
Abstract
The aim of this study was to investigate whether multiple doses of the oral and highly selective dipeptidyl peptidase-4 inhibitor linagliptin affect the steady-state pharmacokinetics of the P-glycoprotein substrate digoxin. This single-center, open-label, two-period cross-over study involved healthy subjects (n = 20), randomized to treatment sequence AB or BA, where A comprised 0.25 mg digoxin qd for 5 days, then 0.25 mg digoxin qd plus 5 mg linagliptin qd for 6 days, and B comprised 0.25 mg digoxin qd for 11 days. A treatment-free period (≥35 days for AB and 14 days for BA) separated each treatment in both sequences. There were no clinically significant changes in steady-state pharmacokinetic parameters of digoxin when it was co-administered with linagliptin. The ratio of the adjusted-by-treatment geometric mean ratios and associated 90% confidence intervals for the AUCτ,ss, C max,ss and renal clearance (CLR,0–24,ss) of digoxin were all within the bioequivalence range 80–125%, which is important as digoxin has a narrow therapeutic range. There was a low incidence of adverse events, which were randomly distributed between treatment groups. In conclusion, linagliptin did not alter the pharmacokinetics of digoxin in this study, indicating that linagliptin does not inhibit P-glycoprotein or other transporters relevant for digoxin pharmacokinetics. These results suggest that linagliptin and digoxin can be co-administered without dose adjustment. Administration of digoxin alone and with linagliptin was well tolerated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Linagliptin is a highly selective orally available dipeptidyl peptidase 4 (DPP-4) inhibitor with a long terminal half-life and high volume of distribution that leads to a potent and long-lasting DPP-4 inhibitory effect in vivo (Eckhardt et al. 2007). The pharmacokinetics of linagliptin are non-linear as a result of the high affinity binding to the target DPP-4 in plasma and tissues. Results from dose-ranging studies indicate that the therapeutic window is likely to be >100-fold from the proposed therapeutic dose of 5 mg (Deacon and Holst 2010). The safety and efficacy of linagliptin has been demonstrated in healthy volunteers (Hüttner et al. 2008; Graefe-Mody et al. 2009, 2010a) and in patients with diabetes (Graefe-Mody et al. 2010b; Heise et al. 2009; Forst et al. 2010; Lewin et al. 2010; Barnett et al. 2010; Gomis et al. 2010; Del Prato et al. 2011; Taskinen et al. 2011; Owens et al. 2010). Initial results from an ongoing program of Phase III studies show that linagliptin 5 mg once daily significantly reduces blood glucose when administered as monotherapy or in combination with other commonly prescribed oral anti-diabetic drugs in type 2 diabetes patients (Gomis et al. 2010; Del Prato et al. 2011; Taskinen et al. 2011; Owens et al. 2010; Kawamori et al. 2010a, b).
Modification of P-glycoprotein function is an important underlying mechanism of drug interactions in humans (Fromm 2000). Inhibitors of P-glycoprotein can increase plasma drug concentrations by enhancing intestinal absorption and/or by reducing drug clearance in the bile or urine. Conversely, drugs that induce P-glycoprotein can reduce plasma concentrations by limiting drug absorption from the gastrointestinal tract and/or by increasing the elimination in the bile or urine (Horn and Hansten 2004). Digoxin is well-characterized as a substrate for P-glycoprotein and can be used as a test substance to determine whether other drugs affect P-glycoprotein activity (Balimane and Chong 2005). In vitro studies have indicated that linagliptin is a P-glycoprotein inhibitor with a low inhibitory potential based on IC50 values (linagliptin-inhibited digoxin transport in the Caco-2 cell monolayer model with a calculated IC50 value of 55 μM; unpublished observations).
The narrow recommended therapeutic range for digoxin (0.5–0.8 ng/mL) reflects the significant increase in risk of toxicity that occurs at higher serum concentrations (Rathore et al. 2003). Toxicity is common, occurring in up to 35% of patients receiving digoxin (Hayward 1987). It is therefore important to establish whether new drugs that may be co-administered with digoxin, such as linagliptin, alter the exposure of digoxin and, if so, to determine whether dose adjustment or contraindication is appropriate. The objective of the present study was to investigate the effects of multiple doses of linagliptin on the steady-state pharmacokinetics, safety and tolerability of digoxin.
No clinically relevant effects on other drugs are described for digoxin and it undergoes mainly renal elimination (Aspen Europe GmbH 2010). Therefore, this drug would not be expected to alter the absorption, clearance or exposure for linagliptin, which has a predominantly non-renal route of excretion following oral dosing (Blech et al. 2010; Graefe-Mody et al. 2010b). Consequently, while we also report here the steady-state pharmacokinetic profile of linagliptin when co-administered with digoxin, a detailed evaluation of the effect of digoxin on linagliptin pharmacokinetics using a treatment arm where subjects receive linagliptin only did not form part of the present study.
2 Methods
2.1 Subjects
The study enrolled 20 healthy male and female volunteers aged ≥18 to ≤50 years. Subjects were excluded if they had any relevant renal, hepatic, cardiovascular, gastrointestinal, neurologic, metabolic or hormonal disorders; if they had donated blood, participated in another clinical trial or taken any prescription or non-prescription drugs with a long (>24 h) half-life within at least 1 month (or 10 half-lives of the respective drug, whichever was longer) prior to study drug administration or during the study; if they had an alcohol or drug abuse problem; if they smoked more than 10 cigarettes, 3 cigars or 3 pipes per day; or if they could not refrain from smoking for the duration of the trial. Female subjects of child-bearing age were excluded unless they and their male partners were willing and able to use appropriate contraception.
Every subject gave written informed consent. The protocol was reviewed and approved by the local ethics committee ‘Ethik-Kommission der Landesärztekammer Baden-Württemberg’ and by the German Competent Authority (BfArM). The study was conducted in compliance with the guidelines on good clinical practice and with ethical standards for human experimentation established by the Declaration of Helsinki (1996 version) and in accordance with applicable regulatory requirements. This trial is registered in the European Union Drug Regulating Authorities Clinical Trials database, registration number 2007-007073-23.
2.2 Study design
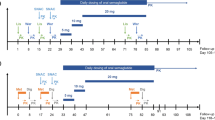
This was a randomized, open-label, two-period cross-over study in healthy male and female subjects with linagliptin (Boehringer Ingelheim Pharma GmbH; 5 mg tablet), and digoxin (Teofarma Srl; 0.25 mg tablet). Two treatments were investigated: 0.25 mg digoxin qd for 5 days, followed by 0.25 mg digoxin qd plus 5 mg linagliptin qd for a further 6 days (Treatment A) and 0.25 mg digoxin qd for 11 days (Treatment B). Subjects were randomized in a 1:1 ratio to receive treatment sequence: A–B (4 male and 6 female subjects) or B–A (5 male and 5 female subjects). Due to the long terminal half-life of linagliptin, a wash-out period of ≥35 days was used to separate each treatment in the AB sequence, and a wash-out period of at least 14 days was used to separate each treatment in the BA sequence (actual wash-out was 39 days for both treatments). A schematic diagram of the study design is shown in Fig. 1.

Schematic diagram of trial design and dosing schedule
Following an overnight fast, study medication was administered with approximately 240 mL of water to subjects in the standing position between 0700 and 0800 h to ensure a dose interval of 24 h. Subjects were not allowed to lie down for 1 h following drug administration, and remained under close medical surveillance on site until 24 h after final drug administration in Treatment A and B. Medical examinations were performed at screening (within 21 days before administration of any study medication) and at the follow-up visit (within 14 days after the last pharmacokinetic blood drawing).
Water was allowed ad libitum except for 1 h before and after drug administration. Standardized meals were served at 2, 4.25, 10.5 and 13 h following drug administration on in-house days.
2.3 Blood and urine sampling
For pharmacokinetic profiling of linagliptin, blood samples were collected into ethylenediaminetetraacetic acid (EDTA) tubes at the following time points during Treatment A: 30 min, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10 and 12 h post-dose on Day 11, prior to drug administration in the morning on Days 1, 9–11 and in the morning on Days 12–17 (following last drug administration on Day 11). For pharmacokinetic profiling of digoxin, blood samples were collected into heparinized tubes at the following time points during Treatment A (digoxin plus linagliptin) and B (digoxin alone): 30 min, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10 and 12 h post-dose on Day 11, and prior to administration of digoxin in the morning on Days 1, 8–11 and in the morning on Days 12–17 (following last drug administration on Day 11). Blood samples were stored on ice and centrifuged within 30 min of collection at 2,000–4,000×g for 10 min at +4°C. Plasma was collected in two aliquots (each containing at least 1.0 mL plasma) and frozen immediately at or below −20°C.
For digoxin, a blank urine sample was collected 15 min before first administration on Day 1 and two 4 mL aliquots were retained to check for analytical interference. All urine voided during the following sampling intervals after drug administration on Day 11 (treatment A and B) was collected: 0–2, 2–4, 4–8, 8–12, 12–24, 24–48, 48–72, 72–96 and 120–144 h. The urine weight (weight was set equal to volume, i.e. 1 kg = 1 L, without correction for specific gravity of urine) for each collection interval was documented. Aliquots of urine were retained for measurement of digoxin concentrations on Days 11–17 of regimens A and B. Urine samples were stored at or below −18°C until analysis.
2.4 Bioanalytical methods
Plasma concentrations of linagliptin were determined using ultra high performance liquid chromatography coupled to tandem mass spectrometry (UHPLC–MS/MS) as described previously (Graefe-Mody et al. 2009). Back-calculation of calibration standard, tabulation of the standard curve fit function parameters and measurement of quality control samples assessed assay performance. No interference of endogenous compounds was observed in the blank plasma of humans. The calibration curve of undiluted plasma samples was linear over the range of concentrations from 0.1 to 20.0 nmol/L using a plasma volume of 150 μL. In-study assay for linagliptin validation at nominal concentrations of 0.25–30.0 nmol/L yielded an assay imprecision of 2.0–4.6%, and an assay inaccuracy of 0.7–4.0%.
Plasma and urine concentrations of digoxin were determined using HPLC-MS/MS. Back-calculation of calibration standards, tabulation of the standard curve fit function parameters and measurements of quality control (QC) samples were performed. No interference of endogenous compounds was observed in the blank plasma of humans. The calibration curve of undiluted plasma samples was linear over the range of concentrations from 0.2–20.0 ng/mL using a sample volume of 500 μL. In-study assay validation at nominal digoxin concentrations of 0.5–16.0 ng/mL in plasma yielded an assay imprecision of 2.7–3.9%, and an assay inaccuracy of 1.25–4.5%. The calibration curve of undiluted urine samples was linear over the range of concentrations from 1–100 ng/mL using a sample volume of 500 μL. In-study assay validation at nominal digoxin concentrations of 3–80 ng/mL in urine yielded an assay imprecision of 2.1–4.4%, and an assay inaccuracy of −4.0 to −7.0%.
2.5 Pharmacokinetic analysis
Pharmacokinetic analyses were carried out by non-compartmental analysis of the plasma concentration–time data using the WinNonlin™ software program (Pharsight Corporation, Mountain View, CA, USA). Steady-state maximum concentration (C max,ss) and time to maximum concentration (t max,ss) values were obtained by inspection of the plasma concentration data. Actual sampling times were used for the pharmacokinetic analysis. Area under the plasma concentration–time curve (AUC) was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. Area under the concentration–time curve in plasma at steady-state over a uniform dosing interval τ (AUCτ,ss) was calculated using the extra- or interpolated concentration at time point τ (24 h).
For digoxin, steady-state renal clearance (\( {\text{CL}}_{{R,t_{1} - t_{2} ,{\text{ss}}}} \)) was determined as the quotient of the quantity of the drug that was excreted unchanged in the urine from time t 1 to time t 2 and the AUC within the same time interval; the apparent clearance of the drug in plasma following extravascular administration at steady-state (CL/F ss); the amount of analyte eliminated from urine over the time interval t 1–t 2 at steady-state (\( Ae_{{t_{1} - t_{2} ,ss}} \)); the apparent volume of distribution during the terminal phase λz following an extravascular dose at steady-state (V z/F ,ss); and the terminal half-life of the analyte in plasma at steady-state (t 1/2,ss) were also determined.
2.6 Safety methods
Physical examinations, vital signs, 12-lead ECGs and safety laboratory measurements comprising hematology, clinical chemistry and urinalysis were performed prior to the first treatment visit, at various time points post-dosing, and at the end-of-study examination. Adverse events (AEs) were monitored throughout the study. The investigator evaluated all clinical AEs in terms of intensity (mild, moderate or severe), duration, severity, outcome and relationship to study drugs. Safety data were described in their entirety and evaluated by descriptive statistical methods; all subjects who received one dose of study drug were included in the safety evaluation.
2.7 Statistical methods
The inferential analysis involved log transforming (natural logarithm) the primary pharmacokinetic endpoints AUCτ,ss, C max,ss and CLR,0–24,ss prior to fitting an analysis of variance (ANOVA). The difference between the expected means for log(T)-log(R) were estimated by the difference in the corresponding least square means (point estimate), where T was the pharmacokinetic parameter following co-administration and R was the parameter during digoxin administration alone. Two-sided 90% confidence intervals (CIs) based on the t-distribution were computed, then back-transformed to the original scale to give the point estimator (geometric mean) and interval estimates for the median intra-subject ratio between response under T and response under R. Descriptive statistics for all other pharmacokinetic parameters were calculated.
Based on experience in similar studies (Sechaud et al. 2008; Schwartz et al. 2008), a sample size of 16 evaluable subjects was considered adequate, and therefore the enrolment of 20 subjects was planned in order to allow for potential drop-outs.
3 Results
3.1 Subject characteristics
Twenty Caucasian subjects (9 male and 11 female) with age ranging from 23 to 49 years gave written informed consent and were entered in the study. All subjects completed the treatment and study procedures as outlined in the study protocol. Mean (standard deviation) height of the study population was 171.8 (8.0) cm, weight was 74.8 (9.7) kg and body mass index was 25.4 (3.2) kg/m2. No relevant medical history or baseline condition was recorded for any subject.
3.2 Effects of linagliptin on the pharmacokinetics of digoxin
The primary objective of the study was to determine if the pharmacokinetics of digoxin were affected by the co-administration of linagliptin. As shown in Fig. 2, digoxin reached a steady-state on day 8 of both treatments. Steady-state plasma concentrations increased rapidly after drug administration in both treatments, reaching a maximum concentration after approximately 1 h. Following maximum concentration, bi-exponential elimination profiles of digoxin were seen, with a rapid disposition phase until approximately 6 h after drug administration followed by a slower disposition phase for both treatments.

Arithmetic mean (±SD) drug plasma concentration–time profiles of digoxin per treatment after multiple oral administration of 0.25 mg digoxin, given alone (filled circles) or in combination with 5 mg linagliptin (open squares) (semi-log scale)
The non-compartmental pharmacokinetic parameters of digoxin at steady-state are shown in Table 1, and the geometric mean ratios (GMRs) and 90% CIs are shown in Table 2. These results show that the GMR between the two treatment periods and the corresponding 90% CIs were within the bioequivalence acceptance range 80–125%.
The C max,ss and AUCτ,ss values were comparable for digoxin administered alone or in combination with linagliptin. The C max,ss decreased slightly from 1.76 ng/mL when digoxin was given alone to 1.66 ng/mL when linagliptin was co-administered. The adjusted-by-treatment GMR between digoxin alone and digoxin with linagliptin was 94.2% (90% CI 86.6; 102.5%) and the difference was not statistically significant. In addition, there was a minimal increase in the AUCτ,ss value from 15.4 ng h/mL for digoxin alone to 15.6 ng h/mL when co-administered with linagliptin. The adjusted GMR between digoxin alone and digoxin with linagliptin was 101.5% (90% CI 96.9; 106.4%); this was also not statistically significant. Renal clearance in the dosing interval (CLR,0–24,ss) was 130 mL/min for both digoxin administered alone and for digoxin co-administered with linagliptin. The adjusted GMR between digoxin alone and for digoxin with linagliptin was 99.5% (90% CI 91.4; 108.4%). These results indicate that co-administration of linagliptin with digoxin has no meaningful effect on the pharmacokinetics of digoxin at steady-state.
3.3 Linagliptin pharmacokinetics
This study also examined the pharmacokinetics of linagliptin following co-administration with digoxin. Adequate exposure of linagliptin was achieved following multiple administration of a 5 mg dose in this study, consistent with findings in previous studies (Graefe-Mody et al. 2009; Heise et al. 2009).
3.4 Safety results
Digoxin was well tolerated, both when given alone or in combination with linagliptin. In total, 17/20 subjects (85%) reported AEs. During Treatment A, 9/20 subjects reported an AE following digoxin administration, and 13/20 subjects reported an AE during co-administration of linagliptin and digoxin. During Treatment B (digoxin alone), 14/20 subjects reported an AE.
The most common class of AE during the study was headache (22 cases during digoxin treatment alone and 7 cases during co-administration of linagliptin and digoxin). There were eight cases of nausea (4 cases during digoxin alone, 4 cases during co-administration) and four cases of fatigue (3 cases during digoxin alone, 1 case during co-administration). All AEs were of mild or moderate intensity with the exception of one severe headache (Treatment B group; digoxin alone). This was judged to be drug-related, but the subject did not require therapy and recovered. Indeed, all AEs resolved completely and there were no discontinuations due to AEs. There were no episodes of hypoglycemia and all clinical laboratory values were stable and within the normal range.
There were no differences between treatment with digoxin alone and treatment with linagliptin and digoxin with regard to the distribution of AE intensities or the distribution of the most frequently occurring AEs. Overall, administration of digoxin alone or in combination with linagliptin was safe and well tolerated.
4 Discussion
Cardiovascular disease is the leading cause of death for patients with diabetes (Haffner et al. 1998) and digoxin is widely used to treat chronic cardiac failure and cardiac arrhythmias. Consequently, linagliptin may be used in patients who are also receiving digoxin. Previous in vitro studies indicated that linagliptin inhibits P-glycoprotein with low potency. Therefore, the present study evaluated the effect of linagliptin on the steady-state pharmacokinetics of digoxin, which serves as both a marker of P-glycoprotein activity and as a likely co-medication for linagliptin.
In adults with normal renal function, a starting dose for digoxin of 0.125–0.25 mg/day may be appropriate to reach the currently recommended therapeutic concentration range 0.5–0.8 ng/mL (an individual’s dosage will reflect age, lean body weight and renal function) (Rathore et al. 2003; Aspen Europe GmbH 2010). The digoxin dose selected for this drug interaction study (0.25 mg qd) was in agreement with regulatory guidance for drug interaction studies, which advocate evaluation of the highest dose likely to be clinically used (FDA 2006). The linagliptin 5 mg qd dose studied here is the expected therapeutic dose (Hüttner et al. 2008; Graefe-Mody et al. 2010b). A multiple-dose design was chosen to investigate the pharmacokinetics of digoxin under steady-state conditions of both drugs, as this best reflects the chronic administration for both drugs in a clinical situation.
The results of this study show that once-daily co-administration of linagliptin 5 mg with digoxin 0.25 mg has no significant effect on the steady-state pharmacokinetics of digoxin drugs in healthy male and female subjects. The ratio of the adjusted-by-treatment GMRs and associated 90% CIs for the AUCτ,ss, C max,ss and renal clearance (CLR,0–24,ss) of digoxin were all contained within the bioequivalence range 80–125% and GMRs were near unity, indicating the lack of drug-drug interaction. The lack of an effect on renal clearance is important for digoxin as the predominant mechanism in the excretion of this drug is glomerular filtration in the kidneys (Aspen Europe GmbH 2010).
Many drugs are known to alter digoxin pharmacokinetics via their effects as P-glycoprotein inhibitors or inducers (Horn and Hansten 2004). Inhibition of P-glycoprotein results in greater digoxin exposure due to increased absorption and/or decreased urinary excretion (Horn and Hansten 2004). Although linagliptin was previously found to be a low potency inhibitor of P-glycoprotein in vitro, the lack of effect of linagliptin on digoxin C max,ss, AUCτ,ss and renal clearance of digoxin suggests that linagliptin does not affect P-glycoprotein activity in the clinical setting. The results further indicate that linagliptin is not an inhibitor of other drug uptake transporters which are relevant for the pharmacokinetics of digoxin, such as the organic anion-transporting polypeptide family (Kim 2003).
In a previous study in healthy subjects, co-administration of digoxin with the first approved DPP-4 inhibitor, sitagliptin (100 mg qd), resulted in a slight increase in both the AUC (11%) and C max (18%) of digoxin (Deacon and Holst 2010). No significant pharmacokinetic interactions were observed during co-administration of digoxin with the other DPP-4 inhibitors alogliptin (Karim et al. 2008), vildagliptin (He et al. 2007) or saxagliptin (Tahrani et al. 2009). Non-renal excretion of the unchanged parent compound is the dominant elimination pathway for linagliptin (Blech et al. 2010). From a clinical perspective, drugs that show an interaction with digoxin when administered concomitantly must be monitored for digoxin toxicity, although this is not expected to be necessary in patients with normal renal function co-treated with linagliptin and digoxin.
Both treatments were well tolerated at exposure to a total of 30 mg of linagliptin and 5.5 mg of digoxin. In previous clinical studies, linagliptin was well tolerated, demonstrating a wide therapeutic window and an AE profile that was similar to placebo (Retlich et al. 2009). The results of this study further support the promising safety profile of linagliptin.
5 Conclusion
Linagliptin does not alter the pharmacokinetics of digoxin in a clinically relevant way. This suggests that linagliptin does not interact with P-glycoprotein mediated transport or with other transporters relevant for digoxin. These results indicate that linagliptin and digoxin can be co-administered without the need for dose adjustment.
References
Aspen Europe GmbH (2010) Lanoxin 125 Tablets, Summary of Product Characteristics (last updated 8 March 2010). http://www.medicines.org.uk/EMC/medicine/2174/SPC/Lanoxin+125+Tablets/. Accessed 17 Nov 2010
Balimane PV, Chong S (2005) A combined cell based approach to identify P-glycoprotein substrates and inhibitors in a single assay. Int J Pharmacol 301:80–88
Barnett AH, Harper R, Toorawa R, Patel S, Woerle H-J (2010) Linagliptin monotherapy improves glycaemic control in type 2 diabetes patients for whom metformin therapy is inappropriate. Diabetologia 53(Suppl 1):S327 (Poster 823, presented at the European Association for the Study of Diabetes 46th Annual Meeting, Stockholm, Sweden. 20–24 September 2010)
Blech S, Ludwig-Schwellinger E, Gräfe-Mody EU, Withopf B, Wagner K (2010) The metabolism and disposition of the oral dipeptidyl peptidase-4 inhibitor, linagliptin, in humans. Drug Metab Dispos 38:667–678
Deacon CF, Holst JJ (2010) Linagliptin, a xanthine-based dipeptidyl peptidase-4 inhibitor with an unusual profile for the treatment of type 2 diabetes. Expert Opin Investig Drugs 19:133–140
Del Prato S, Barnett AH, Huisman H, Neubacher D, Woerle H-J, Dugi KA (2011) Effect of linagliptin monotherapy on glycaemic control and markers of β-cell function in patients with inadequately controlled type 2 diabetes: a randomised controlled trial. Diabetes Obes Metab 13:258–267
Eckhardt M, Langkopf E, Mark M, Tadayyon M, Thomas L, Nar H, Pfrengle W, Guth B, Lotz R, Sieger P, Fuchs H, Himmelsbach F (2007) 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3, 7-dihydropurine-2, 6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J Med Chem 50:6450–6453
FDA Draft Guidance on Drug Interactions (2006) US FDA Center for Biologics Evaluation and Research (CBER), September 2006. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093606.htm. Accessed 17 Nov 2010
Forst T, Uhlig-Laske B, Ring A, Graefe-Mody U, Friedrich C, Herbach K, Woerle H-J, Dugi KA (2010) Linagliptin (BI 1356), a potent and selective DPP-4 inhibitor, is safe and efficacious in combination with metformin in patients with inadequately controlled Type 2 diabetes. Diabet Med 12:1409–1419
Fromm MF (2000) P-glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs. Int J Clin Pharmacol Ther 38:69–74
Gomis R, Espadero R-M, Jones R, Woerle H-J, Dugi KA (2010) Efficacy and safety of initial combination therapy with linagliptin and pioglitazone in patients with inadequately controlled type 2 diabetes. Poster 551-P, presented at the 70th scientific sessions of the American diabetes association, Orlando, Florida, June 25–29, 2010. Abstract available at: http://professional.diabetes.org/Abstracts_Display.aspx?TYP=1&CID=79499. Accessed 17 Nov 2010
Graefe-Mody EU, Padula S, Ring A, Withopf B, Dugi KA (2009) Evaluation of the potential for steady-state pharmacokinetic and pharmacodynamic interactions between the DPP-4 inhibitor linagliptin and metformin in healthy subjects. Curr Med Res Opin 25:1963–1972
Graefe-Mody U, Huettner S, Stähle H, Ring A, Dugi KA (2010a) Effect of linagliptin (BI 1356) on the steady-state pharmacokinetics of simvastatin. Int J Clin Pharmacol Ther 48:367–374
Graefe-Mody U, Friedrich C, Port A, Ring A, Heise T, Halabi A, Woerle H-J (2010b) Linagliptin, a novel DPP-4 inhibitor: no need for dose adjustment in type 2 diabetes patients with renal impairment. Diabetologia, 53(Suppl 1): S326. Poster 822, presented at the European association for the study of diabetes 46th annual meeting, Stockholm, Sweden, 20–24 Sept 2010.
Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M (1998) Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 339:229–234
Hayward R (1987) Digitalis: the present position. In: Hamer J (ed) Drugs for heart disease, 2nd edn. Chapman and Hall, London, pp 145–193
He YL, Sabo R, Sunkara G, Bizot MN, Rivierie GJ, Leon S, Ligueros-Saylan M, Dole WP, Howard D (2007) Evaluation of pharmacokinetic interactions between vildagliptin and digoxin in healthy volunteers. J Clin Pharmacol 47:998–1004
Heise T, Graefe-Mody EU, Hüttner S, Ring A, Trommeshauser D, Dugi KA (2009) Pharmacokinetics, pharmacodynamics and tolerability of multiple oral doses of linagliptin, a dipeptidyl peptidase-4 inhibitor in male type 2 diabetes patients. Diabetes Obes Metab 11:786–794
Horn JR, Hansten PD (2004) Drug interactions with digoxin: the role of P-glycoprotein. Pharmacy Times. http://www.hanstenandhorn.com/hh-article10-04.pdf. Accessed 17 Nov 2010
Hüttner S, Graefe-Mody EU, Withopf B, Ring A, Dugi KA (2008) Safety, tolerability, pharmacokinetics, and pharmacodynamics of single oral doses of BI 1356, an inhibitor of dipeptidyl peptidase 4, in healthy male volunteers. J Clin Pharmacol 48:1171–1178
Karim A, Fleck P, Harris S, Weiss M, Zhang W, Mekki Q (2008) Lack of pharmacokinetic interaction between multiple doses of the dipeptidyl peptidase-4 inhibitor alogliptin and digoxin in healthy subjects. Clin Pharmacol Ther 83:S12–13 (Presented at the American society for clinical pharmacology and therapeutics, Orlando, Florida, April 2–5, 2008)
Kawamori R, Inagaki N, Araki E, Watada H, Hayashi N, Yoshiharu H, Sarashina A, Woerle H-J, Dugi KA (2010a) Linagliptin monotherapy improves glycemic control in Japanese patients with T2DM over 12 weeks. Poster 696-P, presented at the 70th scientific sessions of the American diabetes association, Orlando, Florida, June 25–29, 2010. Abstract available at: http://professional.diabetes.org/Abstracts_Display.aspx?TYP=1&CID=79641. Accessed 17 Nov 2010
Kawamori R, Inagaki N, Araki E, Watada H, Hayashi N, Horie Y, Sarashina A, Woerle H-J, Dugi KA (2010b) Linagliptin provides superior glycemic control compared to voglibose as monotherapy in Japanese patients with type 2 diabetes. Poster 632-P, presented at the 70th scientific sessions of the American diabetes association, Orlando, Florida, June 25–29, 2010. Abstract available at: http://professional.diabetes.org/Abstracts_Display.aspx?TYP=1&CID=79579. Accessed 17 Nov 2010
Kim RB (2003) Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest 33(Suppl 2):1–5
Lewin AJ, Arvay L, Liu D, Patel S, Woerle H-J (2010) Safety and efficacy of linagliptin as add-on therapy to a sulphonylurea in inadequately controlled type 2 diabetes. Diabetologia 53(Suppl 1):S326. Poster 821, presented at the European association for the study of diabetes 46th annual meeting, Stockholm, Sweden, 20–24 Sept 2010
Owens DR, Swallow R, Jones P, Dugi KA, Woerle H-J (2010) Linagliptin improves glycemic control in type 2 diabetes patients inadequately controlled by metformin and sulfonylurea without weight gain or hypoglycemia. Poster 548-P, presented at the 70th scientific sessions of the American diabetes association, Orlando, Florida, June 25–29, 2010. Abstract available at: http://professional.diabetes.org/Abstracts_Display.aspx?TYP=1&CID=79496. Accessed 17 Nov 2010
Rathore SS, Curtis JP, Wang Y, Bristow MR, Krumholz HM (2003) Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA 289:871–878
Retlich S, Withopf B, Greischel A, Staab A, Jaehde U, Fuchs H (2009) Binding to dipeptidyl peptidase-4 determines the disposition of linagliptin (BI 1356)—investigations in DPP-4 deficient and wildtype rats. Biopharm Drug Dispos 30:422–436
Schwartz JI, Agrawal NG, Wehling M, Musser BJ, Gumbs CP, Miechiels N, De Smet M, Wagner JA (2008) Evaluation of the pharmacokinetics of digoxin in healthy subjects receiving etoricoxib. Br J Clin Pharmacol 66:811–817
Sechaud R, Robeva A, Belleli R, Balez S (2008) Absence of an effect of a single-dose deferasirox on the steady-state pharmacokinetics of digoxin. Int J Clin Pharmacol Ther 46:519–526
Tahrani AA, Piya MK, Barnett AH (2009) Saxagliptin: a new DPP-4 inhibitor for the treatment of type 2 diabetes mellitus. Adv Ther 26:249–262
Taskinen M-R, Rosenstock J, Tamminen I, Kubiak R, Patel S, Dugi KA, Woerle H-J (2011) Safety and efficacy of linagliptin as add-on therapy to metformin in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled study. Diabetes Obes Metab 13:65–74
Acknowledgments
The authors would like to thank the volunteers and staff who participated in this study. This study was sponsored and funded by Boehringer Ingelheim. All of the authors are employees of Boehringer Ingelheim. Bioanalytical support for this manuscript was provided by Dr. Frank Runge. Medical writing and editorial support for the manuscript was provided by Alice Walmesley of PHASE II International Ltd with financial support from Boehringer Ingelheim.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Friedrich, C., Ring, A., Brand, T. et al. Evaluation of the pharmacokinetic interaction after multiple oral doses of linagliptin and digoxin in healthy volunteers. Eur J Drug Metab Pharmacokinet 36, 17–24 (2011). https://doi.org/10.1007/s13318-011-0028-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-011-0028-y