
Abstract
(R)-2-Amino-1,3′,3′-trimethyl-7′-(pyrimidin-5-yl)-3′,4′-dihydro-2′H-spiro[imidazole-4,1′-naphthalen]-5(1H)-one (GNE-892) is an orally administered inhibitor of β-secretase 1 (β-site amyloid precursor protein cleaving enzyme 1, BACE1) that was developed as an intervention therapy against Alzheimer’s disease. A clinical microdosing strategy was being considered for de-risking the potential pharmacokinetic liabilities of GNE-892. We tested whether dose-proportionality was observed in cynomolgus monkey as proof-of-concept for a human microdosing study. With cryopreserved monkey hepatocytes, concentration-dependency for substrate turnover and the relative contribution of P450- versus AO-mediated metabolism were observed. Characterization of the kinetics of these metabolic pathways demonstrated differences in the affinities of P450 and AO for GNE-892, which supported the metabolic profiles that had been obtained. To test if this metabolic shift occurred in vivo, mass balance studies in monkeys were conducted at doses of 0.085 and 15 mg/kg. Plasma exposure of GNE-892 following oral administration was more than 20-fold greater than dose proportional at the high-dose. P-gp-mediated efflux was unable to explain the discrepancy. The profiles of metabolites in circulation and excreta were indicative that oxidative metabolism limited the exposure to unchanged GNE-892 at the low dose. Further, the in vivo data supported the concentration-dependent metabolic shift between P450 and AO. In conclusion, microdosing of GNE-892 was not predictive of pharmacokinetics at a more pharmacologically relevant dose due to saturable absorption and metabolism. Therefore, it is important to consider ADME liabilities and their potential concentration-dependency when deciding upon a clinical microdosing strategy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
GNE-892 was a promising candidate for drug development as an oral therapy for slowing the progression of Alzheimer’s disease. Through structure optimization, GNE-892 was discovered to inhibit β-secretase 1 (β-site amyloid precursor protein cleaving enzyme 1, BACE1) (Hunt et al. 2013). BACE1 is an attractive target for pharmacological intervention in Alzheimer’s disease on the basis of its critical role in processing amyloid precursor protein to β-amyloid (Aβ) fibrils (Vassar et al. 1999). Neurodegeneration that manifests as dementia is hypothesized to result from interactions of Aβ fibrils or their aggregates, as neurofibrillary plaques, with neurons (Hardy and Higgins 1992; Hardy and Allsop 1991). GNE-892 is a potent inhibitor of BACE1 with in vitro enzymatic and cellular IC50s of 48 and 27 nM, respectively (Hunt et al. 2013). Following a single oral dose of GNE-892 to cynomolgus monkeys, Aβ levels were significantly reduced in plasma and cerebrospinal fluid (Liu et al. 2013).
The results of in vitro and preclinical assessments indicated that the disposition of GNE-892 was complex and governed by both transport and metabolism. Active efflux by P-glycoprotein (P-gp) was indicated in transfected cells (basolateral to apical (B–A)/apical to basolateral (A–B) ratio of 25 in MDCK-MDR1 cells), which translated to a sevenfold increase in the ratio of areas under the curve from 0 to 1 h (AUC0–1h) in brain versus plasma in Mdr1a/1b knockout relative to wild-type mice (Hunt et al. 2013). The metabolic pathways for GNE-892 in vitro using human liver fractions were mainly oxidative. Though most of the oxidative metabolites appeared to be generated by cytochrome P450 (P450), some formed without NADPH in cytosol-containing matrices, which suggested that aldehyde oxidase (AO) enzymes were responsible. Although the P450 contribution to in vivo clearance could be predicted with confidence, the same could not be said for the contribution of AO. To date, the extrapolation of in vitro clearance for AO substrates has not provided accurate predictions of the clearances that were observed in vivo and has ultimately resulted in failures in the clinic due to inadequate pharmacokinetics (PK) (Pryde et al. 2010; Hutzler et al. 2013).
As a result of these complexities, notable uncertainty existed in the predictions of clinical dose of GNE-892. Microdosing studies of GNE-892 were then considered as a potential strategy to increase confidence in the human PK before pursuing a traditional Phase I clinical study. Microdosing is an approach to obtain early PK that may be especially important for decision making when notable uncertainty exists in the predicted human PK based on in vitro and preclinical species data (Rowland 2012; Lappin 2010; Garner 2010). Because of the low dose (dose strength less than 100 µg or 1/100 of pharmacological dose), an abridged preclinical safety assessment and smaller scale for material requirements would be necessary. This allows microdosing to provide a clinical readout of a drug candidate without the huge resource commitments of full clinical development, albeit without a pharmacodynamic (PD) readout.
The focus of the current study was to test the validity of a microdosing strategy for GNE-892 in monkey as a surrogate for human. These studies were conducted with cynomolgus monkeys given the considerable data and understanding that had been garnered from PK-PD studies of GNE-892 (Liu et al. 2013). Further, monkey was deemed a more relevant non-rodent preclinical species for the metabolic evaluation of GNE-892. Dog was considered not to be appropriate for assessing the ADME liabilities, especially in light of the contribution by AO to metabolism and the absence of functional AO in dogs (Garattini et al. 2008; Beedham et al. 1987). The reported precedence for microdosing studies in preclinical species, though limited, has been largely positive for their scalability to pharmacological dose. The majority of compounds in these studies were dosed in rats and showed linear PK (Balani et al. 2006; Ni et al. 2008). An exception was the investigational compound, MLNX, which showed linearity through two doses (0.01 and 1 mg/kg), but failed to show dose-proportionality through the highest dose (10 mg/kg) (Balani et al. 2006). In larger preclinical species such as dog and monkey, drug candidate molecules tested via a microdosing design were reported to exhibit linear PK (Sandhu et al. 2004; McLean et al. 2007).
Here, we report the results from the in vitro metabolism and a two-dose, in vivo ADME study of GNE-892 in cynomolgus monkeys. We have attempted to rationalize the dose-dependent differences that were observed in the radioactive profiles of circulating and excreted metabolites, and how they aligned with in vitro findings, to demonstrate the rationale in the decision not to pursue a human microdosing strategy for GNE-892.
2 Materials and methods
2.1 General chemicals
Formic acid, acetonitrile (ACN), and methanol were purchased from EMD Millipore (Billerica, MA, USA). Deuterium oxide and methanol-d4 were purchased from Sigma (St Louis, MO, USA). Acetic acid and ammonium hydroxide were purchased from J.T. Baker (Center Valley, PA, USA). Water was filtered using a Barnstead NANOPure® II purification system (Thermo Scientific, Waltham, MA, USA). Liquid scintillation cocktails were purchased from R. J. Harvey Instrument Corporation (Hillsdale, NJ, USA), Beckman Instruments, Inc. (Fullerton, CA, USA), or Amersham Biosciences (Piscataway, NJ, USA).
2.2 Reference compounds
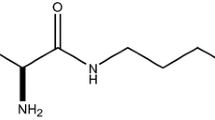
(R)-2-Amino-1,3′,3′-trimethyl-7′-(pyrimidin-5-yl)-3′,4′-dihydro-2′H-spiro[imidazole-4,1′-naphthalen]-5(1H)-one (GNE-892) and M7 were synthesized at Genentech as single stereoisomers with chemical purities of greater than 99 %. [14C]GNE-892 was synthesized by Quotient Bioresearch with a chemical purity of 98.5 % and a radiochemical purity of 99.5 %. The specific activity of the radiolabeled material was 60 mCi/mmol with the radiolabel at carbon 4 of the pyrimidine (Fig. 1).

Chemical structure of [14C]GNE-892 with the position of the radiolabel at carbon 4 of the pyrimidine
2.3 In vitro hepatocyte incubations
Incubations were conducted using cryopreserved pooled male cynomolgus monkey hepatocytes (CellzDirect, Durham, NC, USA). Vials of hepatocytes were thawed in a water bath at 37 °C and diluted with Dulbecco’s modified Eagle’s medium (DMEM), pH 7.4. The cells were collected by centrifugation, media was removed, and the cells were re-suspended in DMEM. For determination of the metabolite distribution at low and high-starting substrate concentrations, GNE-892 was incubated with hepatocytes (final 1 × 106 cells per ml) at 0.05 or 10 µM at 37 °C with gentle shaking. 14C-labeled test article was used so that the generated metabolites could be quantitated based on radiometric analysis. At the low concentration, GNE-892 was incubated entirely as the 14C-test article, whereas for the high concentration, an equivalent amount of 14C was used and the remaining substrate was unlabeled material. At 2 h, incubations were terminated with the addition of three volumes of ACN. The samples were centrifuged and the supernatants were concentrated under vacuum at ambient temperature and analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS) with radiodetection in the same manner as was conducted for monkey excreta and plasma extracts (described below). In separate incubations conducted in the same manner, 1-aminobenzotriazole (ABT), a nonselective P450 inhibitor, was added to hepatocytes at 1 mM final concentration and incubated at 37 °C for 5 min prior to the addition of GNE-892.
2.4 Enzyme kinetics of GNE-892 in hepatocytes
Product formation experiments were conducted with cryopreserved hepatocyte suspension incubations in a similar manner as described above with the following modifications: GNE-892 (0.1–250 µM, n = 4 per concentration) was incubated with thawed hepatocytes at a cell density of 1 × 105 cells per ml in DMEM at a total sample volume of 0.5 ml. Incubations at 37 °C were conducted for 10 min before addition of the substrate, and for 60 min once substrate had been added. Reactions were terminated by addition of 0.5 ml of acetonitrile containing propranolol at 50 nM, which was used as an internal standard. Metabolites were monitored by high-resolution full-scan mass spectrometry following separation by a short (9 min) high-performance liquid chromatography (HPLC) method. Briefly, analytes were resolved on a Kinetex XB-C18, 50 × 2.1 mm, 2.6 µm column (Phenomenex, Torrance, CA, USA) with mobile phases of 0.4 % formic acid in water adjusted to pH 3.2 (mobile phase A) and methanol (B) at a constant flow rate of 0.75 ml/min. The mobile phases were initially 10 % B, which were then linearly changed to 20 % B at 1 min, 40 % B at 6 min, and 100 % at 6.1 min. The column was flushed for 1.4 min before returning to the starting conditions for 1.4 min of column re-equilibration. The formation rates of metabolites were calculated as peak area ratios (normalized to the internal standard) normalized by time and hepatocyte cell density. The metabolite formation profiles were fit with enzyme kinetics models and descriptive parameters were estimated using Prism 6.04 (GraphPad, La Jolla, CA, USA) software. The metabolite generation profiles were best fit by models describing substrate inhibition (Eq. 1) (Shou et al. 2001) or autoactivation (Eq. 2) (Witherow and Houston 1999):
where v is the observed rate of metabolite formation, \({V_{\rm max }}^{\rm app}\) is the apparent maximal rate of metabolism, [S] is the substrate concentration, \({K_{\rm m}}^{\rm app}\) or \({S_{50 }}^{\rm app}\) is the apparent substrate concentration at half \({V_{\rm max }}^{\rm app}\), K i is the dissociation constant for substrate binding, and n is the Hill coefficient. For substrate inhibition, [I] was taken to be equal to [S].
2.5 In vivo studies
All animal husbandry was in accordance with the Guide for the Care and Use of Laboratory Animals. Two male cynomolgus monkeys and a bile-duct cannulated (BDC) male cynomolgus monkey were individually housed in stainless steel metabolism cages. Animals were administered a single oral dose at a target of 15 mg/kg (15 µCi/kg), which was formulated as 1 % carboxymethylcellulose, 0.5 % Tween, and 5 mM sodium citrate in sterile water. Blood (2–4 ml per time point) was collected at pre-dose and at 0.083, 0.25, 0.5, 1, 3, 6, 9, 24, and 48 h post-dose and plasma was prepared. Urine and feces collections were carried out over 336 h post-dose. Bile samples were collected from the BDC animal at pre-dose and at 8, 24, 48, and 72 h post-dose. Following the final sample collection at 336 h post-dose, the bile-duct intact animals were given a 7-day wash-out period and administered a single oral dose at a target of 0.085 mg/kg (15 µCi/kg) in the same formulation as the previous dose. To reach the target dose of radioactivity, the 0.085 mg/kg dosing formulation contained only 14C test article, whereas for the 15 mg/kg dosing formulation, non-radiolabeled test article was added to the 14C test article. When tested in vitro, there was no evidence that GNE-892 was a time-dependent inhibitor of P450 enzymes; therefore, the crossover study design was not expected to have any impact on PK. The BDC animal was not dosed at 0.085 mg/kg. Urine and feces collections, as well as blood collection and plasma preparation, were carried out in the same manner as following the initial dose.
2.6 Determination of radioactivity
Levels of radioactivity in monkey plasma, urine, and bile samples were determined by counting aliquots for 10 min or until the 2-sigma error was less than or equal to 2 %, whichever came first, in a Beckman Model LS6000 liquid scintillation counter (LSC). Fecal samples were combusted in a Model OX-500 biological sample oxidizer (Harvey Instrument Corp, Hillsdale, NJ, USA) and the evolved 14CO2 was counted by LSC. Samples analyzed to produce extraction data were either counted in duplicate (no combustion) or triplicate (combustion). Quench correction was performed using an external standard method. The scintillation spectrometer was programmed to automatically subtract instrument background (including counting cocktail) and convert counts per min (cpm) to disintegrations per min (dpm). Oxidizer efficiencies were validated by combusting a known amount of [14C]mannitol.
2.7 Extraction of metabolites from biological samples
Aliquots of urine, feces, and bile samples were pooled relative to the excreted volume or weight at each time interval, so that each pooled sample represented greater than 95 % of the radioactivity excreted by that route for each animal group. Monkey urine, feces, and bile were pooled from 0–48, 0–72, and 0–48 h post-dose, respectively. Pooled urine and bile samples were centrifuged, and the supernatant was injected onto an HPLC column without further sample treatment. The pooled fecal homogenates were extracted with ACN. The supernatants were separated and the process was repeated. The combined supernatants were evaporated to dryness under nitrogen in an N-Evap Nitrogen Evaporator (Organomation Associates, Inc., Berlin, MA, USA). The residue was reconstituted with 0.1 % formic acid:ACN (1:1, v:v) before injection onto an HPLC column. A pooled plasma sample was prepared for high- and low-dose animals, with pooling volumes proportional to the sampling time interval up to and including 9 h such that the sample concentration reflected the systemic exposures for GNE-892 and its metabolites (Hop et al. 1998). Plasma samples were extracted twice with two volumes of ACN and the extracts were combined, concentrated under nitrogen, and the residue was re-dissolved with 0.1 % formic acid in water:methanol (2:1, v:v) for radiometric analysis.
2.8 Metabolite profiling
Metabolite profiling was conducted on an Alliance 2695 HPLC system (Waters, Milford, MA, USA) with an ACE 3 AR-C18, 3 µm, 4.6 × 150 mm (MAC-MOD Analytical, Chadds Ford, PA, USA) column. The mobile phases were 0.4 % formic acid in water adjusted to pH 3.2 with ammonium hydroxide (mobile phase A), methanol (mobile phase B), and ACN (mobile phase C). The flow rate was 0.7 ml/min. HPLC gradients were initiated with 100 % A for 1.5 min with a shallow gradient to 15 % B at 3.5 min, 45 % B at 68.5 min, and 70 % B at 83.5 min. The column was washed with a gradient to 100 % C over 4 min, then held for 6.5 min before returning to the initial composition of 100 % A within 2 min. The system was allowed to equilibrate for 15 min before the next injection. Plasma and in vitro samples were profiled using an Accela HPLC system (Thermo Fisher Scientific, San Jose, CA, USA) with a modified chromatographic method that used two mobile phases. Mobile phase C was not used and the column was flushed with 100 % B. Experiments for hydrogen–deuterium (H/D) exchange were performed by replacing the water component of the aqueous mobile phase with deuterium oxide and replacing methanol with methanol-d4 to determine the number of exchanged hydrogens.
For radiodetection and construction of metabolite profiles, the column effluent was collected to DeepWell LumaPlate-96 plates (Perkin Elmer, Waltham, MA, USA) by time (15 s/fraction) and subsequently dried under vacuum for up to 8 h. The radioactivity in each fraction was measured for 5 min using a TopCount® NXT™ Microplate Counter (Perkin Elmer). HPLC radiochromatograms were reconstructed using ARC Convert and Evaluation (AIM Research, Newark, DE, USA) or Laura (LabLogic, Brandon, FL, USA) software and the radiopeaks were integrated to determine the percent distribution of radioactivity in each sample.
2.9 Metabolite identification
MS spectra were obtained on an LTQ Orbitrap high-resolution mass spectrometer equipped with an electrospray ionization source (Thermo Fisher Scientific). Solvent flow from the LC column was split such that mass spectrometric data for metabolite detection were collected simultaneously as radiodetection. The electrospray ion source voltage was 5.0 kV. The heated capillary temperature was 350 °C. The scan-event cycle consisted of a full-scan mass spectrum at a mass resolving power of 30,000 (at m/z 400) and the corresponding data-dependent tandem mass spectrometry (MSn) scans were acquired at a resolving power of 7,500. Accurate mass measurements were performed using external calibration.
3 Results
3.1 In vitro metabolite profiles
The in vitro metabolism of GNE-892 was assessed in 2 h incubations with cryopreserved hepatocytes from cynomolgus monkeys at 0.05 and 10 µM substrate concentrations (Table 1). These concentrations were selected at a 200-fold difference to approximate the dose differences for a microdosing study and also approached the maximal systemic concentrations for total radioactivity that were reached in the in vivo study. GNE-892 was extensively metabolized with 6 and 31 % of drug remaining at the end of incubation for the low- and high-concentration, respectively. In the incubation with a low GNE-892 concentration, M3, M5, and M7 accounted for 13, 45, and 8 % of the end sample radioactivity, respectively. At the higher concentration, M3, M5, and M7 accounted for 22, 9, and 14 % of the sample radioactivity, respectively. The formations of M2, M3, and M5 were inhibited by addition of ABT, a broad P450 inactivator, thus showing that these metabolites were generated by P450s. By contrast, generation of M7, the 4-hydroxypyrimidinyl of GNE-892, was not inhibited by ABT. Other in vitro experiments indicated that M7 was generated preferentially by liver cytosolic fractions rather than microsomes and was inhibited by hydralazine, a known chemical inhibitor of AO (Strelevitz et al. 2012) (data not shown). These data were interpreted as indicative that M7 was generated by AO.
The formations of the two major oxidative metabolites of GNE-892 were monitored in hepatocyte incubations to understand the potential for saturation, or partial saturation, of metabolic enzymes. Specifically, M5 was monitored to characterize P450 (retention time 3.6 min), and M7 was monitored to characterize AO (retention time 4.5 min). Incubation conditions (1 × 105 cells per ml and 60 min) were confirmed to provide linear initial reaction rates with less than 10 % depletion of the parent compound. Due to metabolite standards not being available, V max was not determined. A clear difference in the affinities of the enzyme systems for GNE-892 was observed with the estimated Michaelis constants (\({K_{\rm m}}^{\rm app}\) or \({S_{50}}^{\rm app}\)) of 1.4 µM for M5 formation (P450 metabolite) and 95 µM for M7 formation (AO metabolite) (Table 1; Fig. 2). Interestingly, the Eadie–Hoffstee transformation of the kinetic data demonstrated nonconformity to typical Michaelis–Menten kinetics (Fig. 2). In the case of M5 formation, the Eadie–Hoffstee plot showed a convex shape, which fitted with a substrate inhibition model estimated the dissociation constant (K i) to be 276 µM. In the case of M7 formation, the Eadie–Hoffstee plot showed a hook-shaped curve, which fitted with an autoactivation model estimated the Hill coefficient to be 1.2.

Kinetic profiles for formation of a M5 and b M7 by oxidation of GNE-892 in incubations with monkey hepatocytes. Data points and error bars are mean and standard deviation from four separate incubations. The inset plots show the corresponding Eadie–Hoffstee plots (rate versus rate/[S]), which indicated atypical kinetics. Formation of M5 was fit with a model of substrate inhibition and formation of M7 was fit with a model of autoactivation
3.2 Pharmacokinetics and excretion/mass balance
The mean dose-normalized plasma concentration–time profiles of total radioactivity following a single oral dose of [14C]GNE-892 at 0.085 and 15 mg/kg (low- and high-dose, respectively) are presented in Fig. 3. Radioactivity in plasma reached maximal concentrations at 1 h post-dose of 6.6 and 4,289 ng Eq/ml in low- and high-dose animals, respectively. The radioactivity then declined following a log-linear manner with half-lives of 5.7 and 3.0 h following the low and high dose, and was measurable up to 9 h post-dose. The AUC0–9h values of total radioactivity were 35.3 ng Eq h/ml for the low-dose and 19,500 ng Eq h/ml for the high-dose group. Based on radioprofiling results for pooled plasma samples, unchanged GNE-892 accounted for 4.5 % of the circulating radioactivity in the low-dose and 33.8 % in high-dose monkeys. This provided the calculated AUC0–9h for GNE-892 of 1.6 and 6581 ng Eq h/ml for the low- and high-doses, respectively.

Dose-normalized concentration–time profiles of total radioactivity in plasma following a single oral dose of [14C]GNE-892 at 0.085 mg/kg (open squares, dotted line) or 15 mg/kg (filled squares, solid line) in cynomolgus monkeys (n = 2)
Sample collections over 336 h recovered an average of 89.1 % of the administered radioactivity collected in excreta from low-dose and 86.2 % from high-dose monkeys. The average cumulative excretions in urine and feces were 7.8 and 81.2 % of the administered dose, respectively, for low-dose animals. For the high-dose animals, the average cumulative excretions in urine and feces were 28.9 and 57.2 %. Elimination of the administered radioactivity was rapid following dosing with greater than 80 % recovery within the first 48 h post-dose. For the BDC monkey that received an oral dose of 15 mg/kg, 32.3 % of the administered radioactivity was excreted in the bile over 48 h, and 46.0 and 14.9 % were recovered in feces and urine, respectively. On the basis of combined recovery of radioactivity in bile and urine for the high-dose BDC animal, at least 47 % of the oral dose had been absorbed.
3.3 In vivo metabolite profiles
The metabolism of GNE-892 in monkeys was assessed by profiling plasma, urine, and feces. Representative radioprofiles are presented in Fig. 4. The biotransformation pathways of GNE-892 are illustrated in Fig. 5. Radioactivity in plasma samples was determined to be distributed over GNE-892 and its metabolites (Table 2). Unchanged GNE-892 accounted for 4.5 % of the circulating radioactivity in the low-dose and 33.8 % in high-dose monkeys. For low-dose monkeys, M1, M5, and M8 were the most abundant plasma metabolites, accounting for 9.7, 25.0, and 24.2 % of the total radioactivity exposure, respectively. Other metabolites each accounted for 5.6 % or less of the plasma radioactivity. For high-dose monkeys, M5 and M7 were the most abundant plasma metabolites, accounting for 18.4 and 19.1 % of the total radioactivity exposure, respectively. Other minor metabolites each accounted for 2.7 % or less of the plasma radioactivity at this dose.

Representative radiochromatograms of GNE-892 and its metabolites in urine, feces, and plasma. Profiles are shown for cynomolgus monkeys that received a single oral dose at 15 mg/kg

Proposed metabolic pathways for GNE-892 in cynomolgus monkeys
GNE-892 and seven identified metabolites accounted for the majority of the radioactivity in excreta (Table 3) with the remaining radioactivity distributed over minor metabolites that individually accounted for less than 2 % of the dose. Excretion of unchanged GNE-892 in urine was low, accounting for 0.3 and 2.5 % of the dose in low- and high-dose animals, respectively. The major drug-related analytes in urine were M5 in low-dose animals, which accounted for 3.0 % of the dose, and M5 and M7 in high-dose animals, which accounted for 11.0 and 6.6 % of the dose, respectively. Unchanged GNE-892 was the major radioactive peak observed in feces, accounting for 49.4 and 27.0 % of the dose in low- and high-dose animals, respectively. For low-dose animals, M2 and M4 were the major drug-related analytes identified in feces and accounted for 12.5 and 6.7 % of the dose, respectively. In feces collected from high-dose animals, M2 accounted for 5.0 % and M7 accounted for 10.9 % of the dose.
In bile, unchanged drug was a minor component, while M1, M5, and M7 were the major biliary analytes. The low levels of M1 and M5 in feces from intact animals suggested that these metabolites were labile to further biotransformation after secretion into the gut lumen in bile. Structure characterization of M1 and M5 based on mass spectrometric data (see below) indicated that these metabolites were N-oxides. These data indicated that following biliary secretion, N-oxide metabolites (M1 and M5) were reduced in transit through the gastrointestinal tract before fecal excretion. This reduction of N-oxides by the gut flora has been previously reported (Powis et al. 1979). Reduction of M1 and M5 in the gut may contribute to levels of M2/M3 and unchanged GNE-892, respectively, in feces from intact animals.
3.4 Metabolite identification
GNE-892 and its metabolites in plasma and excreta were characterized based on full scan and multistage collision-induced dissociation (CID) product ion spectra. The proposed structures and fragmentation schemes are shown in Table 4. GNE-892 eluted at 73 min and had a protonated molecular ion ([M+H]+) at m/z 336.1820 (mass accuracy = 0.30 ppm). The product ion spectrum of GNE-892 is shown in Fig. 6. The product ion at m/z 126 corresponded to the intact oxo-imidazole moiety, while product ions at m/z 263 and 252 were formed as the result of cleavages of the oxo-imidazole moiety and corresponded to the pyrimidyl-tetrahydronaphthalene moiety. The product ions at m/z 308, 294, and 276 resulted from the loss of CO, NH2CN, and its sequential loss of water, respectively.

The collision-induced dissociation product ion spectrum obtained for GNE-892 at m/z 336
Metabolite M1 had an [M+H]+ at m/z 368.1722, consistent with a molecular formula of C19H22O3N5 (1.25 ppm), which was indicative of di-oxidation. Using H/D exchange, M1 was shown to have three exchangeable hydrogens, which were assigned as two protons at the primary amine of the oxo-imidazole moiety and a single proton at the methyl-hydroxy group. The second oxidation did not introduce an exchangeable proton and was proposed to be N-oxidation of the pyrimidine. This was further supported by an observed product ion at m/z 351, a neutral loss of 17 amu, which can be accounted for by loss of a hydroxyl radical, a known fragmentation for N-oxides. Product ions at m/z 295 and 284 corresponded to 32 amu shifts, showing that both oxidations occurred on the pyrimidyl-tetrahydronaphthalene (m/z 263–295 and m/z 252–284). The product ion at m/z 336 corresponded to the neutral loss of CH3OH (32 amu), suggesting that oxidation had occurred at one of the methyl groups.
Metabolites M2, M3, M5, and M7 had [M+H]+ at m/z 352.1771–352.1773, consistent with a molecular formula of C19H22O2N5 (<1.40 ppm) and indicative that they were generated through mono-oxidation reactions. The product ion spectra for M2 and M3 showed mass shifts of 16 amu on the pyrimidyl-tetrahydronaphthalene (m/z 263–279 and m/z 252–268), and both metabolites were observed to have three exchangeable hydrogens in H/D experiments, which was consistent with addition of a hydroxyl group. The facile losses of water were observed from several product ions arising from cleavages of the oxo-imidazole moiety (m/z 261, 250, and 221), which indicated that the oxidation had occurred at an aliphatic carbon. Hence, M2 and M3 were identified as oxidation on the aliphatic carbons of the dimethyl tetrahydronaphthalene moiety. M5 was identified as an N-oxide of GNE-892 on the basis of a product ion at m/z 335, resulting from the loss of a hydroxyl radical, its reduction to GNE-892 with the addition of titanium (III) chloride (data not shown), and two exchangeable hydrogens present at the primary amine of the oxo-imidazole moiety by H/D experiments. In addition, product ions at m/z 279, 268, and 251 were observed for M5, which supported that oxidation had occurred at the pyrimidine. M7 was identified as the oxidative product at carbon-4 of the pyrimidine moiety on the basis of a comparison to an authentic synthetic standard. The abundant product ions at m/z 324 and 268 resulted from cleavages across the oxo-imidazole, corresponding to modification of the pyrimidyl-tetrahydronaphthalene.
Metabolite M4 had an [M+H]+ at m/z 366.1565, consistent with a molecular formula of C19H20O3N5 (1.16 ppm). The product ion at m/z 282 for this metabolite by cleavage at the oxo-imidazole moiety indicated the addition of 30 amu at the pyrimidyl-tetrahydronaphthalene. Losses of water and formic acid were also observed with product ions at m/z 348 and 320, respectively. Further fragmentation of the ion at m/z 282 generated a base peak ion at m/z 236, a neutral loss of 46 amu, which was rationalized as the loss of formic acid. The losses of formic acid from the molecular and product ion were consistent with a carboxylic acid moiety at a methyl group at the cyclohexane ring. This structure assignment was supported by three exchangeable hydrogens identified in H/D experiments.
Metabolite M6 had an [M+H]+ at m/z 322.1667, consistent with a molecular formula of C18H20ON5 (1.56 ppm) and the loss of one methyl group. The product ion spectra identified an unchanged pyrimidyl-tetrahydronaphthalene moiety (m/z 263 and 252) and showed that N-demethylation occurred at the oxo-imidazole. In H/D experiments, three exchangeable hydrogens were identified, which was consistent with the structure assignment. For some samples, M6 and M7 coeluted, and the radiometric response was calculated from the mass spectrometric response for the individual metabolites. This calculation used a relative response ratio (radiometric to mass spectrometric) that was determined experimentally using a sample of the same matrix where the metabolites were chromatographically resolved to account for differences between metabolites in the mass spectrometric response.
Metabolite M8 was an early eluting analyte (retention time ~3 min) using the profiling chromatography. Efforts to isolate and chromatograph this metabolite were unable to provide a molecular ion and this metabolite remained unidentified.
3.5 Estimating the fraction of dose absorbed
The fraction of the administered oral dose absorbed was estimated as the sum of total radioactivity recovered in urine and the radioactivity from metabolites recovered in feces. These estimates were considered the minimum fraction of the oral dose that had been absorbed since part of the unchanged drug recovered in the feces could come from biliary excretion and any metabolite formed from absorbed drug that transformed back to parent drug during transit through the gut lumen would not be accounted. For example, M5 was observed in bile from the BDC animal, but was absent in feces from bile duct-intact animals presumably due to its reduction during gut transit. Hence, the contribution of M5 to the absorbed dose was not accounted and this contributed some uncertainty to the estimates. Despite this complication, the estimated fraction of dose absorbed supported that differences existed between the dose groups. For the low and high doses, it was estimated that 39.6 and 59.1 % of the dose had been absorbed, respectively. Consistent with this difference between dose groups, there was nearly two times more unchanged drug in feces from low-dose animals (49.4 % of the dose compared to 27.0 % for high-dose), and this was unlikely to be fully explained by M5 reduction in the gut.
4 Discussion
The current studies were conducted to facilitate decision making around the microdosing strategy for GNE-892 in the clinic. Since important ADME liabilities were identified early for this compound, it was unknown whether the assumption of linearity between the microdose and clinical dose was valid. Cynomolgus monkeys were selected as an appropriate surrogate for humans and were evaluated with in vitro and in vivo assessments that spanned the concentration and dose range that would be targeted for a microdosing study. The choice of monkey was made with consideration that it was the preclinical species used for efficacy studies (Liu et al. 2013) and AO was involved in the human metabolism of GNE-892 in vitro; thus, dog was an unsuitable choice for assessing metabolism.
With incubation of GNE-892 with cryopreserved cynomolgus monkey hepatocytes at substrate concentrations that differed by 200-fold, the turnover was less extensive at high concentration (69 %) than at low concentration (94 %) with the metabolites observed predominantly formed by P450-mediated pathways. However, the relative AO contribution to metabolism was greater at high concentration (M7 = 14 %) than at low concentration (M7 = 8 %). We further investigated the primary metabolic pathways for GNE-892 in vitro by characterizing the kinetics for P450- and AO-mediated formations of M5 and M7, respectively. It was apparent that P450s had greater affinity for GNE-892 and may be expected to be a major metabolic pathway at lower substrate concentrations, whilst AO had lower affinity, but higher capacity. Hence, saturation of P450, but not AO, could occur at the concentrations for GNE-892 that were attained in vivo following the high dose. These data were consistent with the in vivo metabolic profiles where metabolite formation was predominantly P450-mediated at low dose; but shifted to AO-mediated oxidation at higher dose as a consequence of P450 saturation. The hepatocyte data also suggested that atypical enzyme kinetics could act to increase the observed metabolic shift between P450 and AO. However, it would be complex to extrapolate these in vitro data to predict if substrate inhibition or cooperativity contributed to the dose-dependent differences seen with the in vivo metabolic profiles.
It was unknown whether the concentration-dependent metabolism of GNE-892 would occur in vivo. In addition, there was no precedence for the interplay between P450 and AO. Either factor, if recapitulated in the clinic, could reduce the translation of the PK obtained from microdosing to a higher, pharmacologically relevant dose. Hence, GNE-892 was administered to cynomolgus monkeys in a crossover design with single oral administration with a 176-fold difference in doses (0.085 and 15 mg/kg). A marked dose-dependence in exposure to GNE-892 was observed after oral administration (Fig. 3). The dose-normalized exposures to drug-related material (measured by total radioactivity) were greater than dose-proportional at 3.7- and 3.1-fold for maximum plasma concentration (C max) and total exposure (AUC0–9h), respectively. This was partially explained with the estimated 1.5-fold greater absorption after dosing 15 mg/kg GNE-892 [percent absorbed: 40 % (low dose) versus 59 % (high dose)]. A more dramatic difference was observed when considering only GNE-892 in plasma and in urine. Radioprofiling of plasma samples indicated that unchanged GNE-892 accounted for 5 and 34 % of the plasma radioactivity in low- and high-dose animals, respectively, which corresponded to drug exposures more than 20-fold greater than proportional to the dose increase. The large difference in systemic GNE-892 between the oral doses was confirmed by a ninefold difference in the fraction of unchanged drug that was excreted in urine between the two dose groups (0.29 % for low dose versus 2.5 % for high dose).
The source of the non-linearity in oral exposures could be explained by saturation of P-gp-mediated efflux or P450- or AO-mediated metabolism. Since GNE-892 is a substrate for P-gp, efflux transport at the gut wall could be effectively effluxing absorbed drug to the gut lumen, potentially having a negative impact on absorption. At the higher dose, this process could be saturated allowing more drug to reach the systemic circulation. Evidence for the active transport was observed in the mouse brain. There was a sevenfold increase in brain-to-plasma AUC0–1h ratios in Mdr1a/1b knockout mice compared to wild-type controls, whereas no differences were observed between the Mdr1a/1b knockout and Mdr1a/1b/Bcrp knockout mice (Liu et al. 2013). Although P-gp is clearly playing a role for GNE-892 at the blood–brain barrier, the magnitude of the difference for fraction of drug absorbed between low and high dose was only 1.5-fold. This was consistent with previous studies where dose-proportional PK has been observed for P-gp substrates following microdosing and pharmacological dosing (Ni et al. 2008; Yamazaki et al. 2010). Further, GNE-892 demonstrated high solubility (>490 µg/ml at pH 4.5 and 7.4) and medium to high membrane permeability (Papp AB >5 × 10−6 cm/s in LLC-PK1 cell line), which may have acted to limit the role of P-gp in impacting overall absorption. Our results for GNE-892 indicated that P-gp did not play a major role in limiting absorption and did not explain the more than 20-fold greater than dose proportional difference in GNE-892 exposures.
The underlying mechanism of the non-linearity in oral exposures is supported by concentration-dependency of metabolism for GNE-892. The main metabolic pathways for GNE-892 were catalyzed by P450s generating M2, M3, M5, and M6, and by AO generating M7. Phase II metabolism of GNE-892 did not play a role in elimination of this drug. First, greater substrate turnover was measured at low substrate concentration in vitro. Second, more extensive metabolism following the low dose was consistent with the majority of radioactivity in plasma being quantitated as metabolites, whereas, following the high-dose, unchanged drug was a major circulating analyte. This suggested that nonlinear metabolic clearance of GNE-892 contributed to the disproportionality observed in drug exposures. The identity of the abundant metabolites (circulating and excreted) in the low-dose animals indicated that metabolism was primarily mediated by P450s. This was also consistent with the in vitro metabolism of GNE-892, where P450s had greater affinity. In contrast, M7, a metabolite generated by AO, was more prominent in plasma and excreta for the high-dose animals and the relative proportions of the P450-mediated metabolites were lower, which suggested that a metabolic shift from P450 to AO-mediated metabolism had occurred in vivo at the high dose. Since GNE-892 metabolism was mediated by P450 and AO in vitro for human, a similar shift in metabolic pathways may exist when comparing microdosing and a traditional clinical study.
In summary, GNE-892 demonstrated nonlinear pharmacokinetics following oral administration in cynomolgus monkeys with less drug absorbed at low dose. It seems unlikely that P-gp-mediated efflux contributed to this nonlinearity. To a greater extent, the low systemic exposure at low dose appeared to be determined by metabolism, mediated primarily by P450s, which was saturated at the high dose. We recognize that the full description of GNE-892 disposition may be more complex and involve efflux, reabsorption, and metabolism; however, additional experiments are necessary to reveal the relative importance of these interactions. Interestingly, a metabolic shift from P450 to AO at higher GNE-892 concentration was observed in vitro and in vivo. This interplay and potential compensatory relationship between P450 and AO will be of interest for further investigations to reveal any implications for metabolite identification, drug–drug interactions, and the predictions of drug clearance. The results of this study contributed to the decision against pursuing microdosing of GNE-892 to obtain estimates of human PK and identified ADME processes for which assumptions of linearity were not valid.
Abbreviations
- ADME:
-
Absorption, distribution, metabolism, excretion
- AO:
-
Aldehyde oxidase
- AUC:
-
Area under concentration–time curve
- BACE:
-
β-Site amyloid precursor protein cleaving enzyme
- BDC:
-
Bile-duct cannulated
- C max :
-
Maximum plasma concentration
- MDCK:
-
Madin–Darby canine kidney cell line
- MDR1:
-
Human multidrug resistance gene
- P450:
-
Cytochrome P450
- P-gp:
-
P-glycoprotein
- PK:
-
Pharmacokinetics
References
Balani SK, Nagaraja NV, Qian MG, Costa AO, Daniels JS, Yang H, Shimoga PR, Wu JT, Gan LS, Lee FW, Miwa GT (2006) Evaluation of microdosing to assess pharmacokinetic linearity in rats using liquid chromatography–tandem mass spectrometry. Drug Metab Dispos 34(3):384–388. doi:10.1124/dmd.105.007195
Beedham C, Bruce SE, Critchley DJ, al-Tayib Y, Rance DJ (1987) Species variation in hepatic aldehyde oxidase activity. Eur J Drug Metab Pharmacokinet 12(4):307–310
Garattini E, Fratelli M, Terao M (2008) Mammalian aldehyde oxidases: genetics, evolution and biochemistry. Cell Mol Life Sci 65(7–8):1019–1048. doi:10.1007/s00018-007-7398-y
Garner RC (2010) Practical experience of using human microdosing with AMS analysis to obtain early human drug metabolism and PK data. Bioanalysis 2(3):429–440. doi:10.4155/bio.10.6
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12(10):383–388
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256(5054):184–185
Hop CE, Wang Z, Chen Q, Kwei G (1998) Plasma-pooling methods to increase throughput for in vivo pharmacokinetic screening. J Pharm Sci 87(7):901–903. doi:10.1021/js970486q
Hunt KW, Cook AW, Watts RJ, Clark CT, Vigers G, Smith D, Metcalf AT, Gunawardana IW, Burkard M, Cox AA, Geck Do MK, Dutcher D, Thomas AA, Rana S, Kallan NC, DeLisle RK, Rizzi JP, Regal K, Sammond D, Groneberg R, Siu M, Purkey H, Lyssikatos JP, Marlow A, Liu X, Tang TP (2013) Spirocyclic beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors: from hit to lowering of cerebrospinal fluid (CSF) amyloid beta in a higher species. J Med Chem 56(8):3379–3403. doi:10.1021/jm4002154
Hutzler JM, Obach RS, Dalvie D, Zientek MA (2013) Strategies for a comprehensive understanding of metabolism by aldehyde oxidase. Expert Opin Drug Metab Toxicol 9(2):153–168. doi:10.1517/17425255.2013.738668
Lappin G (2010) Microdosing: current and the future. Bioanalysis 2(3):509–517. doi:10.4155/bio.09.177
Liu X, Wong H, Scearce-Levie K, Watts RJ, Coraggio M, Shin YG, Peng K, Wildsmith KR, Atwal JK, Mango J, Schauer SP, Regal K, Hunt KW, Thomas AA, Siu M, Lyssikatos J, Deshmukh G, Hop CE (2013) Mechanistic pharmacokinetic-pharmacodynamic modeling of BACE1 inhibition in monkeys: development of a predictive model for amyloid precursor protein processing. Drug Metab Dispos 41(7):1319–1328. doi:10.1124/dmd.112.050864
McLean MA, Tam CYJ, Baratta MT, Holliman CL, Ings RM, Galluppi GR (2007) Accelerating drug development: methodology to support first-in-man pharmacokinetic studies by the use of drug candidate microdosing. Drug Dev Res 68(1):14–22. doi:10.1002/Ddr.20160
Ni J, Ouyang H, Aiello M, Seto C, Borbridge L, Sakuma T, Ellis R, Welty D, Acheampong A (2008) Microdosing assessment to evaluate pharmacokinetics and drug metabolism in rats using liquid chromatography–tandem mass spectrometry. Pharm Res 25(7):1572–1582. doi:10.1007/s11095-008-9555-x
Powis G, Ames MM, Kovach JS (1979) Metabolic conversion of indicine N-oxide to indicine in rabbits and humans. Cancer Res 39(9):3564–3570
Pryde DC, Dalvie D, Hu Q, Jones P, Obach RS, Tran TD (2010) Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J Med Chem 53(24):8441–8460. doi:10.1021/jm100888d
Rowland M (2012) Microdosing: a critical assessment of human data. J Pharm Sci 101(11):4067–4074. doi:10.1002/jps.23290
Sandhu P, Vogel JS, Rose MJ, Ubick EA, Brunner JE, Wallace MA, Adelsberger JK, Baker MP, Henderson PT, Pearson PG, Baillie TA (2004) Evaluation of microdosing strategies for studies in preclinical drug development: demonstration of linear pharmacokinetics in dogs of a nucleoside analog over a 50-fold dose range. Drug Metab Dispos 32(11):1254–1259. doi:10.1124/dmd.104.000422
Shou M, Lin Y, Lu P, Tang C, Mei Q, Cui D, Tang W, Ngui JS, Lin CC, Singh R, Wong BK, Yergey JA, Lin JH, Pearson PG, Baillie TA, Rodrigues AD, Rushmore TH (2001) Enzyme kinetics of cytochrome P450-mediated reactions. Curr Drug Metab 2(1):17–36
Strelevitz TJ, Orozco CC, Obach RS (2012) Hydralazine as a selective probe inactivator of aldehyde oxidase in human hepatocytes: estimation of the contribution of aldehyde oxidase to metabolic clearance. Drug Metab Dispos 40(7):1441–1448. doi:10.1124/dmd.112.045195
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286(5440):735–741
Witherow LE, Houston JB (1999) Sigmoidal kinetics of CYP3A substrates: an approach for scaling dextromethorphan metabolism in hepatic microsomes and isolated hepatocytes to predict in vivo clearance in rat. J Pharmacol Exp Ther 290(1):58–65
Yamazaki A, Kumagai Y, Yamane N, Tozuka Z, Sugiyama Y, Fujita T, Yokota S, Maeda M (2010) Microdose study of a P-glycoprotein substrate, fexofenadine, using a non-radioisotope-labelled drug and LC/MS/MS. J clin pharm ther 35:169–175
Acknowledgments
We thank Matthew Durk, Jialin Mao, and Sophie Mukadam for helpful discussions and Ronitte Libedinsky for editorial contributions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takahashi, R., Ma, S., Yue, Q. et al. Dose-dependent exposure and metabolism of GNE-892, a β-secretase inhibitor, in monkeys: contributions by P450, AO, and P-gp. Eur J Drug Metab Pharmacokinet 40, 171–185 (2015). https://doi.org/10.1007/s13318-014-0198-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-014-0198-5