
Abstract
Tumorigenesis is a multistep process that attributes to the sequential accumulation of abnormal expression in key oncogenes or tumor suppressors. We aimed to identify stage-specific biomarkers to distinguish lung adenocarcinoma (LAC) stages in cancer progression. RNA-sequencing data of LAC and matched adjacent non-cancer tissues were downloaded from the Cancer Genome Atlas, including 29 pairs of samples from LAC at stage I, 14 from LAC at stage II, 13 from LAC at stage III, and 1 from LAC at stage IV. Differentially expressed genes (DEGs) were analyzed for each case at different stages of LAC. DEGs were further annotated based on transcription factor data information, tumor-associated gene database, and protein–protein interaction database. Functional annotation was performed for genes in PPI network by DAVID online tool. Our analysis identified 11 high-frequency DEGs in the stage I, 29 in the stage II, and 90 in the stage III of LAC. Among them, eight genes were significantly correlated with LAC stages and identified as biomarkers in LAC progression. ANGPTL5, C7orf16, EDN3, LOC150622, HOXA11AS, IL1F5, and USH1G significantly distinguished stage III from stages I and II. GJB6 was significantly enriched in the gap junction trafficking pathway, while C7orf16 and EDN3 were enriched in Wnt signaling pathway, cell cycle, and G protein-coupled receptor (GPCR) signaling. Up-regulated GJB6 especially in LAC stage II and down-regulated C7orf16 and EDN3 specifically in stage III were identified as biomarkers for distinguishing cancer stage in tumor progression through dysregulating gap junction, Wnt signaling, and GPCR signaling pathways.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung adenocarcinoma (LAC) is the most common histological form of lung cancer [1, 2], accounting for nearly 40 % of lung cancers. The incidence of LAC continues to rise [3], especially in China, where the number of smokers is continuing to increase every year [4]. The clinical staging reflects that LAC usually progresses through several pathologic stages in its progression. Pathologic stages of LAC are critical factors for cancer treatment and outcome prediction. A better understanding of LAC staging will pave the way for novel therapies for LAC [5].
Over the past decades, numerous analyses revealed the heterogeneity of LAC [6]. It is well known that tumorigenesis is a multistep process that associated with sequential accumulation of abnormal expression, mutation, or epigenetic silencing by methylation of promoter regions in key oncogenes, tumor suppressor genes, or microRNAs (miRNAs). Previous report has shown that restoration of p53 in lung cancer models results in significant tumor cell loss specifically in LAC but not in adenomas [7]. It indicates that p53 is important in LAC progression, and restoration of it may lead to incomplete tumor regression. Moreover, over-expression of angiogenic growth factor vascular endothelial growth factor (VEGF)-A has been shown in early-stage of prostate cancer, while VEGF-D is associated with advanced stage of metastatic prostate cancer [8]. Notably, primary LACs can be discriminated from metastasis of extra-pulmonary origin by expression profiling [9]. With the development of clinical treatment, personalized cancer therapy is widely put forward in modern society. The strategy is based on targeting the underlying genetic disorders resulting from inactivation of tumor suppressors and activation of oncogenes in cancer progression. However, there are still no molecular biomarkers available for distinguishing patients subjected to LAC at various stages.
In this study, we characterized global expression changes by analyzing gene expression data of tumor specimens and matched normal control from patients with stages I, II, III, or IV LAC. After identifying differentially expressed genes (DEGs) in different stages of LAC, potential stage-specific biomarkers were screened out based on gene annotation and correlation analysis, followed by protein–protein interaction (PPI) network construction and pathway enrichment analysis of biomarkers. Our purpose was to identify molecular biomarkers that can distinguish stages of LAC and be potential targets in personalized treatment.
Materials and methods
Data source
Annotated RNA-sequencing (RNA-seq) data were downloaded from the Cancer Genome Atlas (TCGA; https://tcga-data.nci.nih.gov/tcga/) for our computational analysis. A total of 57 pairs of lung cancer and matched adjacent non-cancer tissues were obtained and eligible for gene expression analysis. Of the 57 pairs, 29 pairs of samples came from LAC at stage I (13 from stage IA and 16 from stage IB), 14 from LAC at stage II (5 from stage IIA and 9 from stage IIB), 13 from LAC at stage III (12 from stage IIIA and 1 from stage IIIB), and 1 from LAC at stage IV.
Analysis of gene expression levels
Following alignment of RNA-seq reads to the reference human genome, the digital expression levels of each annotated genes from pairs of LAC and matched adjacent non-cancer tissues were calculated using reads per base per kilo million (RPKM) method by RNA-Seq by Expectation Maximization (RSEM) package [10]. First, a set of reference transcript sequences were generated and preprocessed later by RSEM steps. Then, a set of RNA-seq reads were aligned to the reference transcripts, and the resulting alignments were used to estimate abundances and their credibility intervals.
Analysis of abnormally expressed genes
The gene expression levels may be different between cancer and adjacent non-cancer tissue. DEGs between cancer and adjacent non-cancer tissue of each case were identified using the NOISeq program [11] which is a novel nonparametric approach for identification of abnormally expressed genes. The NOISeq q value of more than 0.8 was demanded in each DEG between cancer sample and control. If the gene expression value in LAC is higher than in the control sample, it was defined as up-regulation. On the contrary, the gene was down-regulated in cancer tissues. To search for heterogeneity of identified DEGs from samples of different cancer stages, principle component analysis (PCA) was performed on all 57 cancer tissues samples using log2 fold change (FC) between case and matched control. For DEGs in different stages of LAC, Spearman’s rank correlation coefficients were calculated to determine the correlation between the number of DEGs and cancer stage.
High-frequency DEGs in LAC
To screen out commonly expressed genes in cancer tissues of LAC, high frequent up- and down-regulated proteins were summarized. A gene was defined as high-frequency DEG if its frequency in cancer cases at a given stage was not less than 50 %.
Gene annotation
To search for high-frequency DEGs involved in transcription regulation, we checked the genes encoding transcription factors in cancer samples and matched controls. Furthermore, we also searched the tumor-associated gene (TAG; http://www.binfo.ncku.edu.tw/TAG/) database [12] using abnormally expressed genes at high frequency to retrieve oncogenes and tumor suppressor genes.
Identification of high-frequency stage-specific DEGs contributing to cancer development
Frequency of DEGs was calculated in the LAC cases at stages I, II, and III, respectively. Fisher’s exact test was used to test whether the difference of each high-frequency DEG between either two cancer stages (stage I vs stage II, stage I vs stage III, stage II vs stage III) was significant according to the threshold of P value <0.05. Genes were nominated to be stage-specific genes associated with LAC progression using the following conditions: (i) the gene was abnormally expressed at frequency just at one stage; and (ii) the gene with P value calculated by Fisher’s exact test of less than 0.05. Meanwhile, gene biomarkers of LAC stage also screened from stage-specific genes using correlation analysis of Fisher’s exact test.
Construction of PPI network
To get insight into the role that stage-specific genes play in different stages of LAC, we scanned the STRING database [13] (http://string-db.org/) to retrieve the protein interaction information. The network was visualized by Cytoscape software [14] (http://cytoscapeweb.cytoscape.org/).
Functional annotation of genes in network
Pathway enrichment analysis was performed to derive all the related functions using DAVID (the Database for Annotation, Visualization, and Integrated Discovery) online tool [15] (http://david.abcc.ncifcrf.gov/) based on REACTOME database (http://www.reactome.org/) [16]. P value was calculated by hypergeometric test, and only results with a P value of less than 0.05 were considered to be significant.
Results
DEGs analysis
Based on LAC RNA-seq data, we first analyzed DEGs between cancer and matched adjacent non-cancer tissues in individual case. The analysis showed that the counts of DEGs in individual case were highly variable, ranging from 206 to 1244 with the difference of gene counts between the maximal and minimal counts as large as six times (Table 1). Similar results were also observed with regard to abnormally up- and down-regulated proteins (Table 1). To detect and distinguish LAC samples at different stages, we performed a conventional PCA analysis based on log2FC expression data (Fig. 1). The result showed less clear segregation of LAC samples indicating that they were heterogeneous at different stages and not suitable as biological replicates.

Principle component analysis (PCA) of each tissue pair based on the log2 fold change (FC)
To investigate whether the LAC transcriptome is associated with disease stage, we compared counts of DEGs from different stages of cancer and checked out the Spearman correction between counts of DEGs and cancer progression. The result showed there was no correction between counts of DEGs and cancer progression (Spearman correlation coefficient = 0.12, P value = 0.3827) (Fig. 2).

Correlation analysis between counts of differentially expressed (DE) genes and stages in cancer progression
Analysis of high-frequency DEGs in LAC
To search for abnormally and frequently expressed genes in LAC progression, we analyzed the distributions of DEGs among the three-stage LAC (since only one sample at stage IV, it was not involved in next analysis). The gene was defined as a high-frequency DEG if it was observed at minimal 50 % LAC cases at a given stage. The analysis identified 69 (23 up-regulated and 46 down-regulated), 100 (58 up-regulated and 42 down-regulated), and 173 (75 up-regulated and 98 down-regulated) high-frequency DEGs in LAC at stages I, II, and III, respectively. Annotation results found that the products of genes functional as transcription factors paired-like homeodomain 2 (PITX2), diencephalon/mesencephalon homeobox 1 (DMBX1), and BARX homeobox 1 (BARX1) were simultaneously up-regulated in majority of cancer tissue cases at stage I, II, or III (Table 2). Similar result was not observed in the high-frequency down-regulated genes (Table 2).
We also analyzed the expression of TAG by querying TAG database for high-frequency DEGs in LAC [12] and found two TAGs, villin 1 (VIL1) and progestagen-associated endometrial protein (PAEP), which were commonly up-regulated in LAC at stages II and III (Table 2). VIL1 was up-regulated in 57 % (8/14) of cancer tissue cases at stage II and 54 % (7/13) at stage III, while PAEP was up-regulated in 50 % (7/14) of cancer tissue cases at stage II and 54 % (7/13) at stage III. There was no TAG observed in stage II of LAC (Table 2). Since the two proteins are frequently overexpressed at stages II and III of LAC, it suggested that VIL1 and PAEP might be involved in regulation of tumor cells at the late stage of LAC progression.
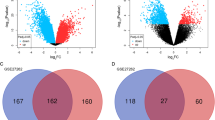
To identify stage-specific genes in LAC progression, Fisher’s exact test was applied to examine significance of high-frequency DEGs between either two stages. From all high-frequency genes, we screened out the stage-specific genes that were abnormally regulated in cancer tissues at each stage. In total, we found 11 (4 up-regulated and 7 down-regulated) specific genes in stage I, 29 specific genes in stage II (22 up-regulated and 7 down-regulated), and 90 specific genes in stage III (40 up-regulated and 50 down-regulated) (Fig. 3).

Venn diagram of high-frequency genes abnormally expressed at various stages of lung adenocarcinoma. a High-frequency down-regulated proteins. b High-frequency up-regulated proteins. Circles with different colors represent different stages of cancer
Screening of biomarkers for diagnosis of LAC stages
To explore genetic biomarkers for diagnosis of LAC stages from stage-specific genes, correlation analysis between expression levels of stage-specific genes and LAC stages were performed. The analysis showed that 8 out of 130 stage-specific genes were significantly correlated with LAC stages I and III. Specifically, gap junction protein, beta 6 (GJB6; 30 kDa) was highly correlated with the stage II of LAC (Table 3). Its protein was up-regulated in 78 % cases of the stage II LAC, whereas there were only 28 % cases of the stage I and 31 % cases of the stage III where GJB6 was overexpressed, respectively (Fig. 4).

Frequency of abnormally expressed genes in the lung adenocarcinoma at stages I, II, and III. Red histogram indicates frequency of up-regulated proteins. Green histogram indicates frequency of down-regulated proteins
In addition, seven genes that frequently occurred abnormal expression in the stage III of LAC significantly distinguished the stage III of LAC from the stages I and II of LAC, including angiopoietin-like 5 (ANGPTL5), C7orf16, endothelin 3 (EDN3), long intergenic non-protein coding RNA 1105 (LOC150622), HOXA11 antisense RNA (HOXA11AS), interleukin 1 family, member 5 (IL1F5), and Usher syndrome 1G (USH1G). Among them, protein expression of ANGPTL5, C7orf16, EDN3, and LOC150622 appeared to be significantly down-regulated in majority of the stage III of LAC, while HOXA11AS, IL1F5, and USH1G showed to be up-regulated in majority of the stage III of LAC. Notably, EDN3 was down-regulated in all the cases at stage III, but there were only 48 % cases at stage I and 43 % cases at stage II. HOXA11AS was up-regulated in 54 % of LAC at stage III, but up-regulated only in 14 % of LAC at stage I and in 7 % of LAC at stage II. However, there was no one gene found to be correlated with the stage I of LAC. Briefly, one stage II-specific gene of LAC and seven stage III-specific genes were identified as biomarkers for diagnosis of LAC progression.
Analysis of PPI network and functional annotation of genetic biomarkers for LAC stages
We queried STRING database of eight potential gene biomarkers for LAC stages. As a result, functional modules of GJB6, C7orf16, EDN3, IL1F5, and USH1G were shown in Fig. 5. But no interaction of ANGPTL5, LOC150622, and HOXA11AS was observed in database. In C7orf16 module, protein kinase, cAMP-dependent, catalytic, alpha (PRKACA), protein kinase, cGMP-dependent, type I (PRKG1), and protein kinase, cGMP-dependent, type II (PRKG2) were interacted with the core protein C7orf16.

Analysis of protein–protein interaction of eight potential genetic biomarkers for stages of lung adenocarcinoma (LAC). Red circle indicates up-regulated protein; green circle indicates down-regulated protein; and purple circle indicates gene that interacted with the potential biomarkers for stages LAC
To explore the molecular functions of five genetic biomarkers, we carried out pathway enrichment analysis for terms in REACTOME (Table 4). Functional annotation showed that GJB6 module was significantly enriched in the pathway of gap junction trafficking and regulation, which interacted proteins gap junction protein, beta 2 (GJB2; 26 kDa) and tight junction protein 1 (TJP1) are also involved in. This result suggests that the pathway of gap junction trafficking and regulation may have relevance to the stage II LAC progression. The module of C7orf16 was mainly enriched in Wnt signaling and cell cycle, and EDN3 was in G protein-coupled receptor (GPCR) signaling pathways. Additionally, PRKACA was also involved in Wnt signaling pathway.
Discussion
In this study, we first analyzed DEGs between LAC and matched adjacent normal tissues based on RNA-seq data obtained from TCGA database. Our results showed that over-expressed GJB6 was significantly correlated to stage II LAC, while seven genes (ANGPTL5, C7orf16, EDN3, LOC150622, HOXA11AS, IL1F5, and USH1G) were specifically correlated to stage III LAC progression. Functional annotation indicated that GJB2 was mainly involved in Gap junction trafficking and regulation pathway. Meanwhile, C7orf16 and EDN3 were significantly enriched in Wnt signaling and cell cycle and GPCR signaling pathways, respectively.
The study showed that different stages of LAC were highly heterogeneous in molecular levels, with great individual differences in proteins abnormally expressed in LAC tissue which was in line with its histological heterogeneity [17]. Up-regulation of PITX2, DMBX1, and BARX1 were observed in majority of LAC at stage I, II, or III. PITX2 is a downstream effector of Wnt/β-catenin signaling. Deregulation of Wnt signaling is strongly associated with LAC progression [18, 19]. More recently, PITX2 can activate the proliferation of cancer cells by inducing the expression of canonical Wnt ligand genes, and in turn activates the signaling pathway [20]. Therefore, it is possible that PITX2 plays a role in the pathogenesis of LAC by activating Wnt signaling. In contrast, PITX2, DMBX1, and BARX1 were first discovered in tumorigenesis of LAC in this study.
VIL1 and PAEP were commonly found to be highly expressed in the late stage of LAC. VIL1 is involved in positive regulation of cell migration [21]. Over-expression of VIL1 in the late stage of LAC suggests it may contribute to LAC metastasis. PAEP is a cell surface glycoprotein. It plays key roles in modulating B cell responses [22], regulating cytokine production in natural killer cells [23], inducing tolerance of dendritic cells [24], T cells apoptosis [25], and Th-2 shift in cytokines [26, 27]. PAEP is expressed in type II pneumocytes, bronchial epithelium cells, and alveolar macrophages which are involved in the pulmonary immune response in the process of asthmatic inflammation [28, 29]. Since over-expression of PAEP in the late stage of LAC, it may accelerate LAC progression through immunosuppression.
Pathway-based enrichment analysis of genetic biomarkers in the PPI network showed that GJB6 module was significantly enriched in the pathway of gap junction trafficking and regulation. Gap junction is a protein channel responsible for intercellular communication which plays important roles in tissue homeostasis, proliferation, differentiation, and death [30]. These channels are formed directly upon the docking of the connexin protein family that contains 21 members in humans [31]. GJB6 has been reported to encode one of connexin proteins, connexin 30 (Cx30). Disruption of gap junction or connexin dysfunction has been indicated to be involved in carcinogenic processes in multiple cancers, including lung carcinogenesis [32–34]. Intriguingly, the expression of GJB6 was increased in the stage II of LAC, suggesting that GJB6 serves as an enhancer in the proliferation of tumor cells [35], which may be involved in the LAC pathogenesis by disrupting intercellular communication. However, inconsistent results are reported in previous studies indicating that its role in tumorigenesis remains to be defined [36, 37].
The C7orf16 module was significantly enriched in Wnt signaling pathway and cell cycle. The Wnt signaling pathway plays a major role in cancer development and progression [38]. Activation of the canonical Wnt/tcf pathway may lead to metastasis to the brain and bone in the progression of LAC [19]. Inhibition of the Wnt/beta-catenin signal transduction pathway in LAC A549 cells has been reported to decrease cell proliferation, clone formation, and migration capacity [39]. Furthermore, C7orf16 is a member of oncology open reading frames that has been found to be associated with multiple solid tumors, including non-small cell lung cancer [40]. Importantly, interacted protein with C7orf16 in the module, PRKACA has been found to be involved in Wnt signaling pathway and mutate in cortisol-producing adrenal tumors [41, 42]. Therefore, we suppose that down-regulation of C7orf16 activates Wnt signaling pathway or disrupts cell cycle, and thus contributes to LAC pathogenesis. In addition, the EDN3 module was significantly enriched in GPCR signaling. Reduced expression of EDN3 has been shown in many solid tumors in arising studies, including but not limited to, colon tumor, lung cancer, and breast cancer [43]. Moreover, GPCRs play important roles in cancer progression from transformation, growth, and survival to metastasis [44]. It is possible that down-regulation of EDN3 in the stage III LAC may disrupt GPCR signaling and lead to poor outcome. PPI network analysis revealed IL1F5 interacted with SMAD family member 3 (SMAD3). It is known that down-regulation of SMAD3 caused by long-term smoking induces resistance to carboplatin in non-small cell lung cancer [45]. Therefore, up-regulation of IL1F5 in the stage III LAC is likely to be implicated in drug resistance in LAC.
In conclusion, GJB6 may contribute to LAC cells proliferation through disrupting intercellular communication via pathway of gap junction trafficking and regulation; down-regulated C7orf16 could activate Wnt signaling pathway or disrupt cell cycle to contribute to LAC pathogenesis, while EDN3 in stage III LAC may disrupt GPCR signaling and lead to poor outcome. Therefore, up-regulated GJB6 especially in stage II LAC and down-regulated C7orf16 and EDN3 specifically in stage III were identified as biomarkers for distinguishing cancer stage in tumor progression through dysregulating biological pathways. These genes may serve as potential biomarkers in personalized treatment of LAC. The study will provide insight into the LAC pathogenesis and directions for future researches. However, more RNA-seq data for stage IV LAC was needed, and experimental validation will be urgent in further studies.
References
Subramanian J, Govindan R. Lung cancer in never smokers: a review. J Clin Oncol. 2007;25:561–70.
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917.
Kadara H, Kabbout M, Wistuba II. Pulmonary adenocarcinoma: a renewed entity in 2011. Respirology. 2012;17:50–65.
Zhang J, Ou JX, Bai CX. Tobacco smoking in China: prevalence, disease burden, challenges and future strategies. Respirology. 2011;16:1165–72.
Huang P, Cao K, Zhao H. Screening of critical genes in lung adenocarcinoma via network analysis of gene expression profile. Pathol Oncol Res. 2014.
Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98:13784–9.
Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature. 2010;468:572–5.
Kaushal V, Mukunyadzi P, Dennis RA, Siegel ER, Johnson DE, Kohli M. Stage-specific characterization of the vascular endothelial growth factor axis in prostate cancer: expression of lymphangiogenic markers is associated with advanced-stage disease. Clin Cancer Res. 2005;11:584–93.
Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–5.
Chen X, Wang S-C, Cao L-H, Yang G-Q, Li M, Su J-C. Comparison between radial head replacement and open reduction and internal fixation in clinical treatment of unstable, multi-fragmented radial head fractures. Int Orthop. 2011;35:1071–6.
Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21:2213–23.
Chen JS, Hung WS, Chan HH, Tsai SJ, Sun HS. In silico identification of oncogenic potential of fyn-related kinase in hepatocellular carcinoma. Bioinformatics. 2013;29:420–7.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–15.
Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. In Data mining in proteomics. Springer; 201. pp. 291–303.
Da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Croft D, O’kelly G, Wu G, Haw R, Gillespie M, Matthews L, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–7.
Solis LM, Behrens C, Raso MG, Lin HY, Kadara H, Yuan P, et al. Histologic patterns and molecular characteristics of lung adenocarcinoma associated with clinical outcome. Cancer. 2012;118:2889–99.
Li B, Ge Z, Song S, Zhang S, Yan H, Huang B, et al. Decreased expression of SOX7 is correlated with poor prognosis in lung adenocarcinoma patients. Pathol Oncol Res. 2012;18:1039–45.
Nguyen DX, Chiang AC, Zhang XH, Kim JY, Kris MG, Ladanyi M, et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138:51–62.
Basu M, Roy SS. Wnt/beta-catenin pathway is regulated by PITX2 homeodomain protein and thus contributes to the proliferation of human ovarian adenocarcinoma cell, SKOV-3. J Biol Chem. 2013;288:4355–67.
Barbazán J, Muinelo‐Romay L, Vieito M, Candamio S, Díaz‐López A, Cano A, et al. A multimarker panel for circulating tumor cells detection predicts patient outcome and therapy response in metastatic colorectal cancer. Int J Cancer. 2014.
Yaniv E, Borovsky Z, Mishan-Eisenberg G, Rachmilewitz J. Placental protein 14 regulates selective B cell responses. Cell Immunol. 2003;222:156–63.
Lee CL, Chiu PC, Lam KK, Chan RW, Chu IK, Koistinen R, et al. Glycodelin-A modulates cytokine production of peripheral blood natural killer cells. Fertil Steril. 2010;94:769–71.
Scholz C, Toth B, Brunnhuber R, Rampf E, Weissenbacher T, Santoso L, et al. Glycodelin A induces a tolerogenic phenotype in monocyte-derived dendritic cells in vitro. Am J Reprod Immunol. 2008;60:501–12.
Lee C-L, Lam KK, Koistinen H, Seppala M, Kurpisz M, Fernandez N, et al. Glycodelin-A as a paracrine regulator in early pregnancy. J Reprod Immunol. 2011;90:29–34.
Mishan-Eisenberg G, Borovsky Z, Weber MC, Gazit R, Tykocinski ML, Rachmilewitz J. Differential regulation of Th1/Th2 cytokine responses by placental protein 14. J Immunol. 2004;173:5524–30.
Lee C-L, Chiu PC, Lam KK, Siu S-O, Chu IK, Koistinen R, et al. Differential actions of glycodelin-A on Th-1 and Th-2 cells: a paracrine mechanism that could produce the Th-2 dominant environment during pregnancy. Hum Reprod. 2011;26:517–26.
Kunert-Keil C, Jeschke U, Simms G, Kasper M. Increased expression of glycodelin mRNA and protein in rat lungs during ovalbumin-induced allergic airway inflammation. Histochem Cell Biol. 2009;131:383–90.
Kunert-Keil C, Steinmüller F, Jeschke U, Gredes T, Gedrange T. Immunolocalization of glycodelin in human adenocarcinoma of the lung, squamous cell carcinoma of the lung and lung metastases of colonic adenocarcinoma. Acta Histochem. 2011;113:798–802.
Czyż J. The stage-specific function of gap junctions during tumourigenesis. Mol Cell Biochem. 2008;13:92–102.
Kar R, Batra N, Riquelme MA, Jiang JX. Biological role of connexin intercellular channels and hemichannels. Arch Biochem Biophys. 2012;524:2–15.
Cesen-Cummings K, Fernstrom MJ, Malkinson AM, Ruch RJ. Frequent reduction of gap junctional intercellular communication and connexin43 expression in human and mouse lung carcinoma cells. Carcinogenesis. 1998;19:61–7.
De Oliveira KD, Tedardi MV, Cogliati B, Dagli ML. Higher incidence of lung adenocarcinomas induced by DMBA in connexin 43 heterozygous knockout mice. Biomed Res Int. 2013;2013:618475.
Harada K, Nonaka T, Hamada N, Sakurai H, Hasegawa M, Funayama T, et al. Heavy-ion-induced bystander killing of human lung cancer cells: role of gap junctional intercellular communication. Cancer Sci. 2009;100:684–8.
Ozawa H, Mutai H, Matsunaga T, Tokumaru Y, Fujii M, Sakamoto K, et al. Promoted cell proliferation by connexin 30 gene transfection to head-and-neck cancer cell line. Anticancer Res. 2009;29:1981–5.
Princen F, Robe P, Gros D, Jarry-Guichard T, Gielen J, Merville M-P, et al. Rat gap junction connexin-30 inhibits proliferation of glioma cell lines. Carcinogenesis. 2001;22:507–13.
Sentani K, Oue N, Sakamoto N, Anami K, Naito Y, Aoyagi K, et al. Upregulation of connexin 30 in intestinal phenotype gastric cancer and its reduction during tumor progression. Pathobiology. 2010;77:241–8.
Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–98.
Teng Y, Wang XW, Wang YW, Wang J. [Effect of siRNA-mediated beta-catenin gene on Wnt signal pathway in lung adenocarcinoma A549 cell]. Zhonghua Yi Xue Za Zhi. 2010;90:988–92.
Delgado AP, Brandao P, Chapado MJ, Hamid S, Narayanan R. Open reading frames associated with cancer in the dark matter of the human genome. Cancer Genomics Proteomics. 2014;11:201–13.
Rennie C, Hulme H, Fisher P, Hall L, Agaba M, Noyes H, et al. A systematic, data-driven approach to the combined analysis of microarray and QTL data. 2010.
Cao Y, He M, Gao Z, Peng Y, Li Y, Li L, et al. Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science. 2014;344:913–7.
Welch A, Jacobs M, Wingo C, Cain B. Early progress in epigenetic regulation of endothelin pathway genes. Br J Pharmacol. 2013;168:327–34.
Kuzumaki N, Suzuki A, Narita M, Hosoya T, Nagasawa A, Imai S, et al. Multiple analyses of G-protein coupled receptor (GPCR) expression in the development of gefitinib-resistance in transforming non-small-cell lung cancer. PLoS One. 2012;7:e44368.
Samanta D, Kaufman J, Carbone DP, Datta PK. Long-term smoking mediated down-regulation of Smad3 induces resistance to carboplatin in non-small cell lung cancer. Neoplasia. 2012;14:644–55.
Acknowledgments
This study was supported by National Natural Science Foundation of China (grant No. 81372632).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liang, J., Lv, J. & Liu, Z. Identification of stage-specific biomarkers in lung adenocarcinoma based on RNA-seq data. Tumor Biol. 36, 6391–6399 (2015). https://doi.org/10.1007/s13277-015-3327-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3327-0