
Abstract
Although three therapeutic modalities (surgical resection, chemotherapy, and radiotherapy) have been established, long-term survival for lung cancer patients is still generally poor. Until now, the mechanisms of lung cancer genesis remain elusive. The JARID1B is a histone demethylase that has been proposed as oncogene in several types of human cancer, but its clinical significance and functional role in human non-small cell lung cancer (NSCLC) remain unclear. In present study, we found that JARID1B was overexpressed in lung cancer cell lines and lung cancer tissues but not in normal lung tissues. The proliferation and invasive potential of lung cancer cells was significantly increased by ectopic expression of JARID1B. Contrarily, RNA interference targeting JARID1B in lung cancer cells significantly decreased the proliferation and invasive potential of cells. Moreover, we also found that the expression of p53 was modulated by JARID1B. Overexpressed JARID1B cell exhibited greatly decreased p53 expression, whereas silencing of JARID1B expression dramatically increased p53 expression at both the messenger RNA (mRNA) and protein levels. Inhibition of p53 by small interfering RNA (siRNA) reversed the shJARID1B-induced suppression of proliferation and invasion. Our results collectively suggested that JARID1B expressed in lung cancer played a role in lung cancer cells proliferation and invasion, which may be partly associated with the p53 expression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lung cancer ranks first of cancer mortality, and non-small cell lung cancer (NSCLC) accounts for more than 80 % of lung cancer [1]. Surgery should be the first choice for patients with early-stage NSCLC. However, 80 % of NSCLC patients are diagnosed in advanced stages [2, 3]. Although improvements in diagnostic and therapeutic are made in lung cancer, the 5-year survival rate of NSCLC patients remains less than 15 % [3]. Stable biomarkers for risk assessment or predication of clinical outcome and further investigations are necessary and urgent.
Chromatin dynamics and cellular functions could be affected by covalent histone modifications, which include acetylation, methylation, phosphorylation, ubiquitination, glycosylation, and sumoylation [4]. Histone lysine methylation is the most common one and control by histone methyltransferase (HMT) enzymes and histone demethylase (HDM) enzymes such as Jumonji (Jmj) and KDM1 [5]. Discriminate mono-, di-, and tri-methylation and respective demethylation of lysines within histones H3 and H4 encompassing euchromatic domains act as a genome-wide epigenetic switch that can either activate or repress transcription [6]. More recently, the expression levels of JARID1B were reported significantly upregulated in cancer tissues, compared with those in corresponding normal tissues in two independent researches [7, 8]. JJARID1B, also known as PLU-1, was identified in silico and shown to be one of the demethylases capable of removing the trimethyl group from histone H3 lysine 9 on pericentric heterochromatin in mammalian cells [9]. A line of recent reports indicated that hypoxic conditions can induce the expression of some JmjC family members, including JARID1B [4, 6, 10–12]. JARID1B has been shown to harbor HIF binding sites in their promoter sequences [13].
However, the significance of JARID1B in lung cancer oncogenesis and cancer progression is not fully understood so far. Here, we revealed the potential role of JARID1B in lung cancer proliferation, invasion, and metastasis.
Material and methods
Patients and tissue samples
A total of 58 lung cancer tissue samples, along with matched normal tissues, were used in this study. All of the samples were obtained from the second affiliated hospitals of Soochow University between 2008 and 2011. For all of the patients who participated in this study, written informed consent was obtained, which was approved by the Ethical Committee of Soochow University School of Medicine.
Immunohistochemistry
Surgically excised specimens were fixed with 10 % neutral formalin and embedded in paraffin, and 4-μm-thick specimen sections were prepared. Immunostaining was performed using the avidin-biotin-peroxidase complex method (UltrasensitiveTM, MaiXin, Fuzhou, China). The sections were deparaffinized in xylene, rehydrated with graded alcohol, and boiled in 0.01 M citrate buffer (pH 6.0) for 2 min with an autoclave. Hydrogen peroxide (0.3 %) was applied to block endogenous peroxide activity, and the sections were incubated with normal goat serum to reduce nonspecific binding. Tissue sections were incubated with JARID1B rabbit polyclonal antibody (1:100 dilution). Mouse immunoglobulin (with the same concentration of the antigen-specific antibody) was used as a negative control. Staining for both antibodies was performed at room temperature for 2 h. Biotinylated goat anti-mouse and goat anti-rabbit serum IgGs were used as a secondary antibodies. The sections were washed and incubated with streptavidin conjugated with horseradish peroxidase. The peroxidase reaction was developed with 3, 30-diaminobenzidine tetrahydrochloride. Two independent blinded investigators randomly examined all tumor slides. Five views were examined per slide, and 100 cells were observed per view at ×400 magnification. Scores for JARID1B membrane and cytoplasmic staining were calculated based on staining intensity (0, below the level of detection; 1, weak; 2, moderate; and 3, strong) and the percentage of cells staining at each intensity level (0–100 %). The final score was calculated by multiplying the intensity score by the percentage, producing a scoring range of 0 to 300. The immunohistochemistry score cutoff point was established as 38 using X-tile software program (version 3.6.3, Yale University School of Medicine, CT, USA).
Cell culture and reagents
The normal lung epithelial cell line (BEAS2B) and lung cancer cell lines (HLAMP, H1299, H1650, A427, H460, and A549) were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). Antibodies that had been raised against JARID1B, p53, and p27 were purchased from Cell Signaling Technology (Beverly, MA, USA), and a mouse anti-PCNA antibody was obtained from Abcam (Cambridge, MA, USA). The signal silence p53 small interfering RNA (siRNA) and its control siRNA were purchased from Cell Signaling Technology. All of the remaining reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise specified.
Plasmid construction and transfection
For overexpression, the cDNA representing the complete open reading frame of JARID1B was cloned into the pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA) to generate the JARID1B expression plasmid. The expression plasmid was verified by sequencing both strands and was used to transfect the H1299 cells to establish the JARID1B overexpression cell line. For JARID1B RNA interference, the control and JARID1B short hairpin RNA (shRNA) plasmids were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and was used to transfect the A549 cells to establish the JARID1B knockdown cell line. The transfection efficiency of JARID1B was confirmed by Western blotting and quantitative reverse-transcription PCR (qRT-PCR) analyses.
MTT assay
A 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium (MTT) assay was used to assess cell proliferation. The cells were seeded, and 20 μl of the MTT solution (5 mg/ml) was then added to each well at the indicated time. The absorbance at 570 nm was measured using a microplate reader (Bio-Rad, Hercules, CA, USA).
Colony formation assay
The cells were seeded in 6-cm dishes at a density of 300 cells per dish. After incubation for 14 days, the colonies were fixed with methanol for 10 min and stained with crystal violet for 15 min, after which point the number of colonies containing more than 50 cells was scored.
Western blot assay
Equal amounts of protein were separated using SDS polyacrylamide gels and were electrotransferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). The membranes were immunoblotted overnight at 4 °C with primary antibodies, followed by their respective secondary antibodies. β-Actin was used as the loading control.
Quantitative reverse-transcription PCR
RNA was extracted using TRIzol reagent, according to the manufacturer’s recommended protocol (Invitrogen). qRT-PCR was performed using Applied Biosystems (Foster City, CA, USA) StepOne and StepOne Plus Real-Time PCR Systems. GAPDH was used as a loading control. The following gene-specific primers were used: JARID1B, forward 5′-TGG ATA CGT GGC GTA AAA TG-3′and reverse 5′-CGA GCA GAC TGG CAT CTG TA-3′; p53, forward 5′- GGC ACT TTT GAA GAT CAT TTT CTC-3′ and reverse 5′-CTG TGT TGA GGG CAA TGA G-3′; and GAPDH, forward 5′-CAA GGT CAT CCA TGA CAA CTT TG-3′ and reverse 5′-GTC CAC CAC CCT GTT GCT GTA G-3′. The experiments were repeated a minimum of three times to confirm the results.
Immunofluorescence staining
The cells were grown on the sterile coverslips, and the cells were fixed with 4 % paraformaldehyde and permeabilized using 0.1 % Triton-X100. Cells were blocked with rabbit anti-PCNA antibody followed by hodamine-conjugated anti-rabbit secondary antibody. Finally, the cells were further stained with 4, 6-diami-dino-2-phenylindole (DAPI).
Chamber assay
Migration and invasion assays were performed as previously described [14]. Invasion assays were performed in 24-well transwell chambers (BD Biosciences, Bedford, MA, USA) containing polycarbonate filters with 8-μm pores coated with Matrigel (BD Biosciences). First, the cells that were suspended in serum-free DMEM were added to the upper compartment of the chamber and the medium containing 10 % FBS was added to the lower compartment of the chamber. At the indicated timepoints, the number of cells that had migrated through the membrane and attached to the lower surface of the membrane was counted under a light microscope for a minimum of ten random visual fields. The migration assay was similar to the invasion assay except that the upper side of the membranes was not coated with the Matrigel.
Tumor formation assay in vivo
The in vivo tumorigenesis and metastasis assays were performed, as previously described [14]. Briefly, 1 × 106 cells were injected subcutaneously into the right flanks of severe combined immunodeficient (SCID) mice. Tumor length (L) and width (W) were measured every week, and tumor volume was calculated using the equation: volume = (W 2 × L) / 2. After 5 weeks, the mice were sacrificed and the tumor volume and weight were measured. All of the animal experiments were performed with the approval of the Soochow University School of Medicine Animal Care and Use Committee.
Statistical analysis
The results were analyzed using SPSS 18.0 software (Chicago, IL, USA). Each experiment was repeated a minimum of three times. A two-tailed t test was used to determine statistical significance. The results were presented as the means ± S.D. P values <0.05 were considered to be statistically significant.
Results
Expression of JARID1B was upregulated in lung cancer tissues and increased JARID1B expression correlates with poor patient survival
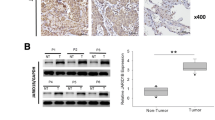
To test whether JARID1B is overexpressed in lung cancer, we first compared the expression of JARID1B in 58 lung cancer tissue samples to those in the adjacent normal tissues using immunohistochemistry. Human normal lung tissues did not show immunostaining. JARID1B protein, which was localized in the nuclei of tumor cells, was expressed in all human lung cancer samples (Fig. 1a). These results indicate significant overexpression of JARID1B in the nuclei of lung cancer cells (Fig. 1a).

Expression of JARID1B in lung cancer tissues. a Representative images of JARID1B immunohistochemical staining in normal lung tissue and lung cancers. b Numbers of JARID1B-positive cells in normal and tumor tissues were analyses. c Kaplan-Meier survival analyses of lung cancer patients. High JARID1B expression correlates with a poorer overall cumulative survival for lung cancer patients (P < 0.001, log-rank test). **P < 0.01 vs normal lung tissues based on Student’s t test. Scale bar, 50 μm
The ratio of JARID1B-positive cells in the lung cancer samples was higher than that in the normal lung tissue samples (Fig. 1b, P < 0.01). To evaluate whether increased JARID1B staining in malignant lung cancer correlates with a worse prognosis, Kaplan-Meier survival curves were constructed using overall cumulative survival to compare the patients with high JARID1B (n = 32) staining to those with low JARID1B (n = 26) staining. Our data revealed that high JARID1B staining correlated with overall poor survival in primary lung cancer (P < 0.001, log-rank test; Fig. 1c). These results suggest a possible role for JARID1B in the development or progression of lung cancer.
Establishment of stable JARID1B transfectants in lung cancer cell lines
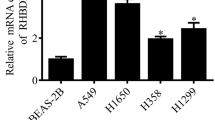
The JARID1B expression levels in six lung cancer cell lines (HLAMP, H1299, H1650, A427, H460, and A549) and one normal lung cell line, BEAS2B, were measured by Western blot (Fig. 2a, b) and qRT-PCR (Fig. 2c). The results show that JARID1B were highly expressed in nearly all tumor cell lines compared with the BEAS2B cell. The H1299 cell line was chosen for establishing a stable cell line because it has lower expression of JARID1B in the six lung cancer cell lines. We also used shRNA to generate a stable JARID1B knockdown in the A549 lung cancer cell line. The transfection efficiency was confirmed using Western blotting and qRT-PCR analyses. As shown in Fig. 2d, e, the H1299 cells transfected with the JARID1B expression plasmid expressed significantly high level of JARID1B at both the messenger RNA (mRNA) and protein levels comparing to the vector cell lines. In addition, the A549 cells transfected with the JARID1B shRNA plasmid expressed significantly lower level of JARID1B at both the mRNA and protein levels comparing to the control cells (Fig. 2f, g).

Transfection efficiency of JARID1B in lung cancer cell lines. a, b JARID1B protein levels in six lung cancer cell lines and one normal lung epithelial cell line, as assessed using Western blotting analyses. β-Actin was used as a loading control. c JARID1B mRNA levels in six lung cancer cell lines and one normal lung epithelial cell line, as assessed using qRT-PCR analyses. d The transfection efficiency of JARID1B in H1299 was analyzed by measuring protein levels by Western blotting. β-Actin was used as a loading control. e The transfection efficiency of JARID1B in H1299 was analyzed by measuring mRNA levels by qRT-PCR. f The transfection efficiency of JARID1B in A549 was analyzed by measuring protein levels by Western blotting. β-Actin was used as a loading control. g The transfection efficiency of JARID1B in A549 was analyzed by measuring mRNA levels by qRT-PCR analyses. The data represent the means ± SD of three independent experiments. **P < 0.01 vs control cells based on Student’s t test
JARID1B promoted lung cancer cell proliferation
We first explored the effects of JARID1B expression on cell growth using the clonogenic assay. As shown in Fig. 3a, JARID1B overexpression significantly increased the colony formation efficiency of H1299 cell line, whereas JARID1B knockdown significantly inhibited the colony formation efficiency of A549 cells (Fig. 3c). Next, we also performed a MTT assay to confirm the effects of JARID1B on proliferation. We found that JARID1B overexpression dramatically enhanced the growth of H1299 cells (Fig. 3b), whereas the growth efficiency was dramatically reduced in the A549 shRNA cell lines (Fig. 3d). As PCNA is an important marker of cell proliferation, we next examined the PCNA by immunofluorescence staining. As shown in Fig. 4, we found that the overexpression of JARID1B in H1299 cells significantly upregulated PCNA staining (Fig. 4a). Also knockdown of JARID1B in A549 cells dramatically downregulated the staining of PCNA (Fig. 4b). These results suggested that JARID1B could significantly promote the proliferation of lung cancer cells.

Effects of JARID1B on proliferation in lung cancer cell lines. a The results of colony formation assays that were conducted in JARID1B-transfected H1299 cell lines. b Cell proliferation after JARID1B overexpression in H1299 cell lines was measured using MTT assays. c The results of colony formation assays that were conducted in JARID1B-knockdown A549 cell lines. d Cell proliferation after JARID1B knockdown in A549 cells was measured using MTT assays. The data represent the means ± SD of three independent experiments. **P < 0.01 vs control cells based on Student’s t test

Effects of JARID1B on PCNA in lung cancer cell lines. a Immunofluorescence staining of PCNA in JARID1B-transfected H1299 cell lines. b Immunofluorescence staining of PCNA in JARID1B-knockdown A549 cell lines. Scale bar, 5 μm
JARID1B enhanced lung cancer cell mobility
We next assessed whether JARID1B could affect the ability of lung cancer cells to migrate and invade using a transwell assay. JARID1B overexpression enhanced both migration and invasion in H1299 cells (Fig. 5a, b). In addition, JARID1B knockdown in A549 cells significantly inhibited cell migration and invasion (Fig. 5c, d). These results indicated that JARID1B significantly promoted the invasion and migration of lung cancer cells.

Effects of JARID1B on invasion and migration in lung cancer cells. a Migration and invasion ability was measured using Transwell assays in JARID1B-transfected H1299 cells. b The summary graphs are presented for the experiment that was outlined in a. c Migration and invasion ability was measured using Transwell assays in JARID1B-knockdown A549 cell lines. d The summary graphs are presented for the experiment that was outlined in c. The data represent the mean number of cells per field and are presented as the means ± SD of three independent experiments. **P < 0.01 vs control cells based on Student’s t test
Knockdown JARID1B significantly inhibited tumorigenesis in vivo
To explore the effects of JARID1B on tumorigenesis in vivo, different cells were injected subcutaneously into the flanks of nude mice. The diameters of the tumors were measured every week. The tumors in the JARID1B knockdown group grew slower compared with those in the mice that had been injected with the control cells during the subsequent days (Fig. 6a, b). We also analyzed the weight of the tumors. As shown in Fig. 6c, the average tumor weight of shJARID1B group was dramatically lower than the control group in A549 cells. These results suggested that JARID1B inhibits lung cancer cell xenograft formation and growth in vivo. These results suggested that knockdown JARID1B inhibit lung cancer cell xenograft formation and growth in vivo.

Knockdown JARID1B inhibited A549 cell line tumorigenesis in vivo. a Representative tumors are presented from the JARID1B-knockdwon A549 and control cells. b Growth curves of lung tumors after the injection of JARID1B-knockdwon A549 and control cells into SCID mice. The error bars represent the means ± SD (n = 4). c Weight of tumors was measured. **P < 0.01 vs control group based on Student’s t test
JARID1B enhanced tumor proliferation and metastasis partly via p53
The p53 has important roles in the proliferation, migration, and invasion of various cancer types, including lung cancer [15, 16]. Thus, we determined whether the p53 was involved in JARID1B-mediated tumor proliferation and metastasis. We evaluated the effects of JARID1B on the p53 and its downstream molecules p27 in H1299 and A549 cells by Western blot and qRT-PCR. As shown in Fig. 7a and Supplemental Figure 1, upregulation of JARID1B significantly inhibited the expression of p53 and p27 in H1299 cells. Knocking down of JARID1B dramatically upregulated the expression of p53 and p27 in A549 cells. To further define a causal relationship between p53 induction and defective foci formation, we transfected A549 cells with both JARID1B shRNA and p53 siRNA (Fig. 7b). p53 inhibition significantly restored the proliferation potential of A549 cells treated with JARID1B shRNA (Fig. 7c, d). We further tested the role of p53 in JARID1B-induced migration and invasion by knocking down p53 expression using siRNA. As shown in Fig. 7e, f, the migration and invasion ability that was inhibited by shJARID1B was reestablished following p53 knockdown using siRNA. Thus, JARID1B siRNA knockdown impaired the proliferation and migration potential of lung cancer cells at least in part through p53 induction.

The effects of JARID1B on the p53. a The expression of p53 and p27 in JARID1B-transfected and knockdown cells were examined using Western blotting. β-Actin was used as a loading control. b The transfection efficiency of p53 siRNA 24 h after transfection was measured using Western blot analyses in JARID1B-knockdown A549 cells. β-Actin was used as a loading control. c, d siRNA-mediated p53 inhibition significantly restored the colony formation (c) and proliferation (d) potential of A549 cells treated with JARID1B shRNA. e The summary graphs show the migration ability of JARID1B knockdown A549 and its control cells after the cells had been transfected with p53 siRNA and its negative control. f The summary graphs are presented for the experiment that was outlined in e. The data represent the mean number of cells per field and are presented as the means ± SD. **P < 0.01 vs control cells based on Student’s t test
Discussion
Lung cancer is one of the most common causes of cancer-related deaths worldwide. And, non-small cell lung cancer (NSCLC) comprises approximately 80 % of all lung cancers [1]. Patients harboring NSCLC are frequently diagnosed at the advanced stage, suffering by metastatically or locally advanced diseases. Nearly 90 % of lung cancer patients die of metastasis [2]. Although great efforts and progressions have been made in the study of the lung cancer in recent decades, the molecular mechanism of lung cancer remains unrevealed. In this manuscript, we identified JARID1B as a candidate target gene accounting for the promotion of lung cancer growth and metastasis.
JARID1 family proteins can promote transcriptional activation, thus affecting important processes such as the hormone response, stem cell renewal, germ cell development, and cellular proliferation and differentiation [17–19]. Recent researches showed that JARID1B is highly expressed in ER-positive primary breast cancers and regulated by both ERα and HIF1α in normoxia and hypoxia [12, 11, 18]. These results imply involvement of JARID1B in tumorigenesis. However, the mechanisms underlying JARID1B-involved carcinogenesis and the roles of JARID1B in lung cancer are largely unknown.
In the present study, we found that JARID1B was overexpressed in both lung cancer cell lines and lung cancer tissues but not in normal lung tissues. The proliferation and invasive potential of lung cancer cells was significantly increased by ectopic expression of JARID1B. Furthermore, interference of JARID1B by siRNA in lung cancer cells significantly decreased the proliferation and invasive potential. Moreover, we also found that the expression of p53 was modulated by JARID1B. Overexpressed JARID1B cell exhibited greatly decreased p53 expression, whereas silencing of JARID1B expression dramatically increased p53 expression at both the mRNA and protein levels. Inhibition of p53 by siRNA reversed the shJARID1B-induced proliferation and invasion. Our results collectively suggested that JARID1B expressed in lung cancer played a role in lung cancer cells proliferation and invasion, which may be partly associated with the p53 expression.
Oncogene enhances tumor growth and invasive and metastatic potential [20, 21]. Overexpression of oncogenes may lead to a malignant cancer phenotype. Previous studies have reported that the expression levels of oncogenes were increased in tumors compared with normal tissues [22, 23]. To confirm the tumor promoter function of JARID1B, we first examined the levels of JARID1B in lung cancer samples and matched normal lung tissue samples using IHC. The results showed that JARID1B was significantly increased in cancers, but not in the normal lung tissues, which suggested that JARID1B was a candidate tumor oncogene in lung cancer. To further explore the role of JARID1B in lung cancer, we transfected lung cancer cells either to ectopically express JARID1B or to inhibit its expression using RNA interference. Our in vitro experiments demonstrated that overexpression of JARID1B significantly enhanced the proliferation, migration, and invasion of lung cancer cells, while knockdown of JARID1B inhibited cell growth and mobility. Our in vivo experiments also demonstrated that JARID1B knockdown markedly inhibited tumorigenesis and growth in nude mice. These data further supported the tumor promoter role of JARID1B in lung cancer.
The p53 protein is a well-known tumor suppressor and has been demonstrated to have an essential role in lung cancer proliferation, motility, and invasion [22]. Lower expression levels of p53 were observed in lung cancer tissues compared with normal samples, and lower levels of p53 were associated with both an increased risk of lymph node metastases and poor prognoses [22, 24, 25]. Therefore, we studied the connection between JARID1B and p53. Our results indicated that the levels of p53 and its downstream molecules p27 were significantly decreased in JARID1B-overexpressing cells and p53 and p27 were correspondingly upregulated in JARID1B knockdown cells. Inhibition of p53 by siRNA reversed the shJARID1B-induced suppression of proliferation and invasion. Previous studies showed that JARID1B-mediated H3K4 demethylation contributes to silencing of retinoblastoma target genes in cells, suggesting a mechanism by which retinoblastoma triggers gene silencing [26, 27]. Therefore, the possible mechanism of JARID1B in regulating p53 and p27 levels maybe via H3K4 demethylation. All of these results demonstrated that JARID1B promotes the proliferation, migration, and invasion activities of lung cancer cells partially via downregulated p53.
In conclusion, we found that JARID1B expression was generally higher in lung cancer lesions compared with matched normal lung tissue samples. Our in vitro and in vivo data demonstrate that JARID1B has a vital function in promoting cell mobility, which is at least partially controlled by the p53. Thus, we propose that the candidate tumor oncogene JARID1B may be an effective novel therapeutic target in the treatment of lung cancer.
References
Nana-Sinkam SP, Powell CA. Molecular biology of lung cancer: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):e30S–9.
Berge EM, Doebele RC. Targeted therapies in non-small cell lung cancer: emerging oncogene targets following the success of epidermal growth factor receptor. Semin Oncol. 2014;41(1):110–25.
Reungwetwattana T, Dy GK. Targeted therapies in development for non-small cell lung cancer. J Carcinog. 2013;12:22.
Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013;73(10):2936–42.
Marmorstein R, Trievel RC. Histone modifying enzymes: structures, mechanisms, and specificities. Biochim Biophys Acta. 2009;1789(1):58–68.
Young LC, Hendzel MJ. The oncogenic potential of Jumonji D2 (JMJD2/KDM4) histone demethylase overexpression. Biochem Cell Biol = Biochimie et biologie cellulaire. 2013;91(6):369–77.
Berry WL, Kim TD, Janknecht R. Stimulation of beta-catenin and colon cancer cell growth by the KDM4B histone demethylase. Int J Oncol. 2014;44(4):1341–8.
Chen L, Fu L, Kong X, Xu J, Wang Z, Ma X, et al. Jumonji domain-containing protein 2B silencing induces DNA damage response via STAT3 pathway in colorectal cancer. Br J Cancer. 2014;110(4):1014–26.
Antony J, Oback F, Chamley LW, Oback B, Laible G. Transient JMJD2B-mediated reduction of H3K9me3 levels improves reprogramming of embryonic stem cells into cloned embryos. Mol Cell Biol. 2013;33(5):974–83.
Coffey K, Rogerson L, Ryan-Munden C, Alkharaif D, Stockley J, Heer R, et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013;41(8):4433–46.
Yang J, Jubb AM, Pike L, Buffa FM, Turley H, Baban D, et al. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010;70(16):6456–66.
Toyokawa G, Cho HS, Iwai Y, Yoshimatsu M, Takawa M, Hayami S, et al. The histone demethylase JMJD2B plays an essential role in human carcinogenesis through positive regulation of cyclin-dependent kinase 6. Cancer Prev Res (Philadelphia, Pa). 2011;4(12):2051–61.
Fu L, Chen L, Yang J, Ye T, Chen Y, Fang J. HIF-1alpha-induced histone demethylase JMJD2B contributes to the malignant phenotype of colorectal cancer cells via an epigenetic mechanism. Carcinogenesis. 2012;33(9):1664–73.
Wang Y, Wen M, Kwon Y, Xu Y, Liu Y, Zhang P, et al. CUL4A induces epithelial-mesenchymal transition and promotes cancer metastasis by regulating ZEB1 expression. Cancer Res. 2014;74(2):520–31.
Qiao Q, Hu W. The association between TP53 Arg72Pro polymorphism and lung cancer susceptibility: evidence from 30,038 subjects. Lung. 2013;191(4):369–77.
Zhang B, Wang E, Dai H, Hu R, Liang Y, Li K, et al. BRIT1 regulates p53 stability and functions as a tumor suppressor in breast cancer. Carcinogenesis. 2013;34(10):2271–80.
Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, et al. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20(12):1557–62.
Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO, et al. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One. 2011;6(3).
Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25(1):1–14.
Xu Y, Wang Y, Ma G, Wang Q, Wei G. CUL4A is overexpressed in human pituitary adenomas and regulates pituitary tumor cell proliferation. J Neuro-Oncol. 2014;116(3):625–32.
Wang Q, Wang Y, Zhang Y, Zhang Y, Xiao W. The role of uPAR in epithelial-mesenchymal transition in small airway epithelium of patients with chronic obstructive pulmonary disease. Respir Res. 2013;14:67.
Yang P, Du CW, Kwan M, Liang SX, Zhang GJ. The impact of p53 in predicting clinical outcome of breast cancer patients with visceral metastasis. Sci Rep. 2013;3:2246.
Wang Y, Ma G, Wang Q, Wen M, Xu Y, He X, et al. Involvement of CUL4A in regulation of multidrug resistance to P-gp substrate drugs in breast cancer cells. Molecules. 2013;19(1):159–76.
Shapira I, Lee A, Vora R, Budman DR. P53 mutations in triple negative breast cancer upregulate endosomal recycling of epidermal growth factor receptor (EGFR) increasing its oncogenic potency. Crit Rev Oncol/Hematol. 2013;88(2):284–92.
Rieber M, Strasberg-Rieber M. p53 inactivation decreases dependence on estrogen/ERK signalling for proliferation but promotes EMT and susceptibility to 3-bromopyruvate in ERalpha + breast cancer MCF-7 cells. Biochem Pharmacol. 2014;88(2):169–77.
Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci U S A. 2012;109(23):8971–6.
Albert M, Schmitz SU, Kooistra SM, Malatesta M, Morales Torres C, Rekling JC, et al. The histone demethylase Jarid1b ensures faithful mouse development by protecting developmental genes from aberrant H3K4me3. PLoS Genet. 2013;9(4):e1003461.
Acknowledgments
This project was supported by National Natural Science Foundation of China (81171488).
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Xudong Shen and Zhixiang Zhuang contributed equally to this work.
Electronic supplementary material
13277_2015_3418_MOESM1_ESM.jpg
The effects of JARID1B on the p53 and p21 mRNA expression. A: The expression of p53 and p27 in JARID1B-transfected H1299 cells and its control cells were examined using qRT-PCR. B: The expression of p53 and p27 in JARID1B knockdown A549 cells and its control cells were examined using qRT-PCR. (JPEG 961 KB)
Rights and permissions
About this article

{kind=link}
Cite this article
Shen, X., Zhuang, Z., Zhang, Y. et al. JARID1B modulates lung cancer cell proliferation and invasion by regulating p53 expression. Tumor Biol. 36, 7133–7142 (2015). https://doi.org/10.1007/s13277-015-3418-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3418-y