufo NA, Berardi F, Cantore M, Contino M, Inglese C, Niso M, Perrone R. Perspectives of P-glycoprotein modulating agents in oncology and neurodegenerative diseases: pharmaceutical, biological, and diagnostic potentials. J Med Chem. 2010;53:1883–97. doi:10.1021/jm900743c.
Fox E, Widemann BC, Pastakia D, Chen CC, Yang SX, Cole D, Balis FM. Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P-glycoproteininhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors. Cancer Chemother Pharmacol. 2015;76:1273–83. doi:10.1007/s00280-015-2845-1.
Srinivas NR. Understanding the role of tariquidar, a potent Pgp inhibitor, in combination trials with cytotoxic drugs: what is missing? Cancer Chemother Pharmacol. 2016;78:1097–8. doi:10.1007/s00280-016-3044-4.
O’Neill AJ, Prencipe M, Dowling C, Fan Y, Mulrane L, Gallagher WM, O’Connor D, O’Connor R, Devery A, Corcoran C, Rani S, O’Driscoll L, Fitzpatrick JM, Watson RW. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol Cancer. 2011;10:126. doi:10.1186/1476-4598-10-126.
Warmann S, Göhring G, Teichmann B, Geerlings H, Fuchs J. MDR1 modulators improve the chemotherapy response of human hepatoblastoma to doxorubicin in vitro. J Pediatr Surg. 2002;37:1579–84. doi:10.1053/jpsu.2002.36188.
Wong HL, Bendayan R, Rauth AM, Wu XY. Simultaneous delivery of doxorubicin and GG918 (Elacridar) by new polymer-lipid hybrid nanoparticles (PLN) for enhanced treatment of multidrug-resistant breast cancer. J Control Release. 2006;116:275–84. doi:10.1016/j.jconrel.2006.09.007.
Marchetti S, Oostendorp RL, Pluim D, van Eijndhoven M, van Tellingen O, Schinkel AH, Versace R, Beijnen JH, Mazzanti R, Schellens JH. In vitro transport of gimatecan (7-t-butoxyiminomethylcamptothecin) by breast cancer resistance protein, P-glycoprotein, and multidrug resistance protein 2. Mol Cancer Ther. 2007;6:3307–13. doi:10.1158/1535-7163.
Takahata T, Ookawa K, Suto K, Tanaka M, Yano H, Nakashima O, Kojiro M, Tamura Y, Tateishi T, Sakata Y, Fukuda S. Chemosensitivity determinants of irinotecan hydrochloride in hepatocellular carcinoma cell lines. Basic Clin Pharmacol Toxicol. 2008;102:399–407. doi:10.1111/j.1742-7843.2007.00199.x.
Luo FR, Paranjpe PV, Guo A, Rubin E, Sinko P. Intestinal transport of irinotecan in Caco-2 cells and MDCK II cells overexpressing efflux transporters Pgp, cMOAT, and MRP1. Drug Metab Dispos. 2002;30:763–70. doi:10.1124/dmd.30.7.763.
Shen H, Lee FY, Gan J. Ixabepilone, a novel microtubule-targeting agent for breast cancer, is a substrate for P-glycoprotein (Pgp/MDR1/ABCB1) but not breast cancer resistance protein (BCRP/ABCG2). J Pharmacol Exp Ther. 2011;337:423–32. doi:10.1124/jpet.110.175604.
Xia CQ, Liu N, Yang D, Miwa G, Gan LS. Expression, localization, and functional characteristics of breast cancer resistance protein in Caco-2 cells. Drug Metab Dispos. 2005;33:637–43. doi:10.1124/dmd.104.003442.
Ceckova M, Libra A, Pavek P, Nachtigal P, Brabec M, Fuchs R, Staud F. Expression and functional activity of breast cancer resistance protein (BCRP, ABCG2) transporter in the human choriocarcinoma cell line BeWo. Clin Exp Pharmacol Physiol. 2006;33:58–65. doi:10.1111/j.1440-1681.2006.04324.x.
Tallkvist J, Yagdiran Y, Danielsson L, Oskarsson A. A model of secreting murine mammary epithelial HC11 cells comprising endogenous Bcrp/Abcg2 expression and function. Cell Biol Toxicol. 2015;31:111–20. doi:10.1007/s10565-015-9298-5.
Stordal B, Hamon M, McEneaney V, Roche S, Gillet JP, O’Leary JJ, Gottesman M, Clynes M. Resistance to paclitaxel in a cisplatin-resistant ovarian cancer cell line is mediated by P-glycoprotein. PLoS One. 2012;7:e40717. doi:10.1371/journal.pone.0040717.
O’Connor R, Ooi MG, Meiller J, Jakubikova J, Klippel S, Delmore J, Richardson P, Anderson K, Clynes M, Mitsiades CS, O’Gorman P. The interaction of bortezomib with multidrug transporters: implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother Pharmacol. 2013;71:1357–68. doi:10.1007/s00280-013-2136-7.
Declèves X, Bihorel S, Debray M, Yousif S, Camenisch G, Scherrmann JM. ABC transporters and the accumulation of imatinib and its active metabolite CGP74588 in rat C6 glioma cells. Pharmacol Res. 2008;57:214–22. doi:10.1016/j.phrs.2008.01.006.
Sato H, Siddig S, Uzu M, Suzuki S, Nomura Y, Kashiba T, Gushimiyagi K, Sekine Y, Uehara T, Arano Y, Yamaura K, Ueno K. Elacridar enhances the cytotoxic effects of sunitinib and prevents multidrug resistance in renal carcinoma cells. Eur J Pharmacol. 2015;746:258–66. doi:10.1016/j.ejphar.2014.11.02.
Neumanova Z, Cerveny L, Greenwood SL, Ceckova M, Staud F. Effect of drug efflux transporters on placental transport of antiretroviral agent abacavir. Reprod Toxicol. 2015;57:176–82. doi:10.1016/j.reprotox.2015.07.070.
de Souza J, Benet LZ, Huang Y, Storpirtis S. Comparison of bidirectional lamivudine and zidovudine transport using MDCK, MDCK-MDR1, and Caco-2 cell monolayers. J Pharm Sci. 2009;98:4413–9. doi:10.1002/jps.21744.
Zhang P, de Gooijer MC, Buil LC, Beijnen JH, Li G, van Tellingen O. ABCB1 and ABCG2 restrict the brain penetration of a panel of novel EZH2-inhibitors. Int J Cancer. 2007;137:2007–18. doi:10.1002/ijc.29566.
An G, Gallegos J, Morris ME. The bioflavonoid kaempferol is an Abcg2 substrate and inhibits Abcg2-mediated quercetin efflux. Drug Metab Dispos. 2011;39:426–32. doi:10.1124/dmd.110.035212.
Sugano T, Seike M, Noro R, Soeno C, Chiba M, Zou F, Nakamichi S, Nishijima N, Matsumoto M, Miyanaga A, Kubota K, Gemma A. Inhibition of ABCB1 overcomes cancer stem cell-like properties and acquired resistance to MET inhibitors in non-small cell lung cancer. Mol Cancer Ther. 2015;14:2433–40. doi:10.1158/1535-7163.MCT-15-0050.
Schrickx J, Lektarau Y, Fink-Gremmels J. Ochratoxin A secretion by ATP-dependent membrane transporters in Caco-2 cells. Arch Toxicol. 2006;80:243–9. doi:10.1007/s00204-005-0041-5.
Schrickx JA, Fink-Gremmels J. Danofloxacin-mesylate is a substrate for ATP-dependent efflux transporters. Br J Pharmacol. 2007;150:463–9. doi:10.1038/sj.bjp.0706974.
Miller DW, Hinton M, Chen F. Evaluation of drug efflux transporter liabilities of darifenacin in cell culture models of the blood-brain and blood-ocular barriers. Neurourol Urodyn. 2011;30:1633–8. doi:10.1002/nau.21110.
Lumen AA, Li L, Li J, Ahmed Z, Meng Z, Owen A, Ellens H, Hidalgo IJ, Bentz J. Transport inhibition of digoxin using several common Pgp expressing cell lines is not necessarily reporting only on inhibitor binding to Pgp. PLoS One. 2013;8:e69394. doi:10.1371/journal.pone.0069394.
Planting AS, Sonneveld P, van der Gaast A, Sparreboom A, van der Burg ME, Luyten GP, de Leeuw K, de Boer-Dennert M, Wissel PS, Jewell RC, Paul EM, Purvis NB Jr, Verweij J. A phase I and pharmacologic study of the MDR converter GF120918 in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2005;55:91–9. doi:10.1007/s00280-004-0854-6.
Lu Y, Slizgi JR, Brouwer KR, Claire RL, Freeman KM, Pan M, Brock WJ, Brouwer KL. Hepatocellular disposition and transporter interactions with tolvaptan and metabolites in sandwich-cultured human hepatocytes. Drug Metab Dispos. 2016;. doi:10.1124/dmd.115.067629.
Nekhayeva IA, Nanovskaya TN, Hankins GD, Ahmed MS. Role of human placental efflux transporter P-glycoprotein in the transfer of buprenorphine, levo-alpha-acetylmethadol, and paclitaxel. Am J Perinatol. 2006;23:423–30. doi:10.1055/s-2006-951301.
Yao HM, Chiou WL. The complexity of intestinal absorption and exsorption of digoxin in rats. Int J Pharm. 2006;322:79–86. doi:10.1016/j.ijpharm.2006.05.030.
Ward KW, Azzarano LM. Preclinical pharmacokinetic properties of the P-glycoprotein inhibitor GF120918A (HCl salt of GF120918, 9,10-dihydro-5-methoxy-9-oxo-N-[4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]phenyl]-4-acridine-carboxamide) in the mouse, rat, dog, and monkey. J Pharmacol Exp Ther. 2004;310:703–9. doi:10.1124/jpet.104.068288.
Sane R, Agarwal S, Elmquist WF. Brain distribution and bioavailability of elacridar after different routes of administration in the mouse. Drug Metab Dispos. 2012;40:1612–9. doi:10.1124/dmd.112.045930.
Sane R, Agarwal S, Mittapalli RK, Elmquist WF. Saturable active efflux by p-glycoprotein and breast cancer resistance protein at the blood-brain barrier leads to nonlinear distribution of elacridar to the central nervous system. J Pharmacol Exp Ther. 2013;345:111–24. doi:10.1124/jpet.112.199786.
Kuppens IE, Witteveen EO, Jewell RC, Radema SA, Paul EM, Mangum SG, Beijnen JH, Voest EE, Schellens JH. A phase I, randomized, open-label, parallel-cohort, dose-finding study of elacridar (GF120918) and oral topotecan in cancer patients. Clin Cancer Res. 2007;13:3276–85. doi:10.1158/1078-0432.CCR-06-2414.
Tang SC, Kort A, Cheung KL, Rosing H, Fukami T, Durmus S, Wagenaar E, Hendrikx JJ, Nakajima M, van Vlijmen BJ, Beijnen JH, Schinkel AH. P-glycoprotein, CYP3A, and plasma carboxylesterase determine brain disposition and oral availability of the novel taxane cabazitaxel (Jevtana) in mice. Mol Pharm. 2015;12:3714–23. doi:10.1021/acs.molpharmaceut.5b00470.
Lin F, Marchetti S, Pluim D, Iusuf D, Mazzanti R, Schellens JH, Beijnen JH, van Tellingen O. Abcc4 together with abcb1 and abcg2 form a robust cooperative drug efflux system that restricts the brain entry of camptothecin analogues. Clin Cancer Res. 2013;19:2084–95. doi:10.1158/1078-0432.CCR-12-3105.
Adane ED, Liu Z, Xiang TX, Anderson BD, Leggas M. Pharmacokinetic modeling to assess factors affecting the oral bioavailability of the lactone and carboxylate forms of the lipophilic camptothecin analogue AR-67 in rats. Pharm Res. 2012;29:1722–36. doi:10.1007/s11095-011-0617-0.
de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13:6440–9. doi:10.1158/1078-0432.CCR-07-1335.
Jonker JW, Smit JW, Brinkhuis RF, Maliepaard M, Beijnen JH, Schellens JH, Schinkel AH. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst. 2000;92:1651–6. doi:10.1093/jnci/92.20.1651.
Yamasaki T, Fujinaga M, Kawamura K, Hatori A, Yui J, Nengaki N, Ogawa M, Yoshida Y, Wakizaka H, Yanamoto K, Fukumura T, Zhang MR. Evaluation of the P-glycoprotein- and breast cancer resistance protein-mediated brain penetration of 11C-labeled topotecan using small-animal positron emission tomography. Nucl Med Biol. 2011;38:707–14. doi:10.1016/j.nucmedbio.2010.12.012.
Kemper EM, Verheij M, Boogerd W, Beijnen JH, van Tellingen O. Improved penetration of docetaxel into the brain by co-administration of inhibitors of P-glycoprotein. Eur J Cancer. 2004;40:1269–74. doi:10.1016/j.ejca.2004.01.024.
Bardelmeijer HA, Ouwehand M, Beijnen JH, Schellens JH, van Tellingen O. Efficacy of novel P-glycoprotein inhibitors to increase the oral uptake of paclitaxel in mice. Invest New Drug. 2004;22:219–29. doi:10.1023/B:DRUG.0000026248.45084.21.
Kemper EM, van Zandbergen AE, Cleypool C, Mos HA, Boogerd W, Beijnen JH, van Tellingen O. Increased penetration of paclitaxel into the brain by inhibition of P-glycoprotein. Clin Cancer Res. 2003;9:2849–55.
Hendrikx JJ, Lagas JS, Wagenaar E, Rosing H, Schellens JH, Beijnen JH, Schinkel AH. Oral co-administration of elacridar and ritonavir enhances plasma levels of oral paclitaxel and docetaxel without affecting relative brain accumulation. Br J Cancer. 2014;110:2669–76. doi:10.1038/bjc.2014.222.
Minocha M, Khurana V, Qin B, Pal D, Mitra AK. Enhanced brain accumulation of pazopanib by modulating P-gp and Bcrp1 mediated efflux with canertinib or erlotinib. Int J Pharm. 2012;436:127–34. doi:10.1016/j.ijpharm.2012.05.038.
Choo EF, Ly J, Chan J, Shahidi-Latham SK, Messick K, Plise EQuiason CM, Yang L. Role of P-glycoprotein on the brain penetration and brain pharmacodynamic activity of the MEK inhibitor cobimetinib. Mol Pharm. 2014;11:4199–207. doi:10.1021/mp500435s.
Tang SC, Nguyen LN, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Int J Cancer. 2014;134:1484–94. doi:10.1002/ijc.28475.
Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther. 2009;330:956–63. doi:10.1124/jpet.109.154781.
Lagas JS, van Waterschoot RA, van Tilburg VA, Hillebrand MJ, Lankheet N, Rosing H, Beijnen JH, Schinkel AH. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin Cancer Res. 2009;15:2344–51. doi:10.1158/1078-0432.CCR-08-2253.
Mittapalli RK, Chung AH, Parrish KE, Crabtree D, Halvorson KG, Hu G, Elmquist WF, Becher OJ. ABCG2 and ABCB1 limit the efficacy of dasatinib in a PDGF-B-driven brainstem glioma model. Mol Cancer Ther. 2016;15:819–29. doi:10.1158/1535-7163.MCT-15-0093.
Agarwal S, Manchanda P, Vogelbaum MA, Ohlfest JR, Elmquist WF. Function of the blood-brain barrier and restriction of drug delivery to invasive glioma cells: findings in an orthotopic rat xenograft model of glioma. Drug Metab Dispos. 2013;41:33–9. doi:10.1124/dmd.112.048322.
Traxl A, Wanek T, Mairinger S, Stanek J, Filip T, Sauberer M, Müller M, Kuntner C, Langer O. Breast cancer resistance protein and P-glycoprotein influence in vivo disposition of 11C-erlotinib. J Nucl Med. 2015;56:1930–6. doi:10.2967/jnumed.115.161273.
Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther. 2010;334:147–55. doi:10.1124/jpet.110.167601.
Kawamura K, Yamasaki T, Yui J, Hatori A, Konno F, Kumata K, Irie T, Fukumura T, Suzuki K, Kanno I, Zhang MR. In vivo evaluation of P-glycoprotein and breast cancer resistance protein modulation in the brain using [(11)C]gefitinib. Nucl Med Biol. 2009;36:239–46. doi:10.1016/j.nucmedbio.2008.12.006.
Bihorel S, Camenisch G, Lemaire M, Scherrmann JM. Influence of breast cancer resistance protein (Abcg2) and P-glycoprotein (Abcb1a) on the transport of imatinib mesylate (Gleevec) across the mouse blood-brain barrier. J Neurochem. 2007;102:1749–57. doi:10.1111/j.1471-4159.2007.04808.x.
Breedveld P, Pluim D, Cipriani G, Wielinga P, van Tellingen O, Schinkel AH, Schellens JH. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005;65:2577–82. doi:10.1158/0008-5472.CAN-04-2416.
Bihorel S, Camenisch G, Lemaire M, Scherrmann JM. Modulation of the brain distribution of imatinib and its metabolites in mice by valspodar, zosuquidar and elacridar. Pharm Res. 2007;24:1720–8. doi:10.1007/s11095-007-9278-4.
Oostendorp RL, Buckle T, Beijnen JH, van Tellingen O, Schellens JH. The effect of Pgp (Mdr1a/1b), BCRP (Bcrp1) and Pgp/BCRP inhibitors on the in vivo absorption, distribution, metabolism and excretion of imatinib. Invest New Drugs. 2009;27:31–40. doi:10.1007/s10637-008-9138-z.
Tang SC, Lagas JS, Lankheet NA, Poller B, Hillebrand MJ, Rosing H, Beijnen JH, Schinkel AH. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib co-administration. Int J Cancer. 2012;130:223–33. doi:10.1002/ijc.26000.
Tang SC, Lankheet NA, Poller B, Wagenaar E, Beijnen JH, Schinkel AH. P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) restrict brain accumulation of the active sunitinib metabolite N-desethyl sunitinib. J Pharmacol Exp Ther. 2012;341:164–73. doi:10.1124/jpet.111.186908.
Oberoi RK, Mittapalli RK, Elmquist WF. Pharmacokinetic assessment of efflux transport in sunitinib distribution to the brain. J Pharmacol Exp Ther. 2013;347:755–64. doi:10.1124/jpet.113.208959.
Parrish KE, Pokorny J, Mittapalli RK, Bakken K, Sarkaria JN, Elmquist WF. Efflux transporters at the blood-brain barrier limit delivery and efficacy of cyclin-dependent kinase 4/6 inhibitor palbociclib (PD-0332991) in an orthotopic brain tumor model. J Pharmacol Exp Ther. 2015;355:264–71. doi:10.1124/jpet.115.228213.
Lagas JS, van Waterschoot RA, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther. 2010;9:319–26. doi:10.1158/1535-7163.MCT-09-066.
Agarwal S, Sane R, Ohlfest JR, Elmquist WF. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther. 2011;336:223–33. doi:10.1124/jpet.110.175034.
Minocha M, Khurana V, Qin B, Pal D, Mitra AK. Co-administration strategy to enhance brain accumulation of vandetanib by modulating P-glycoprotein (Pgp/Abcb1) and breast cancer resistance protein (Bcrp1/Abcg2) mediated efflux with m-TOR inhibitors. Int J Pharm. 2012;434:306–14. doi:10.1016/j.ijpharm.2012.05.028.
Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Oral availability and brain penetration of the B-RAFV600E inhibitor vemurafenib can be enhanced by the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Mol Pharm. 2012;9:3236–45. doi:10.1021/mp3003144.
Edwards JE, Brouwer KR, McNamara PJ. GF120918, a P-glycoprotein modulator, increases the concentration of unbound amprenavir in the central nervous system in rats. Antimicrob Agents Chemother. 2002;46:2284–6. doi:10.1128/AAC.46.7.2284-2286.2002.
Savolainen J, Edwards JE, Morgan ME, McNamara PJ, Anderson BD. Effects of a P-glycoprotein inhibitor on brain and plasma concentrations of anti-human immunodeficiency virus drugs administered in combination in rats. Drug Metab Dispos. 2002;30:479–82. doi:10.1124/dmd.30.5.479.
Robillard KR, Chan GN, Zhang G, la Porte C, Cameron W, Bendayan R. Role of P-glycoprotein in the distribution of the HIV protease inhibitor atazanavir in the brain and male genital tract. Antimicrob Agents Chemother. 2014;58:1713–22. doi:10.1128/AAC.02031-13.
Huisman MT, Smit JW, Wiltshire HR, Beijnen JH, Schinkel AH. Assessing safety and efficacy of directed P-glycoprotein inhibition to improve the pharmacokinetic properties of saquinavir coadministered with ritonavir. J Pharmacol Exp Ther. 2003;304:596–602. doi:10.1124/jpet.102.044388.
Shi J, Cao B, Zha WB, Wu XL, Liu LS, Xiao WJ, Gu RR, Sun RB, Yu XY, Zheng T, Li MJ, Wang XW, Zhou J, Mao Y, Ge C, Ma T, Xia WJ, Aa JY, Wang GJ, Liu CX. Pharmacokinetic interactions between 20(S)-ginsenoside Rh2 and the HIV protease inhibitor ritonavir in vitro and in vivo. Acta Pharmacol Sin. 2013;34:1349–58. doi:10.1038/aps.2013.69.
Gozalpour E, Greupink R, Bilos A, Verweij V, van den Heuvel JJ, Masereeuw R, Russel FG, Koenderink JB. Convallatoxin: a new P-glycoprotein substrate. Eur J Pharmacol. 2014;744:18–27. doi:10.1016/j.ejphar.2014.09.031.
Vaidhyanathan S, Wilken-Resman B, Ma DJ, Parrish KE, Mittapalli RK, Carlson BL, Sarkaria JN, Elmquist WF. Factors influencing the central nervous system distribution of a novel Phosphoinositide 3-Kinase/mammalian target of rapamycin inhibitor GSK2126458: implications for overcoming resistance with combination therapy for melanoma brain metastases. J Pharmacol Exp Ther. 2016;356:251–9. doi:10.1124/jpet.115.229393.
Polli JW, Baughman TM, Humphreys JE, Jordan KH, Mote AL, Webster LO, Barnaby RJ, Vitulli G, Bertolotti L, Read KD, Serabjit-Singh CJ. The systemic exposure of an N-methyl-D-aspartate receptor antagonist is limited in mice by the P-glycoprotein and breast cancer resistance protein efflux transporters. Drug Metab Dispos. 2004;32:722–6. doi:10.1124/dmd.32.7.722.
Liu F, Wang X, Li Z, Li J, Zhuang X, Zhang Z. P-glycoprotein (ABCB1) limits the brain distribution of YQA-14, a novel dopamine D3 receptor antagonist. Chem Pharm Bull (Tokyo). 2015;63:512–8. doi:10.1248/cpb.c15-00089.
Zamek-Gliszczynski MJ, Hoffmaster KA, Tian X, Zhao R, Polli JW, Humphreys JE, Webster LO, Bridges AS, Kalvass JC, Brouwer KL. Multiple mechanisms are involved in the biliary excretion of acetaminophen sulfate in the rat: role of Mrp2 and Bcrp1. Drug Metab Dispos. 2005;33:1158–65. doi:10.1124/dmd.104.002188.
Lee YJ, Kusuhara H, Jonker JW, Schinkel AH, Sugiyama Y. Investigation of efflux transport of dehydroepiandrosterone sulfate and mitoxantrone at the mouse blood-brain barrier: a minor role of breast cancer resistance protein. J Pharmacol Exp Ther. 2005;312:44–52. doi:10.1124/jpet.104.073320.
Kallem R, Kulkarni CP, Patel D, Thakur M, Sinz M, Singh SP, Mahammad SS, Mandlekar S. A simplified protocol employing elacridar in rodents: a screening model in drug discovery to assess Pgp mediated efflux at the blood brain barrier. Drug Metab Lett. 2012;6:134–44. doi:10.2174/1872312811206020134.
Imbert F, Jardin M, Fernandez C, Gantier JC, Dromer F, Baron G, Mentre F, Van Beijsterveldt L, Singlas E, Gimenez F. Effect of efflux inhibition on brain uptake of itraconazole in mice infected with Cryptococcus neoformans. Drug Metab Dispos. 2003;31:319–25. doi:10.1124/dmd.31.3.319.
Montesinos RN, Moulari B, Gromand J, Beduneau A, Lamprecht A, Pellequer Y. Co-administration of P-glycoprotein modulators on loperamide pharmacokinetics and brain distribution. Drug Metab Dispos. 2014;42:700–6. doi:10.1124/dmd.113.055566.
de Barraud Lagerie S, Comets E, Gautrand C, Fernandez C, Auchere D, Singlas E, Mentre F, Gimenez F. Cerebral uptake of mefloquine enantiomers with and without the Pgp inhibitor elacridar (GF1210918) in mice. Br J Pharmacol. 2004;141:1214–22. doi:10.1038/sj.bjp.0705721.
Mariappan TT, Kurawattimath V, Gautam SS, Kulkarni CP, Kallem R, Taskar KS, Marathe PH, Mandlekar S. Estimation of the unbound brain concentration of P-glycoprotein substrates or nonsubstrates by a serial cerebrospinal fluid sampling technique in rats. Mol Pharm. 2014;11:477–85. doi:10.1021/mp400436d.
Letrent SP, Pollack GM, Brouwer KR, Brouwer KL. Effects of a potent and specific P-glycoprotein inhibitor on the blood-brain barrier distribution and antinociceptive effect of morphine in the rat. Drug Metab Dispos. 1999;27:827–34.
Jablonski MR, Markandaiah SS, Jacob D, Meng NJ, Li K, Gennaro V, Lepore AC, Trotti D, Pasinelli P. Inhibiting drug efflux transporters improves efficacy of ALS therapeutics. Ann Clin Transl Neurol. 2014;1:996–1005. doi:10.1002/acn3.141.
Kuntner C, Bankstahl JP, Bankstahl M, Stanek J, Wanek T, Stundner G, Karch R, Brauner R, Meier M, Ding X, Müller M, Löscher W, Langer O. Dose-response assessment of tariquidar and elacridar and regional quantification of P-glycoprotein inhibition at the rat blood-brain barrier using (R)-[(11)C]verapamil PET. Eur J Nucl Med Mol Imaging. 2010;37:942–53. doi:10.1007/s00259-009-1332-5.
Sparreboom A, Planting AS, Jewell RC, van der Burg ME, van der Gaast A, de Bruijn P, Loos WJ, Nooter K, Chandler LH, Paul EM, Wissel PS, Verweij J. Clinical pharmacokinetics of doxorubicin in combination with GF120918, a potent inhibitor of MDR1 P-glycoprotein. Anticancer Drugs. 1999;10:719–28.
Malingré MM, Beijnen JH, Rosing H, Koopman FJ, Jewell RC, Paul EM, Ten Bokkel Huinink WW, Schellens JH. Co-administration of GF120918 significantly increases the systemic exposure to oral paclitaxel in cancer patients. Br J Cancer. 2001;84:42–7. doi:10.1054/bjoc.2000.1543.
Cripe LD, Uno H, Paietta EM, Litzow MR, Ketterling RP, Bennett JM, Rowe JM, Lazarus HM, Luger S, Tallman MS. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood. 2010;116:4077–85. doi:10.1182/blood-2010-04-277269.
Mandery K, Glaeser H, Fromm MF. Interaction of innovative small molecule drugs used for cancer therapy with drug transporters. Br J Pharmacol. 2012;165:345–62. doi:10.1111/j.1476-5381.2011.01618.x.
O’Brien FE, O’Connor RM, Clarke G, Dinan TG, Griffin BT, Cryan JF. P-glycoprotein inhibition increases the brain distribution and antidepressant-like activity of escitalopram in rodents. Neuropsychopharmacology. 2013;38:2209–19. doi:10.1038/npp.2013.120.
Bauer M, Blaickner M, Philippe C, Wadsak W, Hacker M, Zeitlinger M, Langer O. Whole-body distribution and radiation dosimetry of 11C-elacridar and 11C-tariquidar in humans. J Nucl Med. 2016;57:1265–8. doi:10.2967/jnumed.116.175182.
https://clinicaltrials.gov./ct2/show/record/NCT00048633?term=tariquidar&rank=8. Accessed on 18 Dec 2017.
https://clinicaltrials.gov./ct2/show/record/NCT00046930?term=zosuquidar&rank=2. Accessed on 18 Dec 2017
https://clinicaltrials.gov./ct2/show/record/NCT00028873?term=laniquidar&rank=1. Accessed on 18 Dec 2017.
Bauer M, Zeitlinger M, Todorut D, Böhmdorfer M, Müller M, Langer O, Jäger W. Pharmacokinetics of single ascending doses of the P-glycoprotein inhibitor tariquidar in healthy subjects. Pharmacology. 2013;91:12–9. doi:10.1159/000343243.
van Zuylen L, Sparreboom A, van der Gaast A, van der Burg ME, van Beurden V, Bol CJ, Woestenborghs R, Palmer PA, Verweij J. The orally administered P-glycoprotein inhibitor R101933 does not alter the plasma pharmacokinetics of docetaxel. Clin Cancer Res. 2000;6:1365–71.
Chi KN, Chia SK, Dixon R, Newman MJ, Wacher VJ, Sikic B, Gelmon KA. A phase I pharmacokinetic study of the P-glycoprotein inhibitor, ONT-093, in combination with paclitaxel in patients with advanced cancer. Invest New Drugs. 2005;23:311–5. doi:10.1007/s10637-005-1439-x.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for preparation of this manuscript.
Conflicts of interest
Ranjeet Prasad Dash, R. Jayachandra Babu and Nuggehally R Srinivas have no conflicts of interest or competing interests relevant to the contents of the review article.
Rights and permissions
About this article

Cite this article
Dash, R.P., Jayachandra Babu, R. & Srinivas, N.R. Therapeutic Potential and Utility of Elacridar with Respect to P-glycoprotein Inhibition: An Insight from the Published In Vitro, Preclinical and Clinical Studies. Eur J Drug Metab Pharmacokinet 42, 915–933 (2017). https://doi.org/10.1007/s13318-017-0411-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-017-0411-4