
Abstract
Bladder cancer (BC) is one of the most common human malignancies that account for major death in the world. Apoptin that is derived from chicken anemia virus (CAV) has displayed tumor-specific cytotoxic activity in a variety of human carcinomas. However, the magical function of apoptin in bladder carcinoma cell lines has not been identified yet. In our study, we delivered apoptin into bladder-originating T24, EJ, and HCV29 cell lines by adenovirus system. The selective cytotoxic effect of apoptin was determined by cell viability assay, active caspase-3 measurement, and annexin V/PI double staining. Importantly, we have examined the differential expression patterns of tumor-associated genes including Ki67, C-erbB-2, Rb, and nm23 by flow cytometry and western blot in vitro. In an animal study, apoptin was infused into animal models by AAV system, and immunohistochemistry and quantitative real-time PCR (qRT-PCR) were employed to validate results in vivo. The results indicated that apoptin could selectively induce apoptosis in bladder tumorigenic cells coupled with tumor-specific nucleus accumulation in vitro. Interestingly, apoptin could downregulate expression levels of Ki67 and C-erbB-2 and upregulate the expression of Rb both in vitro and in vivo. Moreover, the animal models treated with AAV-apoptin have shown smaller tumor volumes and displayed better prognosis than controls. In conclusion, apoptin could selectively induce apoptosis in bladder tumor cells through altering expression profiles of tumor-associated genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bladder cancer (BC) is a common malignant tumor in urinary system and a major cause of death annually. It is the seventh most common cancer in men and 17th in women worldwide [1]. The incidence of BC increases with age, occurring most commonly in individuals between 50 and 70 years old [2], and men are three or four times more likely to be affected by BC than women. Importantly, BC is characterized by a high rate of tumor recurrence and progression despite of local therapy [3, 4]. Patients with BC require long-term follow-up and repeat interventions after surgery. Intravesical chemotherapy and immunotherapy with Bacille Calmette–Guérin (BCG) are considered to be effective treatments that are currently available [4, 5]. However, chemoresistance and significant side effects have limited the wide use of chemotherapy. Immunotherapy with BCG could cause nonspecific inflammation in the bladder and progression of the disease [5, 6]. Therefore, long-lasting and effective approaches with enough safety are required in clinical application.
Apoptin derived from chicken anemia virus (CAV) is known to induce apoptosis in a variety of tumors and transformed cells including melanoma, hepatoma, lymphoma, colon carcinoma, breast, and lung cancer [7–9]. The protein is composed of 121 amino acids and shares no homologous sequences with any known cellular proteins. It has a bipartite nuclear localization sequence (NLS) and a putative nuclear export sequence (NES) in the C-terminal end, and an accessible phosphorylation site (Thr-108) is located in the C-terminal end, which allows for its interaction with other proteins and modification by cellular kinases [7, 10, 11]. Interestingly, apoptin is specifically localized within the nucleus of tumor cells after transfection[11]. A number of studies have reported that apoptin could induce selective death of cells from several human tumors and has no apoptotic effects on normal cells [12]. These evidences suggest that the specific accumulation of apoptin in the tumor cell nucleus may be responsible for the selective killing effects. These selective killing effects of apoptin may provide safety assurance in the gene therapy of BC by employing this protein. Unfortunately, the mechanism of the selective cytotoxicity of apoptin has still not been well established. Some studies have indicated that apoptin could induce tumor-specific cell apoptosis independently of p53 pathway [13]. Moreover, it was reported that apoptosis induced by apoptin is typically mediated by intracellular cysteine proteases (caspase) [14]. Bcl-2, which is an antiapoptotic protein, could accelerate cell death mediated by apoptin [15, 16].
In terms of BC, a number of studies have reported that the expression profiles of several genes including Ki67, C-erbB-2, Rb, and nm23 are associated with the stages of BC and 5-year survival rates of patients [17–20]. However, the relationships between apoptin and these tumor-associated genes were still unknown in BC. In this study, we intended to investigate the apoptin-induced selective cell death on bladder-derived cell lines T24, EJ, and HCV-29 in vitro by employing adenovirus system. To further explore the mechanism, the influence of apoptin on the expression profiles of Ki67, C-erbB-2, Rb, and nm23 were evaluated. Additionally, apoptin was delivered into the mouse model of BC by adeno-associated virus 2 (AAV-2). The alteration of BC-associated gene expression patterns were also assayed in vivo. Herein, we aimed to identify the apoptotic effect and mechanism of apoptin in BC, and our study may shed light on gene therapy by using apoptin.
Materials and methods
The AdEasy XL System and AAV Helper-Free System were purchased from Stratagene (CA, USA). All the primers, genes, Taq polymerase, restriction enzymes, and T4 DNA ligase were obtained from TaKaRa (Dalian, China). The gene of apoptin (VP3 of CAV) was synthesized and preserved by the Department of Urology, the Second Affiliated Hospital of Kunming Medical University. The antibody against apoptin was prepared by immunization of rabbit with E. coli-derived apoptin. The cell lines of T24, EJ, and HCV29 were obtained from the Institute of Urology (Yunnan). BALB/C nude mice were purchased from Huafu Technology Company (Beijing, China). All experimental procedures were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH Guidelines). The animal studies and protocol were approved by the Experimental Animal Ethics Committee of Yan’an Affiliated Hospital of Kunming Medical University. All surgeries were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering. Experiments involving human tissues were reviewed and approved by the Bioethics Committee of the Second Affiliated Hospital of Kunming Medical University. Informed consent has been received from all the subjects involved in this study.
Construction of recombinant adenovirus and adeno-associated virus
The gene of apoptin was amplified from pcDNA3.0-apoptin, and the PCR product was purified and cloned into the plasmid pShuttle-CMV. When the recombinant plasmid was confirmed, it was linearized and dephosphorylated sequentially by Pme I and alkaline phosphatase. The gel-purified linearized vector and pAdEasy-1 vector were cotransformed into BJ5183-competent cells by electroporation. Homologous recombination took place in the bacterial cells to generate a recombinant AdEasy plasmid DNA carrying apoptin. The correct recombinant AdEasy plasmid was characterized by Pac I enzyme cutting yielding a small fragment of either 3.0 kb or 4.5 kb and a larger fragment of about 30 kb. Recombinant AdEasy clone was transfected into Ad-293 cell to obtain the recombinant adenovirus-expressing apoptin. pShuttle-CMV without any exogenous DNA fragment was also recombined with pAdEasy-1 to get control virus (Ad-VC). The primary stock of adenoviruses were harvested and amplified in large scale. Then, the product that was ready for infection was tittered. Moreover, the gene of apoptin was also cloned into pAAV-MCS followed by screening of recombinant pAAV-MCS-apoptin. pAAV-MCS-apoptin plus two helper plasmids including pAAV-RC and pHelper were cotransfected into AAV-293 cell line to obtain the recombinant AAV-apoptin. The recombinant AAV were harvested from AAV-293 by four rounds of freeze/thaw by alternating the tubes between the dry ice–ethanol bath. The AAV were separated by chloroform and concentrated by PEG800/sodium chloride precipitation. Finally, AAV were purified by chloroform extraction and tittered by digoxin-labeled DNA dot blotting, which were ready for animal study.
Reverse transcription PCR and quantitative real-time PCR
The T24, EJ, and HCV29 cells were suspended in the growth medium (10 % FBS, RPMI 1640 medium) and plated in the 25-cm2 flask at a density of 1 × 106 cells/ml and incubated in 37 °C, 5 % CO2 for 15 h. Next day, on reaching 90 % confluency, the cells were infected by recombinant adenovirus at multiplicity of infection (MOI) = 5. Several days after infection, the cells were scraped off, and RNA was extracted by using GeneJET RNA Purification Kit (Fermentas, Thermo Fisher Scientific). The first strand of cDNA was synthesized by RevertAid First Strand cDNA Synthesis Kit (Fermentas, Thermo Fisher Scientific), and PCR was performed to identify the transcriptions of target genes. The RNA extraction of clinical samples was initiated by liquid nitrogen grinding and followed by RNA purification described above. Moreover, all the quantitative real-time PCR (qRT-PCR) involved in this study were performed on the LightCycler 480 platform (Roche). The first strand of cDNA was synthesized with adjusted concentration, and corresponding genes were amplified by employing LightCycler 480 SYBR Green I Master.
Immunofluorescence assay
The T24, EJ, and HCV29 cells were suspended in the growth medium (10 % FBS, RPMI 1640 medium) and plated in the six-well plate at a density of 1 × 105 cells/ml and incubated in 37 °C, 5 % CO2 for 15 h. Next day, on reaching 50 % confluency, the cells were infected by recombinant adenovirus at MOI = 5. After several days of infection, the cells were washed three times with PBS and fixed and permeabilized with ice cold buffer (0.2 % Triton X-100, 2 % paraformaldehyde, PBS) for 15 min. Then, the cells were treated with a mixture of acetone and methanol and followed by three times PBS washing. After half an hour’s blocking with 3 % BSA, the primary antibody against apoptin was diluted with blocking buffer and incubated with cells for 2 h with shaking. The cells were washed with PBST (0.05 % Tween 20 PBS) three times and incubated with blocking buffer (3 % BSA, 0.5 % Tween 20 PBS) diluted DyLight488-conjugated goat anti-rabbit IgG secondary antibody for 1 h. After washing with PBST three times, the cells were examined under a fluorescence microscope.
Cell proliferation and active caspase-3 immunoassays
The T24, EJ, and HCV29 proliferations were determined by Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc.). Cells (5 × 103) were seeded into each well of 96-well plates with 200 μl medium and incubated at 37 °C, 5 % CO2 overnight. Next day, the cells were infected with Ad-apoptin or Ad-VC as control, respectively, and cells without any treatment were taken as reference. The growth of the cells was monitored every 24 h by adding 10 μl of the CCK-8 solution to the wells. After incubation for 4 h, absorbance was measured with a plate reader at 490 nm. Each cell line for one time point was calculated by values from five independent samples. Moreover, the levels of active form of caspase-3 were measured by an ELISA-based method (Quantikine™ human active caspase-3 immunoassay, R&D Systems). T24, EJ, and HCV29 were plated into 96-well plates and infected with Ad-apoptin or Ad-VC as control. After 72 h postinfection, the amount of active caspase-3 was measured, and the protocol for active caspased-3 immunoassay was followed according to the manufacturer’s instructions.
Flow cytometry and western blot
The apoptosis of cells infected with Ad-apoptin and Ad-VC were detected by annexin V-FITC apoptosis detection kit (eBioscience). Cells treated with Ad-apoptin or Ad-VC were collected and washed with PBS three times. The cells were resuspended with binding buffer with appropriate density. Then, cells were stained with FITC-linked annexin V and PI sequentially, and washing with binding buffer was necessary. The cell apoptosis were analyzed by flow cytometry (BD FACSCalibur). The differential expression profiles of Ki67 in cell nucleus were measured by flow cytometry. The T24, EJ, and HCV29 were seeded into six-well plates with cell density of 5 × 106 cells/well. Ad-apoptin and Ad-VC were inoculated into each well and incubated for 72 h. After infection, cells in each well were harvested and washed with PBS three times. Then, ice-cold 80 % ethanol was added and incubated at −20 °C for 3 h. The cells were resuspended with staining buffer twice and adjusted to concentration of 1 × 107/ml. FITC-conjugated anti-Ki67 antibody (BD Biosciences) were added into tubes and incubated at room temperature for 30 min in the dark, and FITC-conjugated isotype control IgG were taken as control. After staining, the differential expression of Ki67 was analyzed by flow cytometry. Moreover, the expression patterns of C-erbB-2, Rb, and nm23 were determined by western blot. The T24, EJ, and HCV29 infected with various recombinant adenovirus were treated with cell lysis buffer, and the concentration of total proteins extracted was measured by a Lowry-based method. The samples were analyzed by 12 % SDS-PAGE gel loaded with equal amounts of protein. The proteins were electransferred to PVDF membrane at 40 V for 100 min. Next, the membrane was incubated with 5 % BSA in PBST for blocking. The primary antibodies against c-erbB-2 (Dako, Denmark), Rb (Santa Cruz Biotechnology, Inc., Dallas, TX, USA), nm23 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA), and β-actin (Biosis, China) were added and incubated at 4 °C overnight. The HRP-conjugated secondary antibodies were added after three-time PBST washing. After incubation, the membranes were washed thoroughly with PBST three times and then the bands were visualized by an enhanced chemiluminescence kit (Millipore, Billerica, MA, USA).
Immunohistochemistry
The animals were anesthetized with sodium pentobarbital (10 mg/kg) and killed. The tumors were removed and fixed with 4 % paraformaldehyde PBS for 7 days. After fixation, the tissues were dehydrated and embedded in paraffin. The tissues were sectioned in paraffin (5 μm) by a sliding microtome, and the tissue sections were baked at 56 °C for 48 h. Dewaxing and antigen retrieval procedures were necessary before immunohistochemistry was performed. The sections were treated with 3 % hydrogen peroxide and blocked by goat serum for 10 min, respectively. Then, the sections were incubated with the primary antibodies overnight, followed by treatments with biotin- and streptavidin-conjugated antibodies sequentially. After 3× PBS washing, the color was developed using 3′3′-diaminobenzidine as chromogen. Then, the tissue sections were further stained with hematoxylin and eosin.
Results
Expression of apoptin in bladder-derived transformed and nontransformed cell lines and identification of its nuclear targeting effect
Two strains of bladder-derived transformed cell lines T24 and EJ were used as study objects, and HCV29 nonmalignant urothelial cells were employed as control in vitro. To achieve instant and high transfection efficiency, the delivery of apoptin was made by the adenovirus system (AdEasy™ System, USA). The results of RT-PCR have demonstrated that an obvious expression of apoptin was detected at 24 and 48 h after infection of Ad-apoptin, and adenovirus with no exogenous DNA fragment (Ad-VC) and cells without any treatment were taken as control (Fig. 1a). In order to validate the nucleus-targeting effect of apoptin in bladder-derived cells, we have detected the subcellular localization of apoptin by immune staining. As shown in Fig. 1b, nuclear accumulation of apoptin was observed in T24 and EJ that are bladder-derived malignant cells, whereas apoptin was localized in cytoplasm of HCV29 that was nontransformed urothelial cell line.

The detection of apoptin expression by RT-PCR and immunofluorescence dynamically post-transfection (p.t.). a Agarose gel (1 %) electrophoresis analysis of the result of RT-PCR of apoptin transcription. b Immunofluorescence assay of the selective nucleus accumulation in malignant bladder cell lines (T24 and EJ)
Selective cell cytotoxic effect induced by apoptin in bladder-derived cell lines
The antiproliferation effects on bladder carcinoma cells were performed by employing CCK-8 (Cell Counting Kit-8)-based method. Both of the malignant (T24 and EJ) and nonmalignant cell lines (HCV29) were infected with Ad-apoptin or control vector Ad-VC to identify the selective cytotoxic effects of apoptin, and corresponding cells without any treatment were taken as reference to evaluate cell viability. The results have indicated that no proliferation inhibitory effect induced by apoptin was observed in normal urothelial cells HCV29. However, the proliferation of human bladder tumor-derived cells (T24 and EJ) were remarkably inhibited by apoptin with a time-dependent manner compared to cells infected with Ad-VC and cells without any treatment (Fig. 2a). To further identify the cell apoptosis-inducing bias of apoptin on bladder-derived cells, the proportions of apoptotic cells in groups with various treatments were measured by annexin V-FITC/PI double staining. Compared to the control groups, the proportions of apoptotic cells were dramatically increased in apoptin-delivered T24 and EJ cells (75.88 ± 12.44 %, 87.83 ± 20.11 %) 72 h postinfection (Fig. 2b, c). However, there were no obvious apoptotic cells in nonmalignant urothelial cells after infection of Ad-apoptin or other control treatments. Moreover, we also have explored whether the cytotoxic effect on bladder tumor cells caused by apoptin was caspase-3-dependent. An active caspase-3 immunoassay was conducted to measure the active caspase-3 levels in the cell extracts infected with Ad-apoptin or control Ads. A higher level of active form of caspase-3 was detected in Ad-apoptin-infected T24 and EJ cells 72 h postinfection compared to the control group (Fig. 2d). In contrast with malignant cells, no significant difference was observed in active caspase-3 levels of HCV29 groups. Based on the assays above, it was evident that apoptin was able to selectively inhibit the proliferation of malignant urothelial cells and induce apoptosis by a caspase-3-dependent pathway.

The validation of the selective cytotoxicity of apoptin in T24, EJ, and HCV29 by different assays. a The measurement of selective cytotoxicity of apoptin to viability of the malignant bladder-derived cells (T24 and EJ) and normal bladder cell (HCV29) by CCK-8. b The detection of apoptosis of T24, EJ, and HCV29 after infection with Ad-apoptin by employing FITC-annexin V/PI-based method. c The results of flow cytometry analyzed and presented by the percentage of apoptotic cells (mean ± SD, n = 3; *p < 0.05 Student's t test). d The measurement of active caspase-3 after delivery of apoptin by ELISA-based method (mean ± SD, n = 3; *p < 0.05 Student's t test)
Selective cytotoxicity coupled with apoptin-coordinated expression profiles of multiple tumor-associated gene
In order to explore the mechanism of the selective cytotoxic effect of apoptin on bladder-derived cells, we have examined the expression profiles of the tumor-associated genes that have been reported by several studies. Ki67 was detected by flow cytometry, and C-erbB-2, Rb, and nm23 were analyzed by western blot. The data obtained from flow cytometry has shown that Ki67 that was required for maintaining cell proliferation has been downregulated in T24 and EJ after infection of Ad-apoptin. However, the expression levels of Ki67 in HVC29 have not been affected by delivery of apoptin (Fig. 3a). As shown in Fig. 3b, the protooncogene of C-erbB-2 was highly expressed in human bladder carcinoma cell lines T24 and EJ and expressed in a low level in normal urothelial cells HCV29. After infection of Ad-apoptin, the expression of C-erbB-2 has been significantly decreased in T24 and EJ; however, no obvious change in C-erbB-2 expression was detected in HCV29 by western blot (Fig. 3b). Retinoblastoma (RB), a tumor suppressor gene, was found to be significantly upregulated in EJ and HCV29 and moderately upregulated in T24 after infection of Ad-apoptin (Fig. 3b). Furthermore, nm23, which was recognized as a metastasis suppressor, was not influenced by apoptin in protein levels (Fig. 3b).

The analysis of apoptin-coordinated tumor-associated gene profiles in malignant and nonmalignant bladder cell lines. a Flow cytometry analysis of the differential protein expression of Ki67 in apoptin-expressing cells. b Comparison of differential expression patterns of c-erbB-2, Rb, and nm23 in T24, EJ, and HCV29 after infection of Ad-apoptin and Ad-VC
Evaluation of tumor toxic effect of apoptin in bladder cancer of BALB/C nude mouse models
Notice that apoptin could selectively induce apoptosis in malignant bladder cells, and we would further validate the results in vivo by employing nude mouse model of BC. Considering the low immunogenicity, we have employed the adeno-associated virus (AAV) system as the vehicle to deliver apoptin in vivo. Two strains of recombinant AAV were prepared in large scale including rAAV-apoptin and rAAV-VC. In the animal study, we have adopted 48 BALB/C nude mice, and 24 mice were implanted with T24 cells and 24 mice were injected with EJ cells. To establish the BC model, 3 × 106 T24 or EJ cells in 0.2 ml of physiological saline were injected subcutaneously into the backs of nude mice. Mice, either implanted with T24 or EJ cells, were randomly divided into three groups (eight mice per group) and treated with rAAV-apoptin, rAAV-VC, or physiological saline, respectively, by multipoint injection intratumorally. The dosage of rAAV was 1 × 1010 v.g. three times a week and injected for a total of 4 weeks. The tumor volume of each mouse was measured with vernier caliper every 3 days. The results of the tumor volume curves have demonstrated that inhibitory effects of tumor growth were obvious in both T24 and EJ apoptin-delivered groups (Fig. 4a and b). The survival of the animal models in all groups was determined by Kaplan–Meier analysis. The results indicated that no matter if the mice were implanted with T24 or EJ, the mice infused with rAAV-apoptin or physiological saline displayed significantly poorer prognosis than those that treated with apoptin (Fig. 4c and d).

The evaluation of tumor growth and survival rate in nude mice allograft model of human bladder cancer. a and b Dynamic measurement of tumor volumes treated with AAV-apoptin, AAV-VC, and physiological saline, respectively, in animal models of bladder cancer inoculated with T24 and EJ. c and d The analysis of survival rates of allografting animal models with different therapies (p < 0.05, Mantel–Cox logrank test)
Apoptin inhibits the proliferation of bladder carcinoma cells by altering the expression profiles of multiple tumor-associated gene in vivo
To investigate the expression patterns of the tumor-associated genes in vivo, we have conducted qRT-PCR and immunohistochemistry by using tissues from animal models. Firstly, the expression of apoptin in target sites was detected 10 and 20 days after injection of the virus by immunohistochemistry assay. Both in T24- and EJ-implanted groups, a considerable amount of apoptin-positive cells could be seen 10 days after virus injection, and the intensities and color reactive areas were dramatically increased in 20 days (Fig. 5a). However, no immunopositive cells were detected in sections isolated from animals in control groups. To determine the expression profiles of tumor-associated genes including Ki67, C-erbB-2, Rb, and nm23, we isolated the tumors from animals in all groups 20 days after treatments and then sectioned these to perform immunohistochemistry assays. It was evident that Ki67- and C-erbB-2-positive cells have been significantly decreased by infusions of apoptin (Fig. 5c). On the contrary, the expressions of Rb and nm23 were upregulated by injection of apoptin compared to the controls (Fig. 5c). Moreover, an investigator who was blinded to the experiment was invited to perform stereological estimation of the number of immunoreactive cells in all tissue sections. The ranges of the target areas were defined under a microscope, and five consecutive sections were used as the counting samples. A physical dissector counts method was performed to evaluate the number of the immunopositive cells under 40× magnification. As shown in Fig. 5b, the results from stereological quantification were consistent with that from immunohistochemistry. Additionally, we performed qRT-PCR to confirm the expression patterns of these genes on mRNA level by using tissues obtained 20 days after virus injection. Evidences from qRT-PCR demonstrated that the expression of apoptin could downregulate Ki67 and C-erbB-2 and upregulate Rb and nm23 in mRNA levels in T24- and EJ-implanted groups.


The evaluation of apoptin- and tumor-associated gene expression profiles in vivo by immunohistochemistry (IHC) and real-time PCR. a IHC assays of the expression of apoptin in tumors treated with AAV-apoptin, AAV-VC, and physiological saline at 10 days and 20 days postinjections. b: A and B Real-time PCR assays for comparisons of the different gene expression levels (apoptin, Ki67, C-erbB-2, Rb, and nm23) in tumors from animal models treated with different methods, which were normalized by GAPDH (*p < 0.05, Student's t test). C and D Stereological quantification of immunopositive cells of apoptin, Ki67, C-erbB-2, and nm23, respectively, in tumors from T24 and EJ allografting animal models (*p < 0.05, Student's t test). c Immunohistochemistry assays for detection of different tumor-associated gene expression profiles in T24 and EJ allografting models injected with AAV-apoptin, AAV-VC, and physiological saline
Ki67, C-erbB-2, Rb, and nm23 were differentially expressed between malignant and nonmalignant bladders
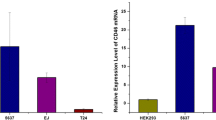
To determine the expression profiles of Ki67, C-erbB-2, Rb, and nm23 in clinical samples, we performed qRT-PCR to detect the expressions of corresponding genes on mRNA level by using clinical bladder carcinoma samples. Ten patients with urothelial BC staging T1 or T2 were recruited, and malignant and nonmalignant tissues were separated as soon as the bladders were resected by surgery. Before applying to qRT-PCR, the tissues were sampled for immunohistochemistry assays to verify malignancy. The results proved that Ki67 and C-erbB-2 were expressed in higher levels in malignant urothelial tissues than in normal tissues (Fig. 6). Conversely, the expression levels of Rb and nm23 were negatively correlated with the malignancies of the tissues involved (Fig. 6).

Relative tumor-associated gene (Ki67, C-erbB-2, Rb, and nm23) expression in clinic bladder tumor samples and tissue from normal bladders, which were normalized by GAPDH (*p < 0.05, Student's t test)
Discussion
In clinical settings, treatment of bladder tumor with chemotherapeutic agents or radiotherapy is a paradoxical practice. On the one hand, we tried to get rid of the tumors that could not be cleared by surgeries as more as possible to prevent tumor recurrence and progression through chemotherapy or radiotherapy. One the other hand, normal tissues also would be injured to lose function by these traditional methods due to their blindness. Alternatively, gene therapies of cancers by viral and nonviral vector-delivering tumor suppressive genes have shown great promise. However, something concerning therapeutic genes and transfer vehicles was still controversial. Taking endogenous genes as the therapeutic agents, for example, p53, may not work as expected [21]. Studies have indicated that tumor-suppressive functions can be harmful under conditions of systemic genotoxic stress, and certain tumors with p53 deficiency were significantly more sensitive to experimental chemo- and radiotherapy than tumors with wild type p53 [22]. Moreover, tumor-oriented vectors were still underdeveloped. The blindness of the vectors may cause severe side effects, which have impaired the progress of clinical trials. Therefore, tumor-specific approaches were needed to ensure safety.
CAV, which is an icosahedral single-stranded DNA virus, can cause depletion of thymocytes and erythroblastoid cell in chickens through apoptosis [23]. The polycistronic mRNA transcribed from CAV genome encodes three proteins including VP1, VP2, and VP3. It was found that the expression of VP3, named apoptin, can induce apoptosis in transformed cells effectively without any cytotoxicity to normal cells [12]. With this unique feature, the treatment of cancer can be more accurate by using blind vectors. Furthermore, it was reported that apoptin shares no significant homogenous sequence with known cellular proteins, which meant that overexpression of apoptin would not lead to hyperfunction of any cellular proteins except for apoptosis compared to employing cellular tumor-suppressive genes. Given the distinct characteristics, apoptin has shown great potential in treatments of cancer with high accuracy and safety. However, the mechanism of the selective cytotoxic effect to tumorigenic cells was still unknown. Notably, accumulating evidences have indicated that the delivery of apoptin could activate the caspase cascade and facilitate the release of cytochrome c through mitochondrial pathway [14]. The results from our study also have reported that an active form of caspase-3 has been significantly upregulated in apoptotic bladder carcinoma cells induced by apoptin, which was consistent with previous studies. Moreover, we have also observed that apoptin could specifically accumulate in nucleus of bladder tumor-derived cells. We speculated that the selective cytotoxic effect to transformed cell lines was correlated with the tumor-specific nucleus localization.
In order to investigate the mechanism of apoptosis in BC cells induced by apoptin, we have examined the expression profiles of multiple tumor-associated genes both in vitro and in vivo. Ki67 expressed in nucleus of cycling cells is a biomarker of cell proliferation. Numerous studies have demonstrated that Ki67 was an important indicator to progression and predictor for recurrence of bladder malignancy [18, 24]. In our studies, the expression of Ki67 was significantly downregulated after infusion of apoptin, which indicated that cell proliferation was stopped. Consistently, Teodoro et al. have found that apoptin could induce G2/M arrest and apoptosis in transformed cells in favor of an anaphase-promoting complex [25]. RB that was a key regulator of cell cycle progression was associated with bladder tumorigenesis [17, 26]. The results from our study have shown that the expression level of Rb was increased after expression of apoptin both in vitro and in vivo. To interpret the results, we hypothesized that the antiproliferation effect in the apoptin-delivered BC cells referred above were attributed to enhanced cell cycle arresting function of Rb. C-erbB-2, also known as HER2, was associated with approximately 30 % of breast cancers. In term of bladder carcinoma, the expression levels of C-erbB-2 were varied between different groups [19, 27, 28]. However, some clinical studies have reported that anti-her-2 therapies have shown large potential in BC patients who were her-2 positive [29]. Interestingly, results from our study have indicated that a higher mRNA level of C-erbB-2 was observed in malignant tissues than benign tissues. Furthermore, we have observed decreased expression levels of C-erbB-2 in vitro after infection of Ad-apoptin and diminished quantities of C-erbB-2 immunopositive cells in an animal study. The metastasis suppressor nm23 has been known to associate the volume and grade of bladder carcinomas [20, 30]. Despite of the negative results in vitro, we have noticed that the nm23-positive cells have been increased after infusion of apoptin through stereological cell counting assay. It is also evident that the mRNA expression level of nm23 in benign tissues was higher than tumorigenic tissues. We speculated that certain molecules or biological events have been involved in the upregulation of nm23 in vivo, besides apoptin.
References
Burger M, Catto J W F, Dalbagni G, Grossman H B, Herr H, Karakiewicz P, Kassouf W, Kiemeney L A, La Vecchia CShariat S. Epidemiology and risk factors of urothelial bladder cancer. European urology. 2012;
Philips BJ, Coyle CH, Morrisroe SN, Chancellor M, BYoshimura N. Induction of apoptosis in human bladder cancer cells by green tea catechins. Biomed Res. 2009;30:207–15.
Malkowicz SB. Intravesical therapy for superficial bladder cancer. Semin Urol Oncol. 2000;18:280–8.
Pan JG, Zhou X, Zeng G, WHan RF. Suppression of bladder cancer growth in mice by adeno-associated virus vector-mediated endostatin expression. Tumor Biology. 2011;32:301–10.
Brandau S, Suttmann H. Thirty years of BCG immunotherapy for non-muscle invasive bladder cancer: a success story with room for improvement. Biomed Pharmacother. 2007;61:299–305.
Matsumoto K, Kikuchi E, Horinaga M, Takeda T, Miyajima A, Nakagawa KOya M. Intravesical interleukin-15 gene therapy in an orthotopic bladder cancer model. Human Gene Therapy. 2011;22:1423–32.
Los M, Panigrahi S, Rashedi I, Mandal S, Stetefeld J, Essmann FSchulze-Osthoff K. Apoptin, a tumor-selective killer. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2009;1793:1335–42.
Rohn JNoteborn M. The viral death effector apoptin reveals tumor-specific processes. Apoptosis. 2004;9:315–22.
Zhou S, Zhang M, Zhang J, Shen H, Tangsakar EWang J. Mechanisms of apoptin-induced cell death. Medical Oncology. 2011;1–7
Pietersen ANoteborn M H M. Apoptin®. Cancer Gene Therapy. 2002;153–161
Poon I, Oro C, Dias M, Zhang J, PJans D. A tumor cell-specific nuclear targeting signal within chicken anemia virus VP3/apoptin. J Virol. 2005;79:1339–41.
Danen-Van Oorschot A, Fischer D, Grimbergen JM, Klein B, Zhuang SM, Falkenburg J, et al. Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells. Proc Natl Acad Sci. 1997;94:5843–7.
Zhuang SM, Shvarts A, van Ormondt H, Jochemsen AG, van der Eb A, JNoteborn MHM. Apoptin, a protein derived from chicken anemia virus, induces p53-independent apoptosis in human osteosarcoma cells. Cancer Res. 1995;55:486–9.
Danen-van Oorschot A, van Der Eb ANoteborn M. The chicken anemia virus-derived protein apoptin requires activation of caspases for induction of apoptosis in human tumor cells. J Virol. 2000;74:7072–8.
Danen-Van Oorschot AA, van der Eb AJ, Noteborn MH. BCL-2 stimulates apoptin-induced apoptosis. Adv Exp Med Biol. 1999;457:245–9.
Danen-Van Oorschot AA, Zhang Y, Erkeland SJ, Fischer DF, van der Eb AJ, Noteborn MH. The effect of Bcl-2 on Apoptin in ‘normal’ vs. transformed human cells. Leukemia. 1999;13(1):S75–7.
Takahashi R, Hashimoto T, Xu HJ, Hu SX, Matsui T, Miki T, et al. The retinoblastoma gene functions as a growth and tumor suppressor in human bladder carcinoma cells. Proc Natl Acad Sci. 1991;88:5257.
Enache M, SIMIONESCU CLASCU LC. Ki67 and Bcl-2 immunoexpression in primitive urothelial bladder carcinoma. Romanian journal of morphology and embryology. Revue Roumaine de Morphologie et Embryologie. 2012;53:521.
Fleischmann A, Rotzer D, Seiler R, Studer U, EThalmann GN. Her2 amplification is significantly more frequent in lymph node metastases from urothelial bladder cancer than in the primary tumours. Eur Urol. 2011;60:350–7.
Krause FS, Feil G, Bichler KH. Immunohistochemical examinations (Ki67, p53, nm23) and DNA cytophotometry in bladder cancer. Anticancer Res. 2000;20:5023–8.
Desilet N, Campbell TN, Choy FY. p53-based anti-cancer therapies: an empty promise? Curr Issues Mol Biol. 2010;12:143–6.
Burdelya LG, Komarova EA, Hill JE, Browder T, Tararova ND, Mavrakis L, et al. Inhibition of p53 response in tumor stroma improves efficacy of anticancer treatment by increasing antiangiogenic effects of chemotherapy and radiotherapy in mice. Cancer Res. 2006;66:9356–61.
Noteborn M. Apoptin®-induced apoptosis: a review. Apoptosis. 1999;4:317–9.
Lara PC, Rey A, Santana C, Afonso JL, Diaz JM, González GApolinario R. The role of Ki67 proliferation assessment in predicting local control in bladder cancer patients treated by radical radiation therapy. Radiother Oncol. 1998;49:163–7.
Teodoro JG, Heilman DW, Parker A, EGreen MR. The viral protein apoptin associates with the anaphase-promoting complex to induce G2/M arrest and apoptosis in the absence of p53. Genes Dev. 2004;18:1952–7.
Grossman HB, Liebert M, Antelo M, Dinney C, Hu SX, Palmer J, et al. p53 and RB expression predict progression in T1 bladder cancer. Clin Cancer Res. 1998;4:829–34.
Koga F, Yoshida S, Tatokoro M, Kawakami S, Fujii Y, Kumagai J, et al. ErbB2 and NFκB overexpression as predictors of chemoradiation resistance and putative targets to overcome resistance in muscle-invasive bladder cancer. PLoS One. 2011;6:e27616.
Wright C, Mellon K, Neal D, Johnston P, Corbett I, Horne C. Expression of c-erbB-2 protein product in bladder cancer. Br J Cancer. 1990;62:764.
Marín ÁP, Arranz EE, Sánchez AR, Auñón PZ, Barón MG. Role of anti-Her-2 therapy in bladder carcinoma. J Cancer Res Clin Oncol. 2010;136:1915–20.
Chow NH, Liu HS, Chan SH. The role of nm23-H1 in the progression of transitional cell bladder cancer. Clin Cancer Res. 2000;6:3595–9.
Acknowledgments
This work is supported by grants from the National Natural Science Foundation of China (No. 81260374). We give sincere thanks to Mr. Shi Huaisheng for his efforts in making all the photos in this paper.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Chunhui Wang and Wenju Wang. These authors contributed equally to this work.
Rights and permissions
About this article
Cite this article
Wang, C., Wang, W., Wang, J. et al. Apoptin induces apoptosis in nude mice allograft model of human bladder cancer by altering multiple bladder tumor-associated gene expression profiles. Tumor Biol. 34, 1667–1678 (2013). https://doi.org/10.1007/s13277-013-0700-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-013-0700-8