
Abstract
Breast cancer is characterized by molecular heterogeneity, and four major breast cancer subtypes have been identified, each characterized by significant differences in survival, prognosis, and response to therapy. We have studied the effects of docetaxel treatment on apoptosis and survivin expression in four breast cancer cell lines: MCF7 (luminal A: estrogen receptor-positive and progesterone receptor-positive, ErbB2-negative), BT474 (luminal B: estrogen receptor/progesterone receptor/ErbB2-positive), SKBR3 (HER2-like: estrogen receptor/progesterone receptor-negative, ErbB2-positive), and MDA-MB231 (basal-like: estrogen receptor/progesterone receptor/ErbB2-negative). We demonstrated that docetaxel-induced apoptosis and survivin upregulation (MCF7 p = 0.002, BT474 p = 0.001, SKBR3 p = 0.001) in luminal A/B and HER2-like cells, while it induced mainly necrosis and a lower rate of survivin upregulation (MDA-MB231 p = 0.035) in basal-like cells. Wortmannin, a p-Akt inhibitor, was able to revert surviving upregulation and, at the same time, induced an increase of docetaxel-dependent apoptosis, suggesting that reduced levels of survivin can sensitize tumor cells to apoptosis. These data show that the analyzed breast cancer cell lines respond differently to docetaxel, depending on their receptor expression profile and molecular phenotype. Yet, these data confirm that one of the pathways involved in taxane-related chemoresistance is the upregulation of survivin. Further studies on the molecular mechanisms of chemoresistance and on the different modalities of apoptosis induced by chemotherapeutic agents are requested to better understand how cancer cells evade cell death, in order to design new kind of anticancer agents and survivin could represent a future target for this kind of research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is characterized by an extreme clinical heterogeneity, due to a high variability of its molecular arrangement; this determines a diversified responsivity of patients to treatment. In fact, breast cancer is clinically divided into four groups, based on the receptor profile: luminal A (estrogen receptor [ER]+, progesterone receptor [PgR]+, human epidermal growth factor receptor 2 [HER2]−), luminal B (ER+, PgR+, HER2+), basal-like (ER−, PgR−, HER2−), and HER2-like (ER−, PgR−, HER2+). Luminal A/B tumors correspond to the hormone-responsive breast cancer phenotypes [1].
Taxanes (paclitaxel and docetaxel) represent, together with anthracyclines, the gold standard of chemotherapy for breast cancer. Over the past 10 years, several clinical trials have confirmed the efficacy of taxanes: regimens containing anthracyclines and taxanes are superior in disease-free survival (DFS) and overall survival (OS), as compared to regimens with anthracyclines alone and without taxanes [2]. Yet, regimens containing taxanes without anthracyclines are considered superior in OS with respect to those with anthracyclines alone [3]. The mechanism of taxane consists in a specific binding to microtubular beta-tubulin subunits, which form the mitotic spindle, determining stabilization and depolymerization of the cytoskeleton and resulting in arrest of mitosis in metaphase.
Resistance to chemotherapy is, unfortunately, an unavoidable stage for each type of treatment, which determines disease recurrence and increased mortality. Chemoresistance is multifactorial and can be due to physiological mechanisms (for example, the presence of ischemic areas in large tumors inhibiting the penetration of the drug through the vascular network), to modifications of the cellular phenotype (alteration of the drug cellular uptake or increased extracellular transport mediated by multidrug resistance 1 [MDR]/P-glycoprotein)[4], to alterations in chemotherapeutic metabolism or to dysregulation of inhibitors of apoptosis proteins (IAPs)[5] through an intrinsic or acquired resistance to apoptosis. In fact, it has been demonstrated that IAP expression contributes to apoptosis resistance of breast cancer cells [6]. Apoptosis is controlled by two distinct pathways: the extrinsic and the intrinsic pathways; the first is mediated by death receptors, while the second is activated by cell stress. Caspases are the mediators of apoptosis, and they can be negatively regulated by IAPs: this is one of the mechanisms that cause chemoresistance [5]. Survivin is the most important molecule within the IAP family in relation to breast cancer chemoresistance; in fact, its positive expression correlates with patient worst prognosis [7–10].
All the described mechanisms, sometimes synergistically, cause resistance to therapy and, consequently, facilitate disease progression and metastasis occurrence. Tumor treatment with taxanes can induce resistance; various mechanisms of escape from apoptosis taxane-induced have been described [11], with survivin as the key protein in the resistance to taxanes.
In this study, we analyzed the effects exerted by docetaxel on apoptosis induction and survivin expression in four cell lines representative of the molecular breast cancer subtypes luminal A-B, HER2-like, and basal-like, in order to compare whether docetaxel effects could be different among these four breast cancer subtypes.
Materials and methods
Cell culture and treatments
The following four human breast cancer cell lines have been utilized: MCF7 (luminal A: estrogen receptor-positive and progesterone receptor-positive, ErbB2-negative), BT474 (luminal B: estrogen receptor/progesterone receptor/ErbB2-positive), SKBR3 (HER2-like: estrogen receptor-negative/progesterone receptor-negative, ErbB2-positive), and MDA-MB231 (basal-like: estrogen receptor/progesterone receptor/ErbB2-negative). All cell lines were grown in DMEM medium supplemented with 10 % fetal calf serum (FCS), 2 mM glutamine, and 50 U/mL penicillin-streptomycin and starved, when necessary, in DMEM medium with 2 % dialyzed FCS.
Docetaxel (Taxotere®, Sanofi Aventis) (diluted in polysorbate 80) was prepared in a 3 mM stock solution in DMEM medium and then diluted to the indicated concentrations. Wortmannin (Sigma) was used (100 nM). Staurosporine (Sigma) was dissolved in DMSO (Sigma) in 1-mM stock solution, conserved frozen and used 1 μM.
Western blot
Cell lysates were obtained scraping the cells in lysis buffer 1 % Triton, 0.1 % SDS, 150 mM NaCl, 50 mM Tris-HCl pH 7.4, 2 mM EDTA plus protease inhibitor cocktail tablet (Roche Applied Sciences) for 30 min at 4 °C, lysates were then centrifuged at 12,000 rpm for 15 min at 4 °C. Protein concentration was evaluated by Bio-Rad Protein Concentration Assay. Samples of lysate (50–100 μg) were separated by molecular weight by 10 or 12 % SDS-PAGE and then transferred into a nitrocellulose membrane. Blots were blocked for 1 h at RT in 5 % nonfat dry milk and then incubated with primary antibody, washed in Tris-buffered saline with 0.1 % Tween-20 and then incubated with horseradish peroxidase conjugated anti-mouse or anti-rabbit antibodies (1:5000 diluted) (Sigma-Aldrich). The filters were then developed by enhanced chemiluminescence (Super Signal West Pico Chemiluminescent Substrate, Thermo Scientific) using Kodak X-Omat films.
The primary antibodies were rabbit anti-survivin (1:1000 diluted) (Novus Biologicals, Littleton, CO, USA); rabbit anti-cleaved caspase-8 (1:500 diluted) (Cell Signaling); mouse anti-cleaved caspase-9 (1:500 diluted) (Cell Signaling), mouse anti-PARP1 (1:500 diluted) (Santa Cruz Biotechnology), mouse anti-Bcl2 and rabbit anti-Bax (1: 500 diluted) (BD Transduction Laboratories, USA), and mouse anti-actin (1:1000 diluted) (Sigma-Aldrich).
RT-PCR assay
Total RNA from the breast cancer cell lines was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions; Moloney murine leukemia virus (M-MLV) reverse transcriptase (Biolab) were used to reverse 1 μg of total RNA into complementary DNA (cDNA) at 42 °C. Five grams of each cDNA was then subjected to reverse transcriptase polymerase chain reaction (RT-PCR) in a buffer containing 25 pmol of upstream and downstream and 1.25 U of Platinum Taq polymerase (Euroclone). The amount of amplified products, expressed in arbitrary optical density units, was normalized with glyceraldehyde-3-phosphate dehydrogenase (GADPH) as housekeeping gene. The amplification reaction was carried out in Piko-Thermal Cycler cyclers (Finnzymes Instrument). The resulting PCR products were separated in 2 % agarose gel and visualized with of Gel-Red (GelRed nucleic acid gel stain, Biotium). The sequences of human gene-specific primers (Sigma-Aldrich) with order of forward and reverse, the conditions of amplification, as well as the amplified products size are as follows: Gadph 5-AGATGTTCCAATATGATTCC, 5′-TGGACTCCACGACGTACTCAG, 60 °C, 161 bp; Bcl-2 GTGGAGGAGCTCTTCAGGGA, AGGCACCCAGGGTGATGCAA, 60 °C, 304 bp; Bax GGCCCACCAGCTCTGAGCAGA, GCCACGTGGGCGTCCCAAAGT, 62 °C, 469 bp; and survivin CAGATTTGAATCGCGGGACCC, CCAAGTCTGGCTCGTTCTCAG, 60 °C, 206 bp.
Cell apoptosis assay
The four different cell lines were treated with docetaxel for 24 h. Both detached and adherent cells were harvested by trypsinization and washed with cold phosphate-buffered saline. The cells were double-stained with annexin V-APC (allophycocyanin)/7AAD (7-amino-actinomycin) in a calcium binding buffer (BD Biosciences kit) and analyzed by a FACS scan cytofluorimeter (BD Biosciences).
Immunofluorescence
The cells were grown directly on Labteck chamber slides (Nunc) for 24 h, then treated with 300 nM docetaxel and 100 nM wortmannin for 24 h; the cells were then washed with PBS with Ca/Mg and fixed with 4 % buffered paraformaldheyde (Sigma) for 20 min at 4 °C. The cells were incubated with 3 % bovine serum albumin for 1 h at RT and then with rabbit anti-survivin (1:100 diluted) (Novus Biologicals, Littleton, CO, USA) for 1 h at RT, then washed twice with PBS with Ca/Mg and then incubated with the secondary anti-rabbit antibody Alexa Fluor 594 conjugated (1:400 diluted) for 1 h at RT. The cells were finally washed twice with PBS with Ca/Mg, mounted with Prolong Antifade reagent (Life Technologies) and analyzed by a fluorescence microscope (Olympus BX52); imagine acquisition and processing were conducted by IAS 2000 software. Fluorescence intensity of three randomly selected fields was acquired by densitometric quantitation using ImageJ software, and the mean was determined.
Cytotoxicity assay
To determine cytotoxicity, sulforhodamine B colorimetric assay was performed [12]. Cells (1.5 × 104) were plated on 96-well plate, grown for 24 and 48 h and treated with different concentrations of docetaxel (3 nM, 30 nM, 300 nM, and 3 μM). Cells were then fixed with 50 % trichloroacetic acid for 1 h at 4 °C and stained for 30 min at RT with 0.4 % sulforhodamine B in 1 % acetic acid. Excess dye was removed by washing four times with 1 % acetic acid. Protein-bound dye was dissolved in 10 mM Tris, pH 10, and optical density (OD) was determined at 510 nm using a microplate reader.
Statistical analysis and graphic programs
All results were analyzed by ANOVA, and the significance was evaluated by the Tukey honestly significant difference (HSD) post hoc test. All figures were elaborated by Adobe Photoshop CS5. All graphs were elaborated by GraphPad Prism 5.0.
Results
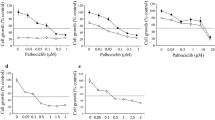
We first investigated docetaxel cytotoxic effect on cell viability: MCF7, MDA-MB231, BT474, and SKBR3 cell lines were treated for 24 and 48 h with 3 nM, 30 nM, 300 nM, and 3 μM docetaxel to determine the effective dose to be utilized for the following experiments. Docetaxel determined a significant decrease of cell viability when used 30 nM, 300 nM, and 3 μM for MDA and SKBR3 cells and at 300 nM and 3 μM for BT474 and MCF7 cells (Fig. 1). The described effects on cell viability were similar when cells were treated with docetaxel for 24 and for 48 h.

Cell viability of four breast cancer cell lines upon 24-h treatment with 3 nM, 30 nM, 300 nM, and 3 μM docetaxel expressed as percentage of alive cells in graph and table
Based on these data, docetaxel was then used for the following experiments at the concentration of 300 nM for 24 h. In order to detect whether docetaxel could induce apoptosis, treated and untreated cells were evaluated for poly-ADP-ribose polymerase (PARP), which is involved in DNA repair and apoptosis in response to stress phenomena and it is cleaved by caspases during the last phases of apoptosis. Docetaxel determined PARP cleavage in three cell lines, but not in MDA-MB231 (Fig. 2), suggesting that only the first three cell lines had an evident apoptosis; the same experiment was performed with higher doses of docetaxel, 1.5 and 3 mM, and similar results were obtained.

Western blot of untreated and 300 nM docetaxel-treated (T) breast cancer cell lines for PARP, Bax, Bcl2, caspase-8, caspase-9, and actin
To analyze the apoptosis induced by docetaxel, Bax (an apoptosis inducer) and Bcl2 (an apoptosis inhibitor) expression was then analyzed. Both are members of the Bcl2 protein family, which controls mitochondria permeability (MOPM-mitochondrial outer permeabilization membrane) and cytochrome C release; an increase in Bax/Bcl2 ratio is often described during apoptosis. An increase of Bax was evident in all treated cell lines, although less evident in MDA-MB231; in parallel, Bcl2 decreased in MDA-MB231 and MCF7, while it was not visible in BT474 and in SKBR3 (Fig. 2). The RT-PCR confirmed that Bax RNA was increased by docetaxel treatment of 29 % in MDA-MB231, 67 % in MCF7, 22 % in BT474 and 79 % in SKBR3 (Fig. 3). Bcl2 RNA was decreased of 22 % only in MCF7, while it was unmodified in the other cell lines (data not shown). The increased Bax/Bcl2 ratio obtained in our experiments in all treated cell lines confirmed the induction of apoptosis by docetaxel.

RT-PCR of Bax mRNA normalized to levels of GAPDH mRNA in docetaxel-treated (Doc) and untreated cells (Ctrl). The histogram reports the densitometric quantification of Bax RNA upon normalization to levels of GAPDH mRNA; the percentual positive differences of Bax between treated and untreated cells are indicated
The cleavage of caspase-8 and caspase-9 was analyzed (Fig. 2): caspase-8 was cleaved in three treated cell lines, but not in MDA-MB321, while caspase-9 was cleaved in all four treated cell lines, characterized by an evident increase of proteolitic fragments of caspase-8 and caspase-9, as index of caspase activation and consequent apoptosis. These data suggested that MDA-MB231 follows an intrinsic pathway, characterized by caspase-9 cleavage, while the other three cell lines have both intrinsic and extrinsic pathways activated.
In order to evaluate whether the cell loss induced by docetaxel was due also to necrosis in addition to apoptosis, we double-stained untreated and docetaxel-treated cells with APC-conjugated annexin V and with 7AAD and analyzed by fluorescence-activated cell sorter (FACS). Docetaxel treatment determined in three cell lines a high rate of annexin V staining, indicative of apoptosis: MCF7 cells resulted the best responsive, SKBR3 and BT474 had an intermediate response, while MDA-MB231 remained practically negative for annexin V (Fig. 4a). In fact, docetaxel determined a rate of necrosis higher than the rate of apoptosis in MDA-MB231, as compared to the other three cell lines, which responded to docetaxel mainly with apoptotic death than with necrosis (Fig. 4b). As positive control of apoptosis, all four cell lines were treated with staurosporine (an apoptosis inducer) and all four staurosporine treated lines resulted highly positive for annexin V expression (data not shown).

Annexin V-APC and 7AAD expression evaluated by flow cytometry. a The upper dot blots represent untreated cells (C); the lower dot blots, docetaxel-treated cells (T). Quadrant location for the representative dot blots: lower left, living cells; lower and upper right, apoptosis cells; upper left, necrotic cells. b Each bar represents the percentage of apoptotic (gray) and necrotic (black) untreated (C) and docetaxel-treated (T) cells expressed as mean ± SD of three different experiments
Finally, we investigated survivin, which is a member of the IAP family: docetaxel treatment determined an upregulation of survivin, as evidenced by Western blot (Fig. 5a), with statistically significant data in MCF7 (p = 0.002), BT474 (p = 0.001), and SKBR3 (p = 0.001), while the increase was less evident but still significant in MDA-MB231 (p = 0.035) (Fig. 5b). These data were confirmed by RT-PCR, demonstrating an evident increase in survivin RNA in three cell lines, but less evident in MDA-MB231, when treated with 300 nM docetaxel (Fig. 5c).

Regulation of survivin expression and transcription by docetaxel. a Western blot of survivin on untreated and 300 nM docetaxel-treated breast cancer cell lines. b Densitometric quantification of survivin expressed as mean values from three different Western blots. c Densitometric quantification of survivin RNA from RT-PCR
We also analyzed whether the positive regulation of survivin by docetaxel was dose and time dependent; therefore, the experiments were repeated with docetaxel concentrations of 1.5 and 3 mM for 24 h and concentration of 300 nM for 48 h: survivin upregulation became even more evident at these higher concentrations of docetaxel in luminal and HER2+ cells, while the data obtained with 300 nM docetaxel at 24 h were confirmed at 48 h of treatment. These data demonstrated that luminal cells (MCF7 and BT474) and HER2+ cells (SKBR3) respond to docetaxel with an upregulation of survivin, differently from basal like cells (MDA-MB231).
Summarizing our results, the four cell lines responded differently to docetaxel: luminal A and B and HER2-like cells showed an evident and massive extrinsic and intrinsic apoptosis and no significant necrosis, together to survivin upregulation, while basal-like cells responded mainly with necrosis and a very low rate of intrinsic apoptosis, survivin upregulation was not significative (Table 1).
We then investigated whether the described survivin upregulation docetaxel dependent could negatively interfere with the apoptotic response of the cells to docetaxel: so, we evaluated whether, once survivin upregulation was inhibited, the apoptosis rate was modified. Consequently, MCF7 and SKBR3 cells were treated with 100 nM wortmannin, an inhibitor of phosphoinositide 3-kinase Akt (PI3K/Akt), since it is well known that Akt activation is required for survivin expression [13]. Wortmannin was able to revert the increased survivin expression docetaxel dependent, as showed by the nuclear staining of survin in MCF7 and SKBR3 cells: the fluorescence intensity was low in both basal cell lines, 34 densitometric arbitrary units (DUs) for untreated MCF7 and 20 DUs for untreated SKBR3, and significantly increased upon docetaxel treatment, 90 and 60 DUs, respectively, with a threefold enhancement of survivin expression (p < 0.001), which returned low by treatment with docetaxel together with wortmannin, 39 and 19 DUs for MCF7 and SKBR3, respectively (Fig. 6). The treatment of the same two cell lines with wortmannin determined a decrease of survivin expression at basal condition, as evidenced by Western blot (untreated vs wortmannin-treated cells); furthermore, the increase of survivin determined by docetaxel treatment was reversed by wortmannin addition (docetaxel vs docetaxel + wortmannin-treated cells) (Fig. 7); at the same time, the treatment with docetaxel together with wortmannin determined an increase of cleaved PARP, as compared to the treatment with docetaxel alone, suggesting an increased apoptosis (Fig. 7). These data suggest that the reduced levels of survivin obtained by wortmannin can sensitize tumor cells to decetaxel-dependent apoptosis.

Immunofluorescent staining of survivin on untreated (a, d), docetaxel-treated (b, e) and docetaxel plus wortmannin-treated (c, f), MCF7 (a–c), and SKBR3 cells (d–f)

Western blot of survivin and PARP on MCF7 and SKBR3 cells untreated and treated with wortmannin (WT), docetaxel (DOC), and wortmannin and docetaxel (WT + DOC)
Discussion
In our study, we have initially evaluated the ratio of apoptosis and necrosis induced by docetaxel in four different breast cancer cell lines, each corresponding to a molecular phenotype, and found a wide variability in responsivity to taxanes by the different molecular phenotypes, in line with the heterogeneity of responses to therapy observed in breast cancer patients.
We then studied survivin expression, since it is a key protein among IAPs, a family of inhibitor of apoptosis [10, 14], which covers an important role in cancer initiation, tumor progression, and chemoresistance to various chemotherapeutics including taxanes. Survivin expression is often described in several tumors, such as in breast cancer, in which survivin upregulation usually correlates with worst prognosis [15]. The overexpression of survivin has been demonstrated also in endothelial cells surrounding the tumors, as a protective mechanism that defends the vasculature from chemotherapy [16]. Survivin main activation pathway is PI3K/Akt/mitogen-activated protein kinase (MAPK), which may cross-react and strengthen other pathways; for example, a survivin correlated pathway is the estrogen pathway PI3K/Akt/mammalian target of rapamycin (mTOR) [17], which reinforces the signal transduction mediated by hormone receptors, with the onset of continuous stimulation on hormone-responsive tumor cell proliferation. Survivin inhibits apoptosis directly and indirectly by interfering with caspase-3, caspase-7, and caspase-9 [5, 6]. Other correlated proteins with survivin pathways are c-IAP-1, HER-2, leptin, signal transducer and activator of transcription 3 (Stat3), progesterone, P53, apoptosis-inducing factor (AIF), heat shock protein 90 (HSP90), extracellular signal-regulated kinases (ERKs), and the Smac/DIABLO complex [18–21]. The most important pathways involved in docetaxel chemoresistance in breast cancer cells are PI3K/Akt and leptin/STAT3 pathways [19, 21].
Our data demonstrated a significative upregulation of survivin in docetaxel-treated luminal A and B and HER2+ cells, while survivin was not significatively modified by docetaxel in basal-like cells. One of the possible explanations for survivin enhancement in luminal B and HER2+ breast cancer cells can involve ERK pathway; in fact, survivin upregulation has been recently correlated with ERK pathway [22] and the presence of HER2 on breast cancer cells (expressed only in luminal B and HER2+ cells) can activate both ERK and PI3K/Akt signaling, inducing a phosphorylation cascade that increases survivin expression [23]. At this regard, Lu showed a correlation between HER2 overexpression and breast cancer cell resistance to taxol mediated by upregulation of survivin [24]. Luminal A cells do not express HER2 receptors; therefore, other mechanisms should be involved in the described survivin upregulation; probably, the presence of hormonal receptors is an important factor, since these play an important role in chemoresistance [25, 26], although the mechanism is still not completely understood.
Our in vitro results correlate with the different responses to taxane-based chemotherapy given by breast cancer patients: luminal A and B patients usually develop higher rates of chemoresistance, as compared to cancer patients negative for hormonal receptors (HER2+ and basal-like patients) [27]. Breast cancer cells with HER2 amplification show more chemoresistance to taxanes, both in vitro and in vivo, as compared to HER2-negative cells, and this chemoresistance is revertible only with anti-HER2 agents [28].
Upregulation of antiapoptotic proteins of the IAP family and of other antiapoptotic molecules has been associated with the development of chemoresistance [29]. Our results underline the importance of survivin in taxane-related chemoresistance, in accordance with other studies that have correlated survivin with both prognosis and chemoresistance in cancer patients [30, 31]. Therefore, survivin could be considered a potential target to inhibit the survival and proliferation of cancer cells, though its activation could involve several collateral pathways, switched on in case of interruption of the principal transduction way. We showed that the negative regulation of survivin expression could sensitize cells to docetaxel, ending with an increase in apoptosis, as has been recently proposed also by other authors [29].
Further studies on the molecular mechanisms of chemoresistance and on the different modalities of apoptosis induced by chemotherapeutic agents can be helpful to physicians to better understand how cancer cells evade cell death in order to design new anticancer agents. For this reason, more investigations are needed to study in deep chemoresistance and to find alternative mechanisms that can downregulate survivin on multiple fronts.
Conclusions
Our data demonstrate a significant difference among breast cancer cell lines in their response to docetaxel, which reflects their receptor expression and molecular phenotype. Luminal A and B and HER2+ breast cancer cell lines show massive apoptosis and survivin upregulation upon docetaxel treatment, both less evident in basal-like cancer cell line. Yet, survivin inhibition can restore chemosensitivity, increasing the apoptosis rate in docetaxel-treated cells. These results taken together suggest that the genetic variability of breast cancer cells should be taken in consideration in order to choose a more tailored therapy and that survivin could be a potential future target for treatment of this kind of disease.
References
Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature. 2012;490(7418):61–70.
Peto R, Davies C, Godwin J, Gray R, Pan HC, Clarke M, et al. Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet. 2012;379(9814):432–44.
Jones SE, Savin MA, Holmes FA, O’Shaughnessy JA, Blum JL, Vukelja S, et al. Phase III trial comparing doxorubicin plus cyclophosphamide with docetaxel plus cyclophosphamide as adjuvant therapy for operable breast cancer. J Clin Oncol. 2006;24(34):5381–7.
Dalton WS. Mechanisms of drug resistance in breast cancer. Semin Oncol. 1990;17(7):37–9.
Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12(9):1543–68.
Foster FM, Owens TW, Tanianis-Hughes J, Clarke RB, Brennan K, Bundred NJ, et al. Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Res. 2009;11(3):R41.
Byun SS, Yeo WG, Lee SE, Lee E. Expression of survivin in renal cell carcinomas: association with pathologic features and clinical out come. Urology. 2007;69(1):34–7.
Hinnis AR, Luckett JC, Walker RA. Survivin is an independent predictor of short-term survival in poor prognostic breast cancer patients. Br J Cancer. 2007;96(4):639–45.
Vucic D, Fairbrother WJ. The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin Cancer Res. 2007;13(20):5995–6000.
LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27(48):6252–75.
Murray S, Briasoulis E, Linardou H, Bafaloukos D, Papadimitriou C. Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat Rev. 2012;38(7):890–903.
Vichaj V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–6.
Papapetropoulos A, Fulton D, Mahboui K, Kalb RG, O’Connor D, Li F, et al. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J Biol Chem. 2000;275(13):9102–5.
Altieri DC. The case for survivin as a regulator of microtubule dynamics and cell-death decisions. Curr Opin Cell Biol. 2006;18(6):609–15.
Lv YG, Yu F, Yao Q, Chen JH, Wang L. The role of survivin in diagnosis, prognosis and treatment of breast cancer. J Thorac Dis. 2010;2(2):100–10.
Virrey JJ, Guan S, Li W, Schontal AH, Chen TC, Hofman FM. Increased surviving expression confers chemoresistance to tumor associated endothelial cells. Am J Pathol. 2008;173(2):575–85.
Siddiqa A, Long LM, Li L, Marciniak RA, Kazhdan I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic protein sSurvivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signaling pathways. BMC Cancer. 2008;8:129.
Ceballos-Cancino G, Espinosa M, Maldonado V, Melendez-Zajgla J. Regulation of mitochondrial Smac/DIABLO-selective release by survivin. Oncogene. 2007;26(54):7569–75.
Asanuma H, Torigoe T, Kamiguchi K, Hirohashi Y, Ohmura T, Hirata K, et al. Survivin expression is regulated by coexpression of human epidermal growth factor receptor2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005;65(23):11018–25.
Xia W, Bisi J, Strum J, Liu L, Carrick K, Graham KM, et al. Regulation of survivin by ErbB2 signaling: therapeutic implications for ErbB2-overexpressing breast cancers. Cancer Res. 2006;66(3):1640–7.
Jiang H, Yu J, Guo H, Song H, Chen S. Upregulation of survivin by leptin/STAT3 signaling in MCF-7 cells. Biochem Biophys Res Commun. 2008;368(1):1–5.
Ju JH, Yang W, Oh S, Nam K, Lee KM, Noh DY, et al. HER2 stabilizes surviving while concomitantly down-regulating survivin gene transcription by suppressing Notch cleavage. Biochem J. 2013;451(1):123–34.
Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of surviving phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003;63(1):230–5.
Lu J, Tan M, Huang WC, Li P, Guo H, Tseng LM, et al. Mitotic deregulation by survivin in ErbB2-overexpressing breast cancer cells contributes to Taxol resistance. Clin Cancer Res. 2009;15(4):1326–34.
Lips EH, Mulder L, De Ronde JJ, Mandjes IA, Vincent A, Vrancken Peeters MT, et al. Neoadjuvant chemotherapy in ER+ HER2- breast cancer: response prediction based on immunohistochemical and molecular characteristics. Breast Cancer Res Treat. 2012;131(3):827–36.
Sui M, Huang Y, Park BH, Davidson NE, Fan W. Estrogen receptor alpha mediates breast cancer cell resistance to paclitaxel through inhibition of apoptotic cell death. Cancer Res. 2007;67(11):5337–44.
Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA. 2011;305(18):1873–81.
Yu D, Liu B, Tan M, Li J, Wang SS, Hung MC. Overexpression of c-erbB-2/neu in breast cancer cells confers increased resistance to Taxol via mdr-1-independent mechanisms. Oncogene. 1996;13(6):1359–65.
Ghanbari P, Mohseni M, Tabasinezhad M, Yousefi B, Saei AA, Sharifi S, et al. Inhibition of survivin restores the sensitivity of breast cancer cells to docetaxel and vinblastine. Appl Biochem Biotechnol. 2014;174(2):667–81.
Cheung CH, Huang CC, Tsai FY, Lee JY, Cheng SM, Chang YC, et al. Survivin-biology and potential as a therapeutic target in oncology. Onco Targets Ther. 2013;6:1453–62.
Lv YG, Yu F, Yao Q, Chen JH, Wang L. The role of survivin in diagnosis, prognosis and treatment of breast cancer. J Thorac Dis. 2010;2(2):100–10.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Rights and permissions
About this article

Cite this article
De Iuliis, F., Salerno, G., Giuffrida, A. et al. Breast cancer cells respond differently to docetaxel depending on their phenotype and on survivin upregulation. Tumor Biol. 37, 2603–2611 (2016). https://doi.org/10.1007/s13277-015-4075-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-4075-x