
Abstract
We analyzed the impact of nine previously identified missense PKD1 variants from our studies, including c.6928G > A p.G2310R, c.8809G > A p.E2937K, c.2899 T > C p.W967R, c.6284A > G p.D2095G, c.6644G > A p.R2215Q, c.7810G > A p.D2604N, c.11249G > C p.R3750P, c.1001C > T p.T334M, and c.3101A > G p.N1034S on RNA structures and PC1 protein structure dynamics utilizing computational tools. RNA structure analysis was done using short RNA snippets of 41 nucleotides with the variant position at the 21st nucleotide, ensuring 20 bases on both sides. The secondary structures of these RNA snippets were predicted using RNAstructure. Structural changes of the mutants compared to the wild type were analyzed using the MutaRNA webserver. Molecular dynamics (MD) simulation of PC1 wild-type and mutant protein regions were performed using GROMACS 2018 (GROMOS96 54a7 force field). Findings revealed that five variants including c.8809G > A (p.E2937K), c.11249G > C (p.R3750P), c.3101A > G (p.N1034S), c.6928G > A (p.G2310R), c.6644G > A (p.R2215Q) exhibited major alterations in RNA structures and thereby their interactions with other proteins or RNAs affecting protein structure dynamics. While certain variants have minimal impact on RNA conformations, their observed alterations in MD simulations indicate impact on protein structure dynamics highlighting the importance of evaluating the functional consequences of genetic variants by considering both RNA and protein levels. The study also emphasizes that each missense variant exerts a unique impact on RNA stability, and protein structure dynamics, potentially contributing to the heterogeneous clinical manifestations and progression observed in Autosomal Dominant Polycystic Kidney Disease (ADPKD) patients offering a novel perspective in this direction. Thus, the utility of studying the structure dynamics through computational tools can help in prioritizing the variants for their functional implications, understanding the molecular mechanisms underlying variability in ADPKD presentation and developing targeted therapeutic interventions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polycystic kidney disease (PKD) stands as a formidable challenge in the realm of renal disorders, with its hallmark manifestation being the formation of fluid-filled cysts in the kidneys, often leading to progressive renal dysfunction and, eventually, end-stage renal disease (ESRD) in severe cases. Autosomal dominant PKD (ADPKD) is the most common form of PKD, affecting approximately 1 in 1000 individuals worldwide. The key clinical features of the disease include renal cysts, which lead to progressive kidney enlargement and failure, liver cysts causing hepatomegaly, and intracranial aneurysms in certain cases, increasing stroke risk. The disease also manifests with extra-renal symptoms such as hypertension, urinary tract infections, kidney stones, and cardiovascular abnormalities. Other organs like the pancreas, spleen, and reproductive system can be affected (Nishio et al. 2021). With approximately 7–10% genetically unresolved cases, ADPKD is primarily caused by mutations in two genes, PKD1 (~ 70% cases) and PKD2 (~ 15% cases). PKD1 and PKD2 encode for the proteins polycystin-1 (PC1) and polycystin-2 (PC2), respectively. ADPKD is genetically heterogeneous, and the majority of cases are attributed to mutations in the PKD1 (Hopp et al. 2020; Yeung et al. 2024). The full function of this protein is not entirely clear, but PC1 is known to be a large transmembrane protein (4303 aa) with a complex structure and crucial role in regulating cellular processes such as differentiation, cell proliferation, and apoptosis (Peintner and Borner 2017). It comprises an N-terminal extracellular domain, several transmembrane domains, and a cytoplasmic C-terminal tail. The leucine-rich repeat (LRR) domains are implicated in signal transduction pathways and mediate PC1's involvement in cell–cell and cell–matrix interactions. The C-type lectin domain suggests roles in protein–protein interactions and cell adhesion. The sixteen PKD repeats are considered vital for cell–cell interactions and normal renal development. The REJ domain regulates PC1’s ion transport, while the PLAT domain facilitates protein–protein and protein–lipid interactions in signaling pathways. The 11 transmembrane domains likely serve as channels for ion transport, and the C-terminal tail regulates downstream signaling pathways through interactions with G protein subunits, highlighting the important functional roles of PC1 in cellular physiology. PC1 present in primary cilia in the renal tubules is believed to perceive fluid movement within these tubules, contributing to the maintenance of their size and structure. The PC1 and PC2 complex within renal tubules facilitates the typical development and operation of the kidneys (Weston et al. 2003; Wang et al. 2019). Mutations in PKD1 are responsible for around 78% of ADPKD cases, making it a primary focus for understanding disease pathogenesis (Hopp et al. 2020). Due to the limited understanding of the disease, effective cure remains a challenge. Understanding the structural and functional consequences of specific PKD1 variants is essential for elucidating genotype–phenotype correlations and guiding personalized treatment approaches.
The experimental determination of its full thermodynamic structural changes at RNA and protein level, and the complete protein’s function remain challenging due to its large complex structure and transmembrane localization. Molecular dynamics (MD) simulation techniques provide valuable insights into studying the dynamic behavior, structural stability of proteins at the atomic level, and interactions within the cellular milieu like how they undergo conformational changes (Hollingsworth and Dror 2018; Vander Meersche et al. 2024). By computationally modeling the interactions between atoms and molecules over time, MD simulations can help understand the impact of mutations on protein structure, conformational dynamics, and interactions with ligands or other biomolecules, making them plausible targets for drug development after understanding the disease mechanisms (Hollingsworth and Dror 2018; Salo-Ahen et al. 2020).
PKD is a complex disease and its complexity can be seen at every scientific level, be it genetic, proteomic, or how the disease develops and affects patients. While much research is being focused on understanding the genetics and protein-level consequences of the mutations, understanding their impact on RNA structure and dynamics remains comparatively less explored. There is an urgent need to deepen our understanding of each level of the molecular processes underlying PKD pathogenesis.
RNA serves as the intermediary between DNA and protein and plays a crucial role in the regulation of gene expression and hence cellular processes. RNA molecules not only fold into secondary structures but also in three dimensions. The way RNA works depends a lot on its shape, which is influenced by its sequence. The RNA often has to fold many times to get the structure which is a complex process. Usually, RNA starts by folding into simpler shapes that are most energetically favorable, then it forms a paired double helix by folding on itself (Draper et al. 2005; Holbrook 2008; Butcher and Pyle 2011; Ganser et al. 2019). Mutations in genes, subsequently within RNA sequences, can disrupt its folding and hence structure, affecting its function and potentially contributing to disease pathogenesis (Halvorsen et al. 2010; Sauna and Kimchi-Sarfaty 2011; Salari et al. 2013; Hunt et al. 2014; Diederichs et al. 2016). Understanding how genetic variants affect RNA could also provide insights into the molecular mechanisms driving PKD progression and potential therapeutic targets. In this study, we explored RNA behavior affected by the missense PKD1 variants previously identified in ADPKD patients, uncovering another layer of complexity, and highlighting RNA's importance in understanding the disease more thoroughly. We also utilized MD simulation to analyze their structure dynamics and functional effects at protein level. Our results contribute to enhancing our understanding of PKD1-related disease mechanisms and could potentially guide further experimental investigations and the development of new therapeutic approaches for managing ADPKD.
Methodology
PKD1 variants
The wild-type and missense variants of PKD1 c.6928G > A p.G2310R, c.8809G > A p.E2937K, c.2899 T > C p.W967R, c.6284A > G p.D2095G, c.6644G > A p.R2215Q, c.7810G > A p.D2604N, c.11249G > C p.R3750P, c.1001C > T p.T334M, and c.3101A > G p.N1034S, identified in our previous studies using Sanger sequencing and whole exome sequencing were studied for RNA structure using available online tools and protein dynamics using MD simulation. These identified missense variants were individually found in different patients diagnosed with ADPKD (Raj et al. 2020; DEVI et al. 2024).
RNA sequence selection and extraction
The RNA sequences of interest were derived from the PKD1 NM_001009944 transcript ID. Short RNA snippets comprising 41 nucleotides were extracted for each wild type and mutant, with the mutation locus precisely positioned at the 21st nucleotide, ensuring 20 bases on both sides of the nucleotide change.
Prediction of RNA secondary structure
The secondary structures of each wild-type and mutant RNA snippets were predicted utilizing the RNAstructure web server (version 6.0.1) (Bellaousov et al. 2013). Using thermodynamic principles, this server employs algorithms to predict RNA secondary structures and provides the folding patterns and base pairing interactions within the RNA molecule.
Effect of mutation on RNA
Mutational analysis of RNA snippets was done using MutaRNA tool (version (5.0.10)) to analyze the structural changes induced by each missense mutation (Miladi et al. 2020). This analysis involved the intra-molecular base pairing potential, base pairing probabilities of the mutant RNA, and assessment of accessibility (single-strandedness) in comparison to the wild-type counterpart (Bernhart et al. 2011). By combining the remuRNA (Salari et al. 2013) and RNAsnp, this tool helps understand mutation-induced alterations in RNA structures. The measure of relative entropy, also known as Kullback–Leibler divergence, in this webserver is used to quantify the difference between two probability distributions. In RNA structural studies, it compares the probability distribution of nucleotide conformations or base-pairing states between wild-type and mutant sequences. This measure is crucial for assessing RNA structural impact because it provides a quantitative way to detect how mutations alter RNA structure.
Secondary and tertiary structure analysis of protein structures
Secondary structure prediction was performed using PSIPRED (McGuffin et al. 2000) and tertiary structure superposition visualization was conducted using Chimera (Pettersen et al. 2004) (Supplementary Figs. 2, 3).
Mutant protein structure creation and preprocessing
Mutant structures of PKD1 protein variants were generated utilizing the Swiss Model (Schwede et al. 2003). Given the substantial size of the PKD1 protein, conventional structure modelling and simulation posed significant challenges. Therefore, in lieu of direct structure determination, we employed motif and domain analysis of PKD1 through the motif scan web server (Sigrist et al. 2010). Then we identified location of the variants and subsequently created mutations within the domain structure of PKD1.
Individually, 9 variant structures were generated based on their specific locations within domains and motifs simultaneously, resulting in the production of 9 distinct wild-type PKD1 structures. To facilitate modeling, different template structures were selected based on the mutation's location within different domains. The selection of template IDs (T.I.) was as follows: p.E2937K (T.I. G9KGT4.1.A_European domestic ferret), p.G2310R (T.I. H3BTE0.1A_Human), p.W967R (T.I. A0A212CYZ4.1.A_European Red deer), p.D2095G (T.I. Q59EY6.1_Human), p.R2215Q (T.I. H3BTE0.1A_Human), p.D2604N (T.I. H3BTE0.1A_Human), p.R3750P (T.I. 6a70.1b_Human), p.T334M (T.I. A0A212CZX9.1.A_ European Red deer), and p.N1034S (T.I. A0A212CYZ4.1A_ European Red deer).
Preprocessing involved the elimination of all non-standard residues, including water molecules, followed by the addition of hydrogen atoms using the Discovery Studio program (Systèmes 2016). Monomeric structures of PC1 were then isolated for further analysis, with additional non-standard residues removed. Energy minimization of mutant structures was subsequently conducted utilizing Modrefiner (Xu and Zhang 2011).
Molecular simulation dynamics
MD simulations of wild-type and mutant PC1 regions were conducted using GROMACS 2018 version (GROMOS96 54a7 force field) (Van Der Spoel et al. 2005; Kumar et al. 2023; Ranjan and Das 2023). The systems were solvated with spc water models in a triclinic box. The systems were neutralized by adding Na + and Cl- ions, and the salt concentration was maintained at 0.15 M. The protein was kept at least 1.0 nm from the box edges. Energy minimization was performed using the steepest descent algorithm, followed by equilibration at 300 K temperature and 1 atm pressure using the NVT (constant number of particles, volume, and temperature) and NPT (constant number of particles, pressure, and temperature) ensembles. MD simulations were extended to 50 ns time frame, and analysis of root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), H bonding, and solvent-accessible surface area (SASA) was conducted using GROMACS tools (2018 version). Visualization of MD trajectory data was performed using the XMGRACE application and MS Excel (Cowan and Grosdidier 2000).
Results
RNA secondary structure
RNA secondary structures predicted using the RNAstructure web server to study the secondary structural changes are depicted in Fig. 1. The analysis of secondary structure changes due to PKD1 missense variants revealed prominent alterations in case of variants c.6928G > A (p.G2310R), c.8809G > A (p.E2937K), c.6644G > A (p.R2215Q), c.11249G > C (p.R3750P), and c.3101A > G (p.N1034S). In these variants, significant deviations from the wild-type secondary structure were observed, indicating impact on RNA folding and stability. Conversely, variants c.2899 T > C (p.W967R), c.6284A > G (p.D2095G), c.7810G > A (p.D2604N), and c.1001C > T (p.T334M), exhibited less pronounced changes in secondary structure, suggesting milder effects on RNA conformation.

Secondary Structures of Original (WT) vs Altered (MT) RNA Snippet for PKD1 Missense Variants. The secondary structures of RNA snippets for each PKD1 missense variants (from panels A–I) predicted using the RNAstructure web server. The original (wild-type) RNA structure is depicted on the left, while the altered (mutant) RNA structure is shown on the right. The nucleotide change is at 21st position in each structure. Different colors of bases indicate different base pairing probabilities
Assessment of structural impact using remuRNA
The PKD1 missense variants were quantitatively assessed for their structural impact on RNA by analyzing the relative entropy H(wt:mu) (Table 1). The greater the entropy value provided by remuRNA, the greater is the structural impact of the variant (Salari et al. 2013; Miladi et al. 2020). This means that mutations with higher relative entropy values exhibit greater deviations in RNA structure compared to mutations with lower values. Variant c.6644G > A (p.R2215Q) exhibited the highest relative entropy value of 4.878, indicating substantial deviation in RNA structure induced by this mutation, followed by variants c.8809G > A (p.E2937K) and c.11249G > C (p.R3750P) with H(wt:mu) values of 4.644 and 4.642, respectively. Conversely, variants c.7810G > A (p.D2604N), c.6284A > G (p.D2095G), and c.2899 T > C (p.W967R) showed relatively lower relative entropy values of 0.446, 0.148, and negligible 0.006, suggesting minimal structural impact on RNA. The remaining variants c.6928G > A (p.G2310R), c.3101A > G (p.N1034S), and c.1001C > T (p.T334M) exhibited moderate relative entropy values of 1.481, 2.912, and 0.192, respectively.
The effect of the variants on RNA structure is illustrated using multiple approaches, including Circos plots (Fig. 2), base pairing probabilities dot plots (Fig. 3), differential base pairing probabilities dot plot (Fig. 4) and RNA accessibility profile analysis (Fig. 5). The Circos plots illustrating changes in base pairing probabilities at different positions in RNA structure due to nucleotide change, depicting both increased and weakened probabilities, are illustrated in Supplementary Fig. 1. These MutaRNA findings are related to the effect of the variant within the RNA snippet affecting the secondary structure and accessibility to its surrounding context.

Circos Plots for Base Pair Probabilities of Original (WT) vs Altered (MT) RNA Snippets. The base pair probabilities are represented using circular plots (circos). The sequence starts from the 5' end at the slight-left bottom and progresses clockwise until reaching the 3' end. Within the circular plot, the analyzed mutation at position 21 is marked by a red color positioned at the top of the mutant circos. In each panel from A to I, the wild-type (WT) RNA structure is depicted on the left, while the mutant (MT) RNA structure is shown on the right. Darker hues of gray indicate higher probabilities of base pairing. Variants c.8809G > A (p.E2937K), c.11249G > C (p.R3750P), and c.3101A > G (p.N1034S) showed prominent structural impacts. Variants c.6928G > A (p.G2310R), c.6284A > G (p.D2095G), and c.7810G > A (p.D2604N) had moderate deviations, while c.2899 T > C (p.W967R) and c.1001C > T (p.T334M) had the least impact on RNA structure

Base Pair Probabilities for Wild-type (WT) and Mutant (MT) PKD1 Variants. The dot plot depicts the base pairing potential of both the WT and MT RNA variants. Darker dots indicate a higher probability of forming a base pair between the respective sequence positions. Base pair probabilities are derived from the Boltzmann distributed energies of all structures that can be formed by RNA, considering folding constraints such as maximal base pair span. The wild-type sequence is represented in the top-right portion of the matrix, while the mutant sequence is shown in the bottom-left. Each tick interval on the axes represents 10 nucleotides, with the evaluated variant at position 21 highlighted by red dotted lines on each axis. Panels A to I, represent each PKD1 missense variants. Variants c.8809G > A (p.E2937K), c.11249G > C (p.R3750P), and c.3101A > G (p.N1034S) showed prominent structural impacts. Variants c.6928G > A (p.G2310R), c.6284A > G (p.D2095G), and c.7810G > A (p.D2604N) had moderate deviations, while c.2899 T > C (p.W967R) and c.1001C > T (p.T334M) had the least impact on RNA structure

Differences in Base Pairing Probabilities between Mutant and Wild-type RNA for PKD1 Missense Variants. The dot plot illustrates the difference in base pairing probabilities (Pr(bp)) between the mutant and wild-type RNA, calculated as Pr(bp in WT)—Pr(bp in mut) [Δ = Pr(bp in WT)—Pr(bp in mut)]. Weakened base pairs resulting from the mutation are represented in blue, while stronger ones are depicted in red. Each tick interval on the axes corresponds to 10 nucleotides. The mutated position is denoted by red dotted lines. Panels A–I represent individual PKD1 missense variants

Comparison of Accessibility Profiles of Wild-Type (wt) and Mutant (mut) RNA Sequences for PKD1 Missense Variants. The accessibility profiles of both wild-type (green) and mutant (orange) sequences and their differences help evaluate how the mutation affects the RNA’s single-strandedness, which is strongly related to its interactions with other proteins or RNAs. Accessibility is evaluated in terms of local single-position unpaired probabilities, comparing the accessibility (i.e., probability of being unpaired) for each nucleotide position of the RNA sequences. The blue line represents the change in accessibility (wt-mut), with negative values indicating positions more likely to be unpaired in the mutant compared to the wild-type. This can be seen as the 'negative drops' in the blue differential accessibility profile. The red line indicates the mutated position for each PKD1 missense variant (from panels A–I)
Circos plots (Fig. 2) were generated to visualize the interplay between different positions of the RNA sequence and their interactions. Variants were represented as arcs connecting the affected nucleotides, providing a view of the positional relationships within the RNA molecule and highlighting potential disruptions caused by sequence change using different hues of grey, with darker grey color showing potentially stronger base pairing probabilities. Prominent deviations from wild-type patterns were observed in variants c.8809G > A (p.E2937K), c.11249G > C (p.R3750P), and c.3101A > G (p.N1034S) indicating significant structural impacts. Similarly, variants c.6928G > A (p.G2310R), c.6284A > G (p.D2095G), and c.7810G > A (p.D2604N), exhibited less pronounced deviations, with c.2899 T > C (p.W967R) and c.1001C > T (p.T334M) being the least, suggesting milder effects on RNA conformation. These base pair probabilities for wild-type (WT) and mutant (MT) sequences can also be visualized in heat maps dot matrices (Fig. 3, 4). Consistent alterations in base pairing patterns were observed in variants indicating significant structural changes. This is also evident in the entropy chart and the secondary structure predictions using 2D structure and Circos plots base pairing probabilities (Table 1, Figs. 1 and 2).
The differences in base pairing probabilities between mutant and wild-type RNA (Δ = Pr(bp in WT)—Pr(bp in mut)) for PKD1 missense variants are depicted as differential heat map dot matrices (Fig. 4) to compare base pairing probabilities between mutant and wild-type RNA sequences with red indicating increased interaction likelihood and blue indicating weakened base pairs.
RNA accessibility
Comparison of accessibility profiles of wild-type (wt) and mutant (mut) RNA sequences for PKD1 missense variants was compared to assess changes in single-strandedness and structure dynamics (Fig. 5), which are strongly related to its interactions with other proteins or RNAs. The accessibility profile indicates the likelihood of nucleotides being unpaired for each position within the RNA sequences. The blue line illustrates the alteration in accessibility (WT-mut), where negative values signify positions more likely to be unpaired in the mutant compared to the wild-type. The “negative drops" in the blue differential accessibility profile signify the reduced accessibility in the folded wild type compared to its mutant. The RNA accessibility profiles varied across different variants, with the prominent accessibility differences observed in variants c.8809G > A (p.E2937K), c.11249G > C (p.R3750P), and c.3101A > G (p.N1034S). Subsequently, c.6644G > A (p.R2215Q), c.6928G > A (p.G2310R), and c.7810G > A (p.D2604N) also exhibited effect on accessibility profile. Conversely, the accessibility was negligible in variants c.2899 T > C (p.W967R), c.6284A > G (p.D2095G), and c.1001C > T (p.T334M). These findings suggest that certain variants, particularly c.8809G > A, c.11249G > C, and c.3101A > G (p.N1034S) may significantly impact RNA accessibility, potentially influencing interactions with other molecules or proteins. In contrast, variants such as c.6284A > G and c.1001C > T may have minimal effects on RNA accessibility, highlighting the variability in the structural consequences of the variants. The structural alterations in the predicted secondary structures are consistent with mutaRNA results.
Secondary and tertiary structure analysis of protein structures
The secondary structure prediction analysis revealed significant changes in the secondary structures of all nine protein variants when compared to the wild-type protein. These changes are primarily observed in the strand and helical structures (Supplementary Fig. 2). Each variant exhibits distinct alterations in these regions, indicating potential impacts on the protein's overall stability and function. Tertiary structure superposition visualization was performed using the Chimera tool. The alignment of mutant and wild-type protein structures showed no visible changes in the overall tertiary structure, indicating similar global folds (Supplementary Fig. 3).
MD simulation of protein structures
The RMSD, RMSF, SASA, Rg, and H-bonding plots for each variant with wild type are depicted in Figs. 6, 7, 8, 10, 11 respectively. The average values of each parameter are mentioned in Table 2. For the c.8809G > A variant (p.E2937K), the mutant structure exhibited higher average RMSD (0.502 nm compared to 0.42 nm) and Rg (2.02 nm compared to 1.61 nm) values compared to the wild-type structure, indicating structural deviations and increased protein compactness. However, RMSF values remained relatively similar between the wild-type and mutant structures. The SASA and the number of hydrogen bonds were slightly higher in the mutant structure, suggesting changes in the protein's surface area and hydrogen bonding interactions.

RMSD Analysis of the Variants: The RMSD plot depicts the average deviation of the backbone atoms of nine PKD1 variants vs wild-type from their initial structures over the 50 ns time frame MD simulation. Each plot A–I represents the RMSD profile for a specific PKD1 variant (Red), with the wild-type (Blue). RMSD values were calculated relative to the initial conformation and plotted against simulation time (ns). The RMSD analysis predicts about the stability and structural changes of PKD1 variants compared to the wild-type protein over time

RMSF Analysis of PKD1 Variants: The RMSF plot illustrates the fluctuation of amino acid residues within the backbone of wild-type and mutant variants during 50 ns time frame MD simulation. Each plat A–I represents the RMSF profile for a specific variant (Red), with the wild-type (Blue). The RMSF analysis provides insights into the individual residue dynamic flexibility and local structural variations of PKD1 variants compared to the wild-type protein highlighting how mutations affect the dynamic behavior and stability of specific regions within the protein

SASA Analysis of PKD1 Variants. The SASA plot displays the solvent accessible surface area of wild-type and mutant PKD1 variants over the 50 ns time frame. Each plot A–I represents the SASA profile for a specific PKD1 variant, with the wild-type depicted in a distinct color (Blue: wild, Red: mutant). The SASA analysis reveals how much of the protein's surface is exposed to solvent molecules, highlighting changes in protein interactions and how accessible the protein is for binding with other molecules. A larger SASA value indicates increased exposure

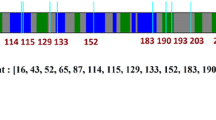
Structural Representation of PKD1 Variants. The figure depicts the protein structure of polycystin-1 with marked missense variants analyzed in this study. Each variant is marked within the protein structure, providing visual representation of their respective locations within the PC1 protein

Radius of Gyration (Rg) Analysis of PKD1 Variants. The Rg plot illustrates the radius of gyration of wild-type and nine mutant PKD1 variants over 50 ns time frame. Each plot A–I represents the Rg profile for a specific PKD1 variant (Red), with corresponding wild-type (Blue). Analysis of Rg profiles provides insights into the overall compactness of PKD1 variant protein compared to the wild-type, with changes in Rg indicating differences in the structural tightness or conformations of the protein
For the c.6928G > A variant (p.G2310R), both wild-type and mutant structures exhibited differences in RMSD, with the mutant structure showing slightly lower RMSD (0.28 nm compared to 0.30 nm). RMSF values were comparable between the wild-type and mutant structures. However, the Rg value increased marginally in the mutant structure (1.41 nm compared to 1.3 nm in the wild type), indicating a slight expansion of the protein structure. Similarly, SASA and the number of hydrogen bonds also increased in the mutant structure, suggesting alterations in the protein's surface characteristics and hydrogen bonding pattern.
For the c.2899 T > C variant (p.W967R), considerable differences were observed between the wild-type and mutant structures. The mutant structure showed higher RMSD (1.51 nm compared to 1.12 nm) and RMSF (0.657 nm compared to 0.487 nm) values, suggesting a more pronounced deviation and greater structural flexibility as compared to the wild-type conformation. This means that the mutant structure is less stable and more prone to changes compared to the normal structure. Rg values were comparable between the wild-type and mutant structures, while SASA and hydrogen bonding patterns showed minor variations.
For the c.6284A > G variant (p.D2095G), both wild-type and mutant structures exhibited similar RMSD and RMSF values. However, the mutant structure displayed a slightly higher Rg value (3.44 nm compared to 3.11 nm) and SASA, indicating a potential expansion of the protein structure and altered surface characteristics. The number of hydrogen bonds remained relatively stable between the wild-type and mutant structures.
For the c.6644G > A variant (p.R2215Q), the mutant structure showed slightly lower RMSD (0.42 nm compared to 0.52 nm) and RMSF values compared to the wild-type structure. Rg values were similar between the two structures, indicating comparable compactness. However, SASA was slightly reduced in the mutant structure, while the number of hydrogen bonds remained consistent.
For the c.7810G > A variant (p.D2604N), both wild-type and mutant structures exhibited similar RMSD and RMSF values. However, the mutant structure displayed a slightly higher Rg value (1.96 nm compared to 2.00 nm) and SASA, suggesting potential structural alterations and changes in surface accessibility. The number of hydrogen bonds was slightly lower in the mutant structure.
For the c.11249G > C variant (p.R3750P), the mutant structure exhibited slightly lower RMSD (1.18 nm compared to 1.32 nm) and RMSF values compared to the wild-type structure. Rg values were similar between the two structures, indicating comparable compactness. However, SASA was slightly reduced in the mutant structure, while the number of hydrogen bonds remained consistent.
For the c.1001C > T variant (p.T334M), the mutant structure displayed slightly higher RMSD (1.35 nm compared to 1.198 nm) and RMSF values compared to the wild-type structure. Rg values were similar between the two structures, indicating comparable compactness. However, SASA was slightly increased in the mutant structure, while the number of hydrogen bonds remained consistent.
For the c.3101A > G variant (p.N1034S), both wild-type and mutant structures exhibited similar RMSD and RMSF values. Rg values were also comparable between the two structures, indicating similar compactness. SASA and the number of hydrogen bonds remained relatively stable between the wild-type and mutant structures.
Discussion
This study involves computational tools to investigate the effects on RNA structure and employed molecular dynamics simulation to explore the protein structure dynamics and functional implications of nine missense variants from the previously identified variants of PC1, the protein product encoded by the PKD1, recognized for its central role in ADPKD. ADPKD is a genotypically and phenotypically heterogeneous disease, meaning there are a wide variety of genetic mutations that can cause it, and the variability in the disease phenotype is observed among the patients, making it a complex disease. The reasons for this phenotypic diversity are not well understood. The PKD1 alone has multiple allelic variants associated with ADPKD. There are many variants identified in PKDI and PKD2. According to the PKDB database, in PKD1, total 2232 variants and in PKD2 total 302 variants have been reported. The missense variants are the most common in the PKD1 after truncating variants in ADPKD (https://pkdb.mayo.edu). Our study focused on these nine specific variants based on their rarity/novelty, and pathogenicity, as determined in our previous studies involving ADPKD patients and families (Raj et al. 2020; DEVI et al. 2024). These missense variants were individually identified in different ADPKD patients. ADPKD stands as the most prevalent hereditary renal disorder characterized by the progressive formation of fluid-filled cysts within the kidneys, resulting in renal failure (Bergmann et al. 2018). PC1, a complex trans-membrane protein, governs crucial cellular processes within renal tubules, considered vital for maintaining renal integrity (Weimbs 2007; Paul and Vanden Heuvel 2014). So far, the exact function of this protein largely remains elusive. The protein's complexity arises from its large structure with multiple domains, each anticipated to contribute distinct functions. These include the leucine-rich repeat (LRR) domains, implicated in signal transduction and cell–matrix interactions; the C-type lectin domain facilitating protein–protein interactions; and the low-density lipoprotein A (LDL-A) region, known for its cysteine-rich nature; however, the presence of LDL-A in PC1 is still a debate. The 16 PKD repeats are considered to play essential roles in mediating cell–cell interactions and normal kidney development. The REJ domain possibly modulates ion transport. Also, the PLAT domain facilitates protein–protein and protein–lipid interactions, and the 11 transmembrane domains likely act as ion transport channels. The cytoplasmic C-terminal tail regulates downstream signaling pathways by interacting with G protein subunits. Studying the dynamics of RNA structures in vitro is challenging, and comprehending the role of PC1 in cellular physiology, particularly in the context of ADPKD, remains a formidable challenge, given its intricate nature as a large trans-membrane protein with diverse domains (Weston et al. 2003; Wang et al. 2019). The analysis of various RNA structural parameters and the MD simulation of protein regions enable us to decipher the impact of mutations on RNA structure and interactions, PC1's stability, dynamics, and interactions, offering helpful insights into the molecular foundations of ADPKD pathogenesis and paving the way for targeted therapeutic interventions.
The MD simulations conducted for various PKD1 variants provided significant insights into the structural consequences of these mutations. As the protein size is large (4303 aa), motif and domain analysis through the motif scan web server aided in identifying variant locations and creating mutations within PKD1’s domain structure for simulation (DEVI et al. 2024). The MD simulations revealed significant differences in their dynamic behavior, including altered flexibility and transient local structural changes affecting function, such as binding and activity. Tertiary structure superposition provides a static snapshot of protein structures, focusing on the overall fold. This method may miss subtle or transient changes. The mutation may shift the dynamic equilibrium of conformations, leading to functional impacts not evident in static views. The MD simulation capture time-dependent behaviors, revealing dynamic differences like flexibility and local structural changes over time in cellular-like environment. Thus, while static analysis indicates structural similarity, MD simulation underscore important dynamic differences, highlighting the necessity of both static and dynamic analyses to fully understand the mutation’s effects. Therefore, similar tertiary structures do not imply functional similarity, as dynamic behavior is crucial. Analysis of MD simulation parameters, such as RMSD, RMSF, Rg, SASA, and hydrogen bonding patterns, revealed deviations between wild type and mutant structures. RMSD measures the average distance between atoms of aligned proteins. RMSD values were calculated relative to the initial conformation and plotted against simulation time (ns). The RMSD analysis predicts the stability and structural changes. The RMSD curve (Fig. 6), calculated using GROMACS, illustrates the changes in backbone stability for each mutant protein (red) as compared to its wild-type counterpart (blue). Higher RMSD values indicate greater deviations from the original protein structure, suggesting significant conformational changes due to mutations.
The RMSF analysis provides insights into the individual residue dynamic flexibility and local structural variations of PKD1 variants compared to the wild-type protein. RMSF measures how much each residue moves from its average position over time. The RMSF curve from MD simulations helps understand the flexibility of different parts of the protein. These RMSF plots typically represent residues that have undergone significant changes during the MD simulation. Higher RMSF values indicate increased flexibility and movement in specific regions.
The radius of gyration (Rg) describes the compactness of the protein structure over time. Smaller Rg values indicate a more compact structure, while larger values suggest an expanded protein. Changes in the Rg curve reflect alterations in the protein's overall shape. It can provide insights into the molecule's structural properties and conformational changes over time.
Hydrogen bonds play a key role in maintaining protein stability. Mutations can change the number and strength of hydrogen bonds, impacting the protein's stability. Changes in hydrogen bonding patterns can lead to significant conformational changes in the protein, affecting its function and interactions.
The SASA analysis helps understand how much of the protein surface is exposed to solvent molecules, reflecting changes in protein interactions. A higher SASA value means a larger surface area is accessible to solvents, which can be more favorable for binding with enzymes, interacting proteins, or molecules.
For the c.6928G > A (p.G2310R), although the RMSD values were slightly lower in the mutant structure compared to the wild type, indicating a degree of stabilization, other parameters such as Rg, SASA, and hydrogen bonding showed modest increases (Table 2; Figs. 6, 8, 10). This suggests that the mutation may induce subtle structural changes in the protein, potentially affecting its stability and surface properties. In contrast, the c.8809G > A (p.E2937K) exhibited notable deviations in RMSD and Rg values (Table 2; Figs. 6, 10), indicating significant structural alterations in the mutant protein. The increased SASA and hydrogen bonding (Figs. 8, 11) observed in the mutant structure further emphasize the substantial impact of this mutation on the protein's conformation and interactions. The c.2899 T > C (p.W967R) showed the most noticeable differences, with substantially higher RMSD and RMSF values (Table 2; Figs. 6 and 7) in the mutant structure, indicating increased flexibility and deviation from the native conformation. This mutation also resulted in a noteworthy increase in Rg and SASA, suggesting a more extended and exposed protein structure. For the c.6284A > G variant (p.D2095G), although the RMSD and RMSF values were comparable between wild-type and mutant structures, minor differences in Rg and SASA were observed, indicating alterations in the protein’s compactness and surface accessibility induced by the mutation. Similarly, the c.6644G > A variant (p.R2215Q) displayed slight differences in RMSD and RMSF values, with relatively stable Rg and SASA. These findings suggest that this mutation may also induce subtle changes in the protein structure without significantly affecting its overall compactness or surface properties. The c.7810G > A variant (p.D2604N) and c.11249G > C variant (p.R3750P) showed similar trends, with minor differences in RMSD and RMSF values but comparable Rg and SASA between wild-type and mutant structures, suggesting that these mutations may have limited effects on the overall structure and surface properties of the protein. In contrast, the c.1001C > T variant (p.T334M) exhibited slightly higher RMSD and RMSF values in the mutant structure, indicating increased flexibility. However, other parameters, such as Rg, SASA, and hydrogen bonding, remained relatively stable, suggesting that this mutation may induce localized structural changes without significantly altering the overall protein conformation. For the c.3101A > G variant (p.N1034S), minimal differences were observed in RMSD, RMSF, Rg, SASA, and hydrogen bonding between wild-type and mutant structures, indicating that this mutation may have limited impact on the protein's structure and dynamics. Thus, MD simulations provided valuable clues about the structural consequences of PKD1 mutations, highlighting their diverse effects on protein stability, flexibility, and surface properties.

Hydrogen bonding analysis of PKD1 Variants. The H-bonding plot illustrates the number of hydrogen bonds formed within the protein and between protein and solvent molecules for wild-type and mutant PKD1 over 50 ns time frame. Each plot A–I represents the H-bonding profile for a specific PKD1 variant (Red), with the corresponding wild-type (Blue). Analysis of hydrogen bonding dynamics reveals how mutations in PKD1 affect the stability and interaction patterns of the protein compared to the wild type, with changes in hydrogen bonds indicating potential alterations in protein stability, conformational changes and function
The studied missense variants in the PKD1 are located in crucial domains involved in various aspects of PC1 function. The p.G2310R, p.R2215Q, p.D2604N, and p.E2937K variants occur within or near the REJ domain (Fig. 9), implicated in ion transport regulation, potentially altering its ability to modulate ion flux across cell membranes. The p.T334M, p.W967R, p.N1034S, and p.D2095G variant in the 1st, 3rd, 5th, and 16th PKD domains may impair PC1's ability to interact with other proteins or structural elements. Similarly, the p.R3750P variant in the PKD cation channel domain may alter ion channel activity, affecting cellular ion homeostasis.
Considering the RNA level, changes in secondary structure suggest prominent or milder effects on RNA conformation and highlight the heterogeneous nature of structural alterations induced by missense variants in PKD1. This emphasizes the importance of considering the specific molecular consequences of each mutation in disease pathogenesis. The study comprehensively analyzed the structural impact of these variants on RNA secondary structure using a combination of computational tools and predictive models. RNA secondary structures were initially predicted using the RNAstructure web server, providing insights into the potential alterations induced by these variants. The analysis revealed prominent deviations from the wild-type RNA secondary structure in some variants, which are c.6928G > A (p.G2310R), c.8809G > A (p.E2937K), c.6284A > G (p.D2095G), c.6644G > A (p.R2215Q), c.11249G > C (p.R3750P) and also c.3101A > G (p.N1034S). These variants exhibited significant impacts on RNA folding and stability, suggesting potential functional consequences at the molecular level. Further assessment of structural impact was analyzed using the relative entropy between wild-type and mutant RNAs to quantify the extent of structural changes induced by each mutation (Table 1). In descending order of relative entropy H(wt:mu) values, the most to the least impactful variants are as follows: c.6644G > A (p.R2215Q) with an H(wt:mu) of 4.878, c.8809G > A (p.E2937K) with 4.644, c.11249G > C (p.R3750P) with 4.642, c.3101A > G (p.N1034S) with 2.912, c.6928G > A (p.G2310R) with 1.481, c.7810G > A (p.D2604N) with 0.446, c.1001C > T (p.T334M) with 0.192, c.6284A > G (p.D2095G) with 0.148, c.2899 T > C (p.W967R) with 0.006. These results indicate that variants, such as c.6644G > A, c.11249G > C, and c.8809G > A, have the most substantial impact on RNA structure, followed by c.3101A > G and c.6928G > A. Variants c.2899 T > C (p.W967R), c.7810G > A, c.1001C > T, and c.6284A > G exhibit minimal effects on RNA conformational stability. These observed structural alterations were further corroborated by multiple approaches, including Circos plots, base pair probabilities dot plots, and differential base pairing probabilities dot plots (Figs. 2, 3, 4, Supplementary Fig. 1). A similar trend was also observed in the RNA accessibility profile analysis, with prominent differences observed in variants such as c.8809G > A (p.E2937K), c.11249G > C (p.R3750P), and c.3101A > G (p.N1034S). Variants like c.6644G > A (p.R2215Q), c.6928G > A (p.G2310R), and c.7810G > A (p.D2604N) also showed notable effects on the accessibility profile. In contrast, variants c.2899 T > C (p.W967R), c.6284A > G (p.D2095G), and c.1001C > T (p.T334M) exhibited almost negligible changes in RNA accessibility. These findings suggest that certain variants, particularly in our study c.8809G > A, c.11249G > C, and c.3101A > G (p.N1034S), may have a significant impact on RNA accessibility, potentially affecting interactions with other RNA or proteins (Fig. 5). The accessibility profile is evaluated in terms of the probability of being unpaired for each nucleotide position of the RNA sequences. The accessibility profiles of both wild-type and mutant sequences and their differences help evaluate how the mutation affects the RNA’s interactions with other proteins or RNAs. These findings suggest the diversity in the structural consequences of the studied PKD1 missense variants, and the variants showing substantial structural alterations in RNA structure represent promising drug targets for precision medicine interventions and can be prioritized for future studies. The variants c.2899 T > C (p.W967R), c.6284A > G (p.D2095G), c.7810G > A (p.D2604N), c.1001C > T (p.T334M) exhibited minimal alterations, suggesting milder effects on RNA level conformations. However, alterations were observed in MD simulations, particularly c.2899 T > C (p.W967R), which exhibited the most noticeable differences, suggesting that these variants may have an effect on protein structure dynamics, emphasizing the importance of considering both RNA and protein levels when assessing the functional consequences of genetic variants, a detail not widely explored in previous studies. Also, it is essential to note that even subtle structural changes induced by mutations can potentially alter RNA dynamics and function.
From this study, we can infer that there is indeed a possibility that different missense variations can intricately influence RNA stability, structure, splicing patterns, translational efficiency, and protein structure dynamics differently and contribute to variability in disease presentation and progression as observed in ADPKD. The study also highlights the heterogeneous nature of structural alterations induced by PKD1 missense variants, drawing attention to the importance of considering the specific molecular consequences of each mutation in disease pathogenesis. We could deduce that the variant may impact RNA either alone or in conjunction with other genetic variants, such as synonymous or frameshift variants. The synonymous variants, although not altering the amino acid sequence, can influence mRNA stability, ribosome binding, and translation kinetics, thereby impacting protein expression levels (Diederichs et al. 2016; Ganser et al. 2019). MutaRNA could come up as a useful tool to have a grasp on the structural consequences of synonymous variants on RNA (Miladi et al. 2020). These variants may disrupt RNA folding, alter splicing regulatory elements, impact translational kinetics, and act as disease modifiers by exacerbating the effects of other mutations, leading to a more severe disease phenotype (Diederichs et al. 2016). Moreover, the complex interplay between genetics, environmental factors, and epigenetic modifications can further modulate the overall impact of missense mutations on disease phenotypes. Understanding the multifaceted effects of missense variants on RNA is also essential for elucidating the molecular mechanisms underlying ADPKD pathogenesis. Insights from MD simulation provide additional grasp of how these missense variants could affect PC1 structure, stability, and interactions, aiding in understanding their functional consequences. While in silico tools cannot replace experiments conducted in vitro or in model organisms, they can help sift through many variants to find a priority, e.g., identifying regions of RNA or protein that undergo significant structural changes due to point mutations and guide experimental investigations. Simulation studies that examine RNA or protein structures in conditions resembling biological environments will improve our grasp of these structures within original physiological conditions.
The study revealed that the PKD1 missense variants caused diverse RNA structural changes. Variants like c.8809G > A (p.E2937K), c.11249G > C (p.R3750P) and c.3101A > G (p.N1034S) led to significant alterations in RNA secondary structure, increasing the likelihood of unpaired nucleotides and potentially affecting RNA stability. These structural changes most plausibly impact RNA's ability to interact with proteins and other RNAs, potentially disrupting normal cellular processes such as splicing, translation, and localization. For example, altered accessibility profiles might hinder the binding of regulatory proteins or splice factors, leading to altered RNA processing or reduced translational efficiency. Targeting these variants for therapeutic intervention offers several potential benefits. For instance, small molecules or antisense oligonucleotides can be designed to stabilize or modify the altered RNA structures due to missense or synonymous variants in a way that can restore normal function. Certain compounds that specifically bind and correct the misfolded or destabilized proteins resulting from these variants could help maintain proper function of the protein progression (Solem et al. 2015; Kirtonia et al. 2020; Yu et al. 2020; Aguilar et al. 2022). Such targeted therapies at the RNA or protein level would aim to mitigate the pathogenic effects of the specific mutations, potentially slowing or halting ADPKD progression. The existing literature often focuses on the direct protein interactions and the resulting phenotypic expressions in ADPKD. Our computational study provides novel insights on RNA and protein structural changes induced by PKD1 variants that could influence gene expression and protein synthesis, further contributing to disease heterogeneity at the phenotypic level. This perspective aligns with recent discussions in the field about the importance of RNA structures in disease mechanisms, as highlighted by research on riboSNitches, which are elements of RNA structure affected by single-nucleotide variants that can impact gene expression and offer insights into disease mechanisms and personalized medicine (Solem et al. 2015; Kirtonia et al. 2020).
Study limitation
By computationally examining the consequences of the missense variants at the RNA and protein level, we hope to deepen our understanding of molecular dynamics underlying ADPKD and identify new avenues for future therapeutic strategies targeting specific mutations. However, this study has several limitations as the scope of our investigation was limited to short snippets of RNA and specific regions/domains of the PC1 protein containing the identified variants. Though MD simulation offers valuable insights into protein dynamics and structure–function relationships, it inherently relies on computational modeling and predictions that are influenced by various factors, including the accuracy of force fields, simulation parameters, and initial protein structures, thus may not perfectly replicate biological reality. Hence, our findings may not fully capture the comprehensive functional consequences of these mutations across the entirety of the protein; however, they certainly give initial close-by insights into the dynamics affected by the point mutations. The experimental validation of our computational findings through biochemical and biophysical assays would fully weigh up the observations and establish the physiological relevance of the identified variants in the context of ADPKD.
Conclusion
Through the systematic integration of computational methodologies, encompassing structural predictions and MD simulations, this study analyzed PKD1 missense variants structural and functional consequences at the RNA and protein levels. The analysis of parameters such as RMSD, RMSF, radius of gyration, SASA, and hydrogen bonding elucidated the effects of these variants on PC1 protein dynamics, stability, and interactions. The findings suggest that these variants may disrupt crucial domains such as the REJ domain, PKD domains, and cation channel domain, potentially compromising protein function. Variants including c.8809G > A (p.E2937K), c.11249G > C (p.R3750P), c.3101A > G (p.N1034S), c.6928G > A (p.G2310R), and c.6644G > A (p.R2215Q) exhibited substantial alterations in RNA structures along with protein dynamics, suggesting prioritization for further functional implications as well as their potential as promising drug targets. We also observed that some variants may not be influencing the RNA structure greatly but can affect the protein structure dynamics, highlighting the importance of considering both RNA and protein levels while assessing their functional implications. The study also highlights the diverse impact of each missense variant on RNA stability, structure, and protein dynamics, potentially contributing to the heterogeneous clinical manifestations and progression as observed in ADPKD.
Translational statement
This computational study on PKD1 missense variants sheds light on the impact of these variants on RNA structure and protein dynamics towards understanding the molecular mechanisms underlying the heterogeneous nature of ADPKD. The findings have great implications for prioritization of the genetic variants for further functional implications as well as their potential as promising drug targets to develop targeted therapeutic interventions aimed at mitigating the progression of ADPKD and likewise utility in other genetic disorders.
Data availaibility
Data supporting the findings of this study are available upon reasonable request from the corresponding author. Due to ethical restrictions and privacy concerns, some data may not be publicly available.
References
Aguilar R et al (2022) Targeting Xist with compounds that disrupt RNA structure and X inactivation. Nature 604(7904):160–166. https://doi.org/10.1038/s41586-022-04537-z
Bellaousov S et al (2013) RNAstructure: web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res 41(W1):W471–W474. https://doi.org/10.1093/nar/gkt290
Bergmann C et al (2018) Polycystic kidney disease. Nat Rev Dis Prim 4(1):50
Bernhart SH, Mückstein U, Hofacker IL (2011) RNA accessibility in cubic time. Algorithms Mol Biol 6:1–7
Butcher SE, Pyle AM (2011) The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Acc Chem Res 44(12):1302–1311
Cowan R, Grosdidier G (2000) ‘Visualization tools for monitoring and evaluation of distributed computing systems. In: Proc of the International Conference on Computing in High Energy and Nuclear Physics, Padova, Italy.
Devi C et al (2024) A whole exome sequencing study of a small indian autosomal dominant polycystic kidney disease patient cohort. medRxiv. https://doi.org/10.1101/2023.04.20.23288719
Diederichs S et al (2016) The dark matter of the cancer genome: aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol Med 8(5):442–457
Draper DE, Grilley D, Soto AM (2005) Ions and RNA folding. Annu Rev Biophys Biomol Struct 34:221–243
Ganser LR et al (2019) The roles of structural dynamics in the cellular functions of RNAs. Nat Rev Mol Cell Biol 20(8):474–489. https://doi.org/10.1038/s41580-019-0136-0
Halvorsen M et al (2010) Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet 6(8):e1001074
Holbrook SR (2008) Structural principles from large RNAs. Annu Rev Biophys 37(1):445–464. https://doi.org/10.1146/annurev.biophys.36.040306.132755
Hollingsworth SA, Dror RO (2018) Molecular dynamics simulation for all. Neuron 99(6):1129–1143
Hopp K et al (2020) Detection and characterization of mosaicism in autosomal dominant polycystic kidney disease. Kidney Int 97(2):370–382
Hunt RC et al (2014) Exposing synonymous mutations. Trends Genet 30(7):308–321
Kirtonia A et al (2020) A comprehensive review of genetic alterations and molecular targeted therapies for the implementation of personalized medicine in acute myeloid leukemia. J Mol Med 98:1069–1091
Kumar S et al (2023) Phloretin and phlorizin mitigates inflammatory stress and alleviate adipose and hepatic insulin resistance by abrogating PPARγ S273-Cdk5 interaction in type 2 diabetic mice. Life Sci 322:121668
McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16(4):404–405
Miladi M et al (2020) MutaRNA: analysis and visualization of mutation-induced changes in RNA structure. Nucleic Acids Res 48(W1):W287–W291
Nishio S et al (2021) A digest from evidence-based clinical practice guideline for polycystic kidney disease 2020. Clin Exp Nephrol 25:1292–1302
Paul BM, Vanden Heuvel GB (2014) Kidney: polycystic kidney disease. Wiley Interdiscip Rev Dev Biol 3(6):465–487
Peintner L, Borner C (2017) Role of apoptosis in the development of autosomal dominant polycystic kidney disease (ADPKD). Cell Tissue Res 369:27–39
Pettersen EF et al (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612
Raj S, Singh RG, Das P (2020) Mutational screening of PKD1 and PKD2 in Indian ADPKD patients identified 95 genetic variants. Mutati Res 821:111718
Ranjan P, Das P (2023) ‘An inclusive study of deleterious missense PAX9 variants using user-friendly tools reveals structural, functional alterations, as well as potential therapeutic targets. Int J Biol Macromolecules 233:123375
Salari R et al (2013) Sensitive measurement of single-nucleotide polymorphism-induced changes of RNA conformation: application to disease studies. Nucleic Acids Res 41(1):44–53. https://doi.org/10.1093/nar/gks1009
Salo-Ahen OMH et al (2020) Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 9(1):71
Sauna ZE, Kimchi-Sarfaty C (2011) Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet 12(10):683–691
Schwede T et al (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31(13):3381–3385
Sigrist CJA et al (2010) PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res 38(suppl_1):D161–D166
Solem AC et al (2015) The potential of the riboSNitch in personalized medicine. Wiley Interdiscip Rev RNA 6(5):517–532
Systèmes D (2016) ‘Biovia, discovery studio modeling environment’, Dassault Systèmes Biovia: San Diego, CA, USA [Preprint].
Van Der Spoel D et al (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718
Vander Meersche Y et al (2024) ATLAS: protein flexibility description from atomistic molecular dynamics simulations. Nucleic Acids Res 52(D1):D384–D392
Wang Z et al (2019) The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO Rep 20(11):e48336
Weimbs T (2007) Polycystic kidney disease and renal injury repair: common pathways, fluid flow, and the function of polycystin-1. Am J Physiol-Renal Physiol 293(5):F1423–F1432
Weston BS, Malhas AN, Price RG (2003) Structure–function relationships of the extracellular domain of the autosomal dominant polycystic kidney disease-associated protein, polycystin-1. FEBS Lett 538(1–3):8–13
Xu D, Zhang Y (2011) Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J 101(10):2525–2534
Yeung KC, Fryml E, Lanktree MB (2024) How does ADPKD severity differ between family members?’. Kidney Int Rep. https://doi.org/10.1016/j.ekir.2024.01.053
Yu A-M, Choi YH, Tu M-J (2020) RNA drugs and RNA targets for small molecules: principles, progress, and challenges. Pharmacol Rev 72(4):862–898
Acknowledgements
We acknowledge the Senior Research Fellowship provided by the Indian Council of Medical Research (ICMR) to first author, India; Banaras Hindu University, Varanasi, India, for providing the internet and computer resources essential for conducting this study.
Funding
No direct funding was received for this study. However, the study findings are derived from secondary analysis conducted on the publically available initial findings of a project funded by the Department for Biotechnology (DBT), Ministry of Science and Technology, Government of India to the Corresponding author.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
The genetic variants in this study were derived from our previous study which received ethics approval (No.: I.Sc./ECM-XII/2021-22) from the Institute Ethical Committee of the Institute of Science, Banaras Hindu University. Participants were recruited with informed consent to participate and to publish. Samples were assigned unique study codes for confidentiality.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Devi, C., Ranjan, P., Raj, S. et al. Computational exploration of protein structure dynamics and RNA structural consequences of PKD1 missense variants: implications in ADPKD pathogenesis. 3 Biotech 14, 211 (2024). https://doi.org/10.1007/s13205-024-04057-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-024-04057-9