
Abstract
Background
Endothelial dysfunction induced by oxidized low-density lipoprotein (ox‐LDL) is implicated in the pathogenesis of atherosclerosis (AS). Activated autophagy was reported to improve endothelial functions and alleviate AS development. AMPK is a key protein in the regulation of autophagy, which can be modulated by Wnt signaling pathway.
Objective
Our study was designed to explore whether the canonical Wnt activator Wnt3a promotes autophagy in AS through in vitro and in vivo assays.
Results
Human umbilical vein endothelial cells (HUVECs) were first treated with ox‐LDL at 50 μg/mL for 24 h and then transfected with Wnt3a-overexpressed plasmid pcDNA3.1/Wnt3a. At 48 h after transfection, HUVECs were treated with 100 μM 3-MA. To investigate the effects of GSK-3β inhibition on the activation of AMPK, HUVECs were treated with 10 μM TWS119. Cell viability was detected via Trypan blue staining. Wnt3a expression, autophagy markers, GSK-3β inhibition and AMPK activation were examined by western blotting. To induce animal model of AS, male apolipoprotein E deficient (ApoE−/−) mice were fed with high-fat diet. pcDNA3.1/Wnt3a was injected into ApoE−/− mice after high-fat diet induction. Oil Red O staining was performed to examine lipid and plaque deposition in atherosclerotic lesions. In this study, ox‐LDL treatment decreased HUVEC viability and downregulated Wnt3a expression. Wnt3a overexpression promoted autophagy, induced GSK-3β phosphorylation at Ser9, and AMPK phosphorylation at Thr172 in ox-LDL-stimulated HUVECs. Overexpressing Wnt3a ameliorated lipid accumulation in aortic plaque of ApoE−/− mice. Furthermore, Wnt3a overexpression also activated autophagy, and induced GSK-3β and AMPK phosphorylation in ApoE−/− mice.
Conclusion
Wnt3a activates AMPK through inhibiting GSK-3β, thereby facilitating autophagy in AS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis (AS), a chronic multifactorial vascular disorder featured by accumulation of lipid and cholesterol in arterial walls, is the prevailing cause of cardiovascular morbidity and mortality (Bäck et al. 2019; Herrington et al. 2016). Vascular endothelial cells play a critical role in maintaining cardiovascular homeostasis (Endemann and Schiffrin 2004). As the precursor of AS, endothelial dysfunction can be triggered by multiple risk factors, including oxidized low-density lipoprotein (ox‐LDL), immune response, mechanical stress and virus infection (Steinberg 2009; Tabas et al. 2007). Ox-LDL increases the level of oxidative stress, enhances endothelial cell adhesion, and induces the release of inflammatory factors in vascular endothelial cells, thereby contributing to endothelial dysfunction and the occurrence of AS (Li et al. 2002; Trpkovic et al. 2015). Thus, the improvement of ox-LDL‐induced damage in endothelial cells is a therapeutic direction for AS.
Autophagy is a fundamental catabolic process in which damaged proteins or organelles are transported to lysosomes for degradation, plays an essential role in maintaining cellular energy homeostasis (Hamasaki et al. 2013; Xie and Klionsky 2007). Growing evidence supports the involvement of autophagy in a variety of physiological processes, including immunity, host defense, cellular metabolism and survival, development, and cancer (Bhattacharya and Eissa 2015; Wu et al. 2013; Xia et al. 2021). The beneficial effects of autupohagy on endothelial functions and the participation of autophagy in cardiovascular diseases have been demonstrated in recent studies (Jiang 2016; Ren and Zhang 2018). The impairment of endothelial cell autophagy can result in multiple adverse consequences, such as decreased vascular permeability and damaged vascular integrity (Zhang et al. 2018a). The activation of endothelial cell autophagy inhibits monocyte invasion and infiltration into the subendothelium byusion, which pushes the pause button on the atherogenic process (Hua et al. 2022). Many studies have proved that enhancing autophagy is an effective measure to delay AS to a certain extent (Meng et al. 2021; Schrijvers et al. 2011). For example, ANGPTL4 enhances endothelial cell proliferation and repressed endothelial cell injury by increasing autophagy, thereby protecting against the development of AS (Zhan et al. 2022). Resveratrol attenuates endothelial inflammation in the pathogenesis of AS by inducing autophagy (Chen et al. 2013) However, current research at home and abroad has not shown effective methods in enhancing the autophagy capacity of endothelial cells.
Wnt signaling mediates multiple biological processes by a canonical or noncanonical pathway, and is involved in a vast spectrum of disorders (Kahn 2014). In the classical pathway, classical Wnt ligands bind to the Frizzled receptor, resulting in GSK-3β inhibition, which allows β-catenin stabilization and nuclear translocation (Angers and Moon 2009). Nevertheless, new players have recently emerged in the classical Wnt pathway, indicating that the biological effects of β-catenin-independent pathways rely on the rapid regulation of GSK-3β in the cytoplasm but don’t require β-catenin accumulation (Acebron et al. 2014; Acebron and Niehrs 2016). Previously, Li et al. demonstrated that the suppression of the wnt/β-catenin/GSK3β pathway inhibited SIRT1 expression to promote autophagy in endothelial progenitor cells, thereby alleviating the coronary atherosclerosis in mice (Li et al. 2022). In addition, Wnt signaling and cellular metabolism can be linked through AMPK (Takatani et al. 2011). AMPK is a key sensor of cellular energetic conditions, which is extensively expressed in various tissues and organs (Hardie et al. 2016). AMPK’s cell signaling pathways participate in a variety of physiological processes, including apoptosis, autophagy, transcriptional control, cytoskeleton construction and metabolism (Kahn et al. 2005). AMPK modulates endothelial cell functions and lipid and carbohydrate metabolism, thereby participating in AS development (Dong et al. 2010; Ewart and Kennedy 2011). Reduced AMPK activity and autophagy dysfunction are associated with AS (Ou et al. 2018). When intracellular ATP production is reduced, AMPK is phosphorylated at Thr172, and the activated AMPK can inhibit mTORC-1 complex, thereby enhancing autophagy and promoting catabolism (Chandramouleeswaran et al. 2020). In 2014, Suzuki and colleagues identified AMPK to be negtaively regulated by GSK-3β, which promotes AMPK phosphorylation at Thr172 by PP2A and subsequent dephosphorylation, leading to the inactivation of AMPK (Suzuki et al. 2013). Previously, Wnt3a, a canonical Wnt ligand, was reported to enhance autophagy in hippocampal neurons via activating AMPK through GSK-3β inhibition (Ríos et al. 2018).
Based on the above knowledge, we inferred whether the GSK-3β-AMPK signaling can be regulated by Wnt3a in mediating autophagy in AS. We examined the autophagy role of Wnt3a-GSK-3β-AMPK axis in ox-LDL-stimulated human umbilical vein endothelial cells (HUVECs) and in apolipoprotein E deficient (ApoE−/−) mice.
Materials and methods
Cell culture, treatment and transfection
HUVECs obtained from Lonza (Walkersville, MD, USA) were maintained in EGM-2 endothelial growth medium (CC-3162; Lonza, Walkersville, MD, USA) containing 2% fetal bovine serum (FBS; Catalog No: 10099141C, Gibco) at 37 °C with 5% CO2. To induce cell damage, HUVECs were first exposed to ox‐LDL (0–100 μg/mL; Catalog No: AY-1501, Shanghai AngYu Biotechnology Co., Ltd., Shanghai, China) for 24 h to examine the cytotoxic effects to HUVECs. Then, empty pcDNA3.1 or pcDNA3.1/Wnt3a were transfected into HUVECs using Lipofectamine 2000 (Catalog No: 11668019, Invitrogen, Carlsbad, CA, USA) for 48 h following 50 μg/mL ox‐LDL stimulation. The pcDNA3.1/Wnt3a-transfected HUVECs were then treated with 100 μM 3-MA (the autophagy inhibitor; Catalog No: M9281, Sigma-Aldrich, St. Louis, MO, USA). The control or ox‐LDL-stimulated HUVECs were treated with 10 μM TWS119 (the inhibitor of GSK-3β; Catalog No: S1590, Selleck, Houston, TX, USA).
Trypan blue staining for cell viability
Cells were seeded in six‐well plates and incubated until about 90% confluence. Cells after different treatment and/or transfection were digested with 0.25% trypsin solution (Catalog No: T4049, Sigma-Aldrich). Then, cells were suspended in PBS (pH 7.4; Catalog No: 806552, Sigma-Aldrich), and 0.4% trypan blue dye (1:1 volume; Catalog No: ST798, Beyotime, Shanghai, China) was added to cell suspension. The unstained living cells were counted by the Countstar automated cell counter (Inno-Alliance Biotech, USA).
Green fluorescent protein (GFP)‑LC3 immunofluorescence
HUVECs following indicated treatment or transfection were seeded (8 × 104 cells/mL) into a Petri dish (35 mm diameter) covered with a glass slide and incubated for 24 h. Cells were then transfected with GFP-LC3 plasmid using Lipofectamine 2000. Forty-eight h after transfection, the slides were rinsed three times with PBS, fixed with 4% paraformaldehyde, washed three times with PBS, and mounted with anti-fluorescence quenching mounting medium. Fluorescence images were captured using laser confocal microscopy (Zeiss LSM700, Carl Zeiss, Canada)).
Animals
Animal experiments were reviewed and approved by the Ethics Committee of Wuhan Myhalic Biotechnology Co., Ltd (Approval number: 202109136; Hubei, China). Forty male ApoE−/− mice and thirty wild-type C57BL/6 J controls (6-week-old, 28–32 g) were purchased from Peking University Animal Center (Beijing, China). All mice were maintained in a specifc pathogen-free environment with the temperature of 24 °C and the humidity of 55% under a 12 h light/dark cycle.
Induction of AS in mice
The in vivo experiments were comprised of 3 parts. The frist part was designed to investigate the effects of Wnt3a overexpression on the lipid accumulation in aortic plaque of AS mice, and included 4 groups: sham + vehicle group (n = 10), sham + Wnt3a group (n = 10), Model + vehicle group (n = 10), and Model + Wnt3a group (n = 10). The second part was designed to assess the influence on the autophagy in AS mice, and included 5 groups: sham + vehicle group (n = 10), sham + Wnt3a group (n = 10), Model + vehicle group (n = 10), Model + Wnt3a group (n = 10), and Model + Wnt3a + 3-MA group (n = 10). The third part was designed to evaluate the effects of Wnt3a overexpression on the GSK-3β/AMPK axis, and included 6 groups: sham + vehicle group (n = 10), sham + Wnt3a group (n = 10), sham + TWS119 group (n = 10), Model + vehicle group (n = 10), Model + Wnt3a group (n = 10), and Model + TWS119 group (n = 10). After one week of feeding acclimation, ApoE−/− mice were fed on high-fat diet (0.15% cholesterol, 21% fat, and 78.85% of basic mice maintain feed) for 12 weeks to establish a visible AS model. C57BL/6J mice fed a high-fat diet served as sham group. After 12 weeks, ApoE−/− mice or C57BL/6J mice were injected via tail vein with Wnt3a-overexpressed plasmid pcDNA3.1/Wnt3a or the empty pcDNA3.1 vector, or intraperitoneally injected with 30 mg/kg TWS119 (the inhibitor of GSK-3β; Catalog No: S1590, Selleck, Houston, TX, USA) or 15 mg/kg 3-MA (the autophagy inhibitor; Catalog No: M9281, Sigma-Aldrich, St. Louis, MO, USA) After 24 h, the blood samples were collected via cardiac puncture and centrifuged to obtain the serum. Then, mice were euthanized via cervical dislocation, and the hearts with aortic arch were resected, fixed in 10% neutral buffered formalin, embedded in paraffin, and cut into 5 µm-thick sections for Oil Red O staining.
Oil red O staining
The slices (5 μm) were dried with excess temperature for 20 min and incubated with 100% isopropanol (Catalog No: 563935, Sigma-Aldrich) for 5 min. Then the slices were incubated with 0.5% oil red O staining solution (Catalog No: B1094, Applygen Technologies Inc., Beijing, China) in a 60 °C oven, washed in 85% isopropanol, dyed with hematoxylin (Catalog No: B21220, Shanghai yuanye Bio-Technology Co., Ltd., Shanghai, China), cleaned and sealed. The nucleus was stained light blue and the lipid was stained orange or red. The area of atherosclerotic plaque in each aortic section was evaluated using Image-Pro Plus software (NIH, USA).
Serum analysis of lipids
Serum levels of triglyceride (TG; Catalog No: ml094956), total cholesterol (TC; Catalog No: ml094952), low-density lipoprotein cholesterol (LDL-C; Catalog No: ml076620) and high-density lipoprotein cholesterol (HDL-C; Catalog No: ml092722) were detected using commercially available enzyme kits (Shanghai Enzyme-linked Biotechnology Co., Ltd., Shanghai, China) with a Hitachi 7600 Automatic Biochemistry Analyzer (Tokyo, Japan).
Western blotting
The total protein was extracted from HUVECs and mouse aortic tissues using RIPA lysis buffer (#P0013B, Beyotime) containing protease and phosphatase inhibitors. Equal amount of protein was separated by 10% SDS-PAGE gels and electro-transferred onto a PVDF membrane (Millipore). The membrane was sealed in 5% non-fat dried milk, and added with primary antibodies against Wnt3a (ab219412, 1: 1000; Abcam, Cambridge, MA, USA), LC3B (ab192890, 1: 2000; Abcam), BECN1 (ab207612, 1: 2000; Abcam), P62 (ab240635, 1: 1000; Abcam), AMPK (#2532, 1:1000; Cell Signaling, Danvers, Massachusetts, USA), pThr172-AMPK (#2535, 1: 1000; Cell Signaling), GSK-3β (#9315, 1: 1000; Cell Signaling), pSer9-GSK-3β (#9336, 1: 1000; Cell Signaling), and β-actin (ab8227, 1: 1000; Abcam) at 4 °C overnight. Next day, the membrane was incubated with corresponding secondary antibodies (1: 1000, Abcam) for 2 h. Protein bands were visualized using an enhanced chemiluminescence kit (#P0018S, Beyotime) and then photographed with a gel imaging system. ImageJ software was used to analyze the gray values of protein bands. Relative quantification of protein was carried out with β-actin as an internal reference.
Statistical analysis
All data from at least three independent experiments were analyzed by SPSS 16.0 software (SPSS, Chicago, IL), and are indicated as mean ± standard deviation. Disparities between two or among multiple groups were formulated by Student’s t test or one-way analysis of variance. Statistical significance was established by p < 0.05.
Results
Ox‐LDL treatment decreases HUVEC viability and downregulates Wnt3a expression
First, HUVECs were stimulated by ox-LDL (0–100 μg/mL) for 24 h and cell viability was assessed by trypan blue staining. The results showed that ≤ 10 μg/mL ox-LDL stimulation did not influence HUVEC viability (Fig. 1A). When the ox-LDL concentration exceeded 25 μg/mL, cell viability started to decrease, and when the cells were stimulated by ox-LDL at 50 μg/mL, cell viability decreased by nearly 50%, so cells stimulated by 50 μg/mL ox-LDL were used in the subsequent experiments. Then, pcDNA3.1/Wnt3a was transfected into the ox‐LDL‐stimulated HUVECs. Western blotting analysis illustrated that ox‐LDL considerably reduced Wnt3a expression in HUVECs, which was restored after pcDNA3.1/Wnt3a transfection (Fig. 1B–C). The ox‐LDL-induced reduction in HUVEC viability was recovered by overexpressing Wnt3a (Fig. 1D).

Influence of ox‐LDL stimulation on HUVEC viability and Wnt3a expression. A The viability of HUVECs stimulated by ox-LDL (0–100 μg/mL) for 24 h was detected by trypan blue staining. B Western blotting analysis of Wnt3a expression in HUVECs following ox‐LDL treatment or pcDNA3.1/Wnt3a transfection. C HUVEC viability influenced by ox‐LDL treatment or pcDNA3.1/Wnt3a transfection was measured by trypan blue staining. *P < 0.05, **P < 0.01, ***P < 0.001
Wnt3a overexpression enhances autophagy in ox‐LDL-exposed HUVECs
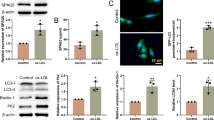
Next, we evaluated the autophagy in ox‐LDL-stimulated HUVECs affected by Wnt3a overexpression. Immunofluorescence staining indicated that Wnt3a overexpression efficiently enhanced the percentage of GFP-LC3 cells, which was reversed by the treatment of 3-MA (Fig. 2A). Furthermore, the expression of autophagy-related proteins (LC3, Beclin1, and P62) in HUVECs after different treatment or transfection was detected by western blotting. The results showed that overexpressing Wnt3a in ox‐LDL-stimulated HUVECs resulted in an increase in the LC3-II/LC3-I ratio and BECN1 protein level and a decrease in P62 protein level, which were also recovered after 3-MA treatment (Fig. 2B–E). Neither ox‐LDL treatment alone nor pcDNA3.1/Wnt3a transfection alone influenced the percentage of GFP-LC3 cells, the LC3-II/LC3-I ratio, and BECN1 and p62 protein levels. Overall, overexpressing Wnt3a can promote autophagy in ox‐LDL-exposed HUVECs.

Autophagy activation in ox‐LDL-stimulated HUVECs caused by Wnt3a overexpression. HUVECs were first stimulated by 50 μg/mL ox-LDL for 24 h, and then transfected with pcDNA3.1/Wnt3a for 48 h and treated with 100 μM 3-MA for 24 h. A HUVECs were stained with LC3-antibody (green) and Hoechst (blue) to visualize LC3 puncta and nuclei by confocal laser scanning microscopy. B–E HUVECs were harvested for western blotting to examine autophagy markers LC3, BECN1 and P62. Relative quantification of protein was carried out with β-actin as an internal reference. *P < 0.05, **P < 0.01
Wnt3a overexpression inhibits GSK-3β activity to promote AMPK activation in ox‐LDL-stimulated HUVECs
We investigated whether Wnt3a overexpression modulates AMPK activity in HUVECs after ox‐LDL stimulation. AMPK and GSK-3β activities were evaluated by assessing Thr172 and Ser9 phosphorylation, respectively. We found that Wnt3a overexpression or TWS119 treatment further enhanced the ox‐LDL-induced AMPK phosphorylation at Thr172 and GSK-3β phosphorylation at Ser9 (Fig. 3A–C). Since AMPK can be activated through the phosphorylation at Thr172, and GSK-3β can be inactivated through the phosphorylation at Ser9, we concluded that overexpressing Wnt3a further stimulated AMPK activation and GSK-3β inhibition in ox-LDL-stimulated HUVECs. These data imply that Wnt3a causes inhibition of GSK-3β, thereby promoting AMPK activation.

GSK-3β inhibition and AMPK activation in ox‐LDL-stimulated HUVECs caused by Wnt3a overexpression. HUVECs were first stimulated by 50 μg/mL ox-LDL for 24 h, and then transfected with pcDNA3.1/Wnt3a for 48 h or treated with 10 μM TWS119 for 24 h. A–C In HUVECs, AMPK phosphorylation at Thr172 and GSK-3β phosphorylation at Ser9 were evaluated using western blotting analysis. *P < 0.05, **P < 0.01
Overexpressing Wnt3a attenuates lipid accumulation in aortic plaque of ApoE−/− mice
The effects of pcDNA3.1-Wnt3a injection on the lipid content in aortic tissues of mice were assessed utilizing Oil red O staining. Blue nucleus and red fat can be observed in the images. There were hardly any visible lipid deposition and atherosclerotic plaque in arterial vessels of C57BL/6J mice injected with empty pcDNA3.1 or pcDNA3.1-Wnt3a. In contrast, accumulated lipids and obvious areas of atherosclerotic plaque can be observed in experimental ApoE-deficient mice injected with empty pcDNA3.1 compared with C57BL/6J mice pre-injected with empty pcDNA3.1. However, injection with pcDNA3.1-Wnt3 substantially attenuated lipid deposition and the atherosclerotic plaque area in mouse aorta (Fig. 4A–B). In addition, at the end of the experiment, serum lipid levels were detected. In the serum of ApoE−/− mice, TG, TC and LDL-C levels were evidently elevated, while HDL-C level was considerably reduced, compared with the control C57BL/6J mice. Nevertheless, injection of pcDNA3.1-Wnt3a into ApoE−/− mice obviously lowered TG, TC and LDL-C levels in serum, but exerted no evident impact on HDL-C level (Fig. 4C–F). Taken together, Wnt3a attenuates lipid deposition in aortic plaque of ApoE-deficient mice.

Improvement of lipid deposition in aortic tissues of ApoE−/− mice by Wnt3a overexpression. After high-fat diet induction for 12 weeks, ApoE−/− mice and C57BL/6J mice were injected with pcDNA3.1/Wnt3a or empty pcDNA3.1. A Lipid deposition and atherosclerotic plaque formation were evaluated in each group of mice adopting Oil Red O staining. B Plaque areas in each group were quantified using ImageJ software. C–F TG, TC, HDL-C and LDL-C levels in serum of mice in each group were assessed by commercial kits. n = 10 in each group. *P < 0.05, **P < 0.01, ***P < 0.001
Wnt3a promotes autophagy in ApoE−/− mice by activating the GSK-3β-AMPK signaling
Finally, whether Wnt3a mediates autophagy and the GSK-3β-AMPK signaling in ApoE-deficient mice was assessed. The expression of key autophagy-related proteins and the activities of AMPK and GSK-3βwere analyzed using western blotting. The findings revealed that injection of pcDNA3.1-Wnt3a upregulated the LC3-II/LC3-I ratio and BECN1 expression, while downregulated P62 expression in the aorta of ApoE−/− mice, which was antagonized by 3-MA injection (Fig. 5A–D). Furthermore, injection of pcDNA3.1-Wnt3a or TWS119 further enhanced AMPK phosphorylation at Thr172 and GSK-3β phosphorylation at Ser9 in ApoE-deficient mice (Fig. 5E–G), indicating that Wnt3a further stimulated AMPK activation and GSK-3β inhibition in ApoE−/− mice. In conclusion, Wnt3a inhibits GSK-3β activity to induce the activation of AMPK, thereby promoting autophagy in ApoE-deficient mice.

On activation of autophagy and GSK-3β-AMPK signaling in ApoE−/− mice by Wnt3a overexpression. After high-fat diet induction for 12 weeks, ApoE−/− mice and C57BL/6J mice were injected with pcDNA3.1/Wnt3a and 15 mg/kg 3-MA to evaluate autophagy, or injected with pcDNA3.1/Wnt3a or 30 mg/kg TWS119 to assess the GSK-3β-AMPK axis. A–D The aortic tissues of ApoE−/− mice were collected for western blotting to assess autophagy markers LC3, BECN1 and p62. Relative quantification of protein was carried out with β-actin as an internal reference. E–G In each group, AMPK phosphorylation at Thr172 and GSK-3β phosphorylation at Ser9 were investigated using western blotting. *P < 0.05, **P < 0.01
Discussion
AS is recognized as a worldwide public health problem, in which ox-LDL-induced endothelial damage is a key factor contributing to its initiation and development (Rajagopalan et al. 2004). Ox-LDL can directly damage the endothelial cells on the surface of blood vessels, which result in the increase of the gap between endothelial cells and the enhancement of permeability, facilitating the passage of lipid components through the endothelial layer into the sub-endothelial layer and leading to lipid deposition (Guo et al. 2021). The present study demonstrated Wnt3a, the canonical Wnt activator, attenuated ox‐LDL‐induced endothelial injury by reinforcing autophagy via inducing GSK-3β inhibition and the subsequent AMPK activation, enriching our understanding of Wnt signaling in AS progression.
In the past decades, both canonical and non-canonical Wnt pathways have been identified to be implicated in the development of AS (Badimon and Borrell-Pages 2017). In particular, the canonical Wnt/β-catenin pathway widely participates in regulating endothelial dysfunction and abnormal lipid metabolism in AS. SIRT1 alleviates AS in ApoE−/− mice by inhibiting autophagy in endothelial progenitor cells through activating Wnt/β-catenin/GSK3β signaling (Li et al. 2022). LncRNA H19 sponges miR-148b to upregulate WNT1 level, thereby facilitating proliferation and repressing apoptosis in ox-LDL-exposed human aorta vascular smooth muscle cells via triggering Wnt/β-catenin pathway (Zhang et al. 2018b). Knockdown of Dickkopf-2 stimulates Wnt/β-catenin pathway, thereby protecting against lipid loading and macrophage polarization and in ApoE knockout mice (Zhang et al. 2021). However, in recent years, the β-catenin-independent Wnt signaling that depends on the modulation of GSK-3β in the cytoplasm has attracted people's attention (Cisternas et al. 2016). Wnt3a is a canonical Wnt activator, and Wnt3a ligand treatment was demonstrated to prevent neurotoxic damage through GSK-3β-mediated inhibition, which emphasized the prominent function of GSK-3β as a potential master regulator of β-catenin-independent Wnt pathway (Alvarez et al. 2004).
Autophagy, an evolutionarily conserved lysosome-mediated biodegradation process, is the recycling and reuse of cells’ own waste and is essential in maintaining cellular homeostasis (Klionsky and Emr 2000). Substantial evidence suggests that autophagy defection is associated with the pathogenesis of various metabolic disorders, including AS, while increased autophagy mitigates the development of AS (Osonoi et al. 2018). Aloe-emodin derivative promotes autophagy in human aortic endothelial cells via enhancing the level of AMBRA1, a key protein associated with autophagosome formation, thereby exerting potent anti-atherosclerosis effects (Tang et al. 2022). Metformin attenuates atherosclerosis and enhances plaque stability in ApoE−/− mice by activating Krueppel-like factor 2-mediated autophagy (Wu et al. 2021). Morin hydrate activates autophagy in HUVECs by inhibiting the NF-κB pathway and ameliorates vascular inflammation in AS mouse models (Meng et al. 2021). AMPK is a key protein in the regulation of autophagy, and mainly acts as an “energy regulator” in eukaryotic cells (Yan et al. 2022). Previously, inhibition of GSK-3β induced by Wnt3a ligand was shown to activate AMPK, thereby facilitating autophagy in hippocampal neurons (Ríos et al. 2018). Therefore, our study was designed to confirm the hypothesis that Wnt3a activates autophagy in AS via the GSK-3β-AMPK axis. We discovered that Wnt3a promoted autophagy, inhibited GSK-3β activity and activated AMPK activity in both ox-LDL-stimulated HUVECs and in ApoE-deficient mice. Since the use of TWS119 further confirmed that the activity of AMPK can be activated after the treatment of TWS119, the inhibitor of GSK-3β, we concluded that Wnt3a inhibited GSK-3β to induce AMPK activation, thereby promoting autophagy in ox-LDL-stimulated HUVECs and in ApoE-deficient mice.
Our paper demonstrated for the first time that Wnt3a enhanced autophagy in AS via modulating GSK-3β-AMPK axis, shedding light on the mechanism underlying the protection of Wnt signaling activation on endothelial dysfunction induced by ox‐LDL and abnormal lipid metabolism. Therefore, the activation of Wnt-GSK-3β-AMPK-autophagy pathway be a promising therapy for AS.
References
Acebron S, Niehrs C (2016) β-catenin-independent roles of Wnt/LRP6 signaling. Trends Cell Biol 26(12):956–967
Acebron S et al (2014) Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol Cell 54(4):663–674
Alvarez AR et al (2004) Wnt-3a overcomes beta-amyloid toxicity in rat hippocampal neurons. Exp Cell Res 297(1):186–196
Angers S, Moon R (2009) Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol 10(7):468–477
Bäck M et al (2019) Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 16(7):389–406
Badimon L, Borrell-Pages M (2017) Wnt signaling in the vessel wall. Curr Opin Hematol 24(3):230–239
Bhattacharya A, Eissa N (2015) Autophagy as a stress response pathway in the immune system. Int Rev Immunol 34(5):382–402
Chandramouleeswaran P et al (2020) Autophagy mitigates ethanol-induced mitochondrial dysfunction and oxidative stress in esophageal keratinocytes. PLoS ONE 15(9):e0239625
Chen M et al (2013) Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 9(12):2033–2045
Cisternas P et al (2016) Activation of Wnt signaling in cortical neurons enhances glucose utilization through glycolysis. J Biol Chem 291(50):25950–25964
Dong Y et al (2010) Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 121(6):792–803
Endemann D, Schiffrin E (2004) Endothelial dysfunction. J Am Soc Nephrol 15(8):1983–1992
Ewart M, Kennedy S (2011) AMPK and vasculoprotection. Pharmacol Ther 131(2):242–253
Guo J et al (2021) LncRNA PVT1 knockdown alleviated ox-LDL-induced vascular endothelial cell injury and atherosclerosis by miR-153-3p/GRB2 axis via ERK/p38 pathway. Nutr Metab Cardiovasc Dis 31(12):3508–3521
Hamasaki M, Shibutani S, Yoshimori T (2013) Up-to-date membrane biogenesis in the autophagosome formation. Curr Opin Cell Biol 25(4):455–460
Hardie D, Schaffer B, Brunet A (2016) AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol 26(3):190–201
Herrington W et al (2016) Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res 118(4):535–546
Hua Y et al (2022) The induction of endothelial autophagy and its role in the development of atherosclerosis. Front Cardiovasc Med 9:831847
Jiang F (2016) Autophagy in vascular endothelial cells. Clin Exp Pharmacol Physiol 43(11):1021–1028
Kahn M (2014) Can we safely target the WNT pathway? Nat Rev Drug Discov 13(7):513–532
Kahn B et al (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1(1):15–25
Klionsky D, Emr S (2000) Autophagy as a regulated pathway of cellular degradation. Science (new York, NY) 290(5497):1717–1721
Li D et al (2002) Statins modulate oxidized low-density lipoprotein-mediated adhesion molecule expression in human coronary artery endothelial cells: role of LOX-1. J Pharmacol Exp Ther 302(2):601–605
Li Y et al (2022) Autophagy-sirtuin1(SIRT1) alleviated the coronary atherosclerosis (AS)in mice through regulating the proliferation and migration of endothelial progenitor cells (EPCs) via wnt/β-catenin/GSK3β signaling pathway. J Nutr Health Aging 26(3):297–306
Meng Q et al (2021) Morin hydrate inhibits atherosclerosis and LPS-induced endothelial cells inflammatory responses by modulating the NFκB signaling-mediated autophagy. Int Immunopharmacol 100:108096
Osonoi Y et al (2018) Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy 14(11):1991–2006
Ou H et al (2018) Role of AMPK in atherosclerosis via autophagy regulation. Sci China Life Sci 61(10):1212–1221
Rajagopalan S et al (2004) Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood 103(10):3677–3683
Ren J, Zhang Y (2018) Targeting autophagy in aging and aging-related cardiovascular diseases. Trends Pharmacol Sci 39(12):1064–1076
Ríos J, Godoy J, Inestrosa N (2018) Wnt3a ligand facilitates autophagy in hippocampal neurons by modulating a novel GSK-3β-AMPK axis. Cell Commun Signal 16(1):15
Schrijvers D, De Meyer G, Martinet W (2011) Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol 31(12):2787–2791
Steinberg D (2009) The LDL modification hypothesis of atherogenesis: an update. J Lipid Res 50:S376–S381
Suzuki T et al (2013) Inhibition of AMPK catabolic action by GSK3. Mol Cell 50(3):407–419
Tabas I, Williams K, Borén J (2007) Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 116(16):1832–1844
Takatani T et al (2011) AMP-activated protein kinase attenuates Wnt/β-catenin signaling in human osteoblastic Saos-2 cells. Mol Cell Endocrinol 339:114–119
Tang X et al (2022) Aloe-emodin derivative produces anti-atherosclerosis effect by reinforcing AMBRA1-mediated endothelial autophagy. Eur J Pharmacol 916:174641
Trpkovic A et al (2015) Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit Rev Clin Lab Sci 52(2):70–85
Wu X, Won H, Rubinsztein D (2013) Autophagy and mammalian development. Biochem Soc Trans 41(6):1489–1494
Wu H et al (2021) Metformin attenuates atherosclerosis and plaque vulnerability by upregulating KLF2-mediated autophagy in apoE mice. Biochem Biophys Res Commun 557:334–341
Xia H, Green D, Zou W (2021) Autophagy in tumour immunity and therapy. Nat Rev Cancer 21(5):281–297
Xie Z, Klionsky D (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9(10):1102–1109
Yan L et al (2022) Schisandrin B mitigates hepatic steatosis and promotes fatty acid oxidation by inducing autophagy through AMPK/mTOR signaling pathway. Metabolism 131:155200
Zhan W et al (2022) ANGPTL4 attenuates palmitic acid-induced endothelial cell injury by increasing autophagy. Cell Signal 98:110410
Zhang D et al (2018a) Autophagy maintains the integrity of endothelial barrier in LPS-induced lung injury. J Cell Physiol 233(1):688–698
Zhang L et al (2018b) H19 knockdown suppresses proliferation and induces apoptosis by regulating miR-148b/WNT/β-catenin in ox-LDL -stimulated vascular smooth muscle cells. J Biomed Sci 25(1):11
Zhang Y et al (2021) Dickkopf-2 knockdown protects against classic macrophage polarization and lipid loading by activation of Wnt/β-catenin signaling. J Cardiol 78(4):328–333
Acknowledgements
Not applicable.
Funding
The work was supported by Wuhan Municipal Health Commission (approval number: WX21B08).
Author information
Authors and Affiliations
Contributions
SQ and BN were the main designers of this study. KZ, NJ, and HL performed the experiments. BN analyzed the data. SQ drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Shifang Qu declares that he/she has no conflict of interest. Kuanxin Zhang declares that he/she has no conflict of interest. Nan Jin declares that he/she has no conflict of interest. Han Li declares that he/she has no conflict of interest. Bin Nie declares that he/she has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. Animal experiments were granted by the Ethics Committee of Wuhan Myhalic Biotechnology Co., Ltd (Approval number: 202109136; Hubei, China).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Qu, S., Zhang, K., Jin, N. et al. The activation of Wnt signaling facilitates autophagy by modulating GSK-3β-AMPK axis in atherosclerosis. Mol. Cell. Toxicol. 19, 721–729 (2023). https://doi.org/10.1007/s13273-022-00298-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13273-022-00298-y