
Abstract
The present study investigated antiviral peptides (AVPs) as inhibitors of SARS-CoV-2 Orf9b protein, a novel target for disrupting the Orf9b–TOM70 complex crucial for viral infection. In silico screening via molecular docking and MD simulations identified AVP1442 and AVP1896 with high binding affinities to Orf9b (− 846.3 kcal mol−1 and − 820 kcal mol−1, respectively), comparable to the Orf9b–TOM70 complex (− 810.99 kcal mol−1). These AVPs interacted with key amino acid residues in Orf9b, including phosphorylation sites. In addition, AVPs also closely interacted with conserved regions in Orf9b. AVP1896 formed a hydrogen bond with Orf9b’s threonine at position 84. AVP1442 interacted with Orf9b’s leucine at position 15. Favorable Ramachandran plots and compactness during MD simulations for up to 100 ns suggest good stability of formed complexes. These non-toxic AVPs warrant further in vitro and in vivo evaluation, potentially as components of drug cocktails with small molecules or interferon-based therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Severe acute respiratory syndrome coronavirus (SARS-CoV-2) is the etiological agent for coronavirus disease 2019 (COVID-19). It is a positive-sense single-stranded RNA virus from the family Coronaviridae. The coronavirus family includes a large group of viruses, once associated with the common cold, which has now produced some of the deadliest pathogens known to human beings. In the last 2 decades, members of this family, such as SARS-CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2 have challenged our current public health systems, causing epidemic and pandemic situations in 2002, 2012, and 2019, respectively. Although SARS-CoV-2 has a low case fatality rate compared with its predecessors, its ability to spread has made it a global public health crisis (Gordon et al. 2020). As of February 18, 2022, more than 418 million COVID-19 cases have been reported, with nearly 6 million deaths worldwide (https://covid19.who.int/). Concerted global efforts to find novel antiviral drugs/vaccines to debilitate the pathogen have resulted in a flood of studies and clinical trials (Armstrong 2021). Despite these efforts, the emergence of various new SARS-CoV-2 strains (Mahase 2021) with only a few available vaccines has placed a tremendous burden on the pharmaceutical industry to find novel treatment options (Batista et al. 2021). These new variants have ability to cause severe infection due to their continuous evolution (by fast mutation rate), with better survival fitness, and wide spread into less vaccinated countries (Thakur et al. 2022a, b), (Thakur et al. 2022a, b*).
Emerging evidence suggests that multifaceted approaches can contribute to short-term goals in COVID-19 management, such as preventing infection (Kumaravel et al. 2020). These approaches include good diet which influences gut microbiota (Rishi et al. 2020), and traditional medicines (Patel et al. 2020, (Ratre et al. 2021). However, a comprehensive understanding of transmission, mortality, and long-term efficacy alongside the multifaceted roles of immunity and health remains crucial. Hence, some novel therapeutics are required to facilitate healthcare sector.
Interferon (IFN) regulation is the crucial event of activating innate immunity in human body. TOM70 is a mitochondrial surface receptor, which helps in the IFN regulation by activating mitochondrial antiviral signaling pathway. Generally, host pattern recognition receptors (PRR), such as Toll-like receptors (TLR) and retinoic acid-inducible gene I (RIG) receptors, are specialized receptors that recognize viral components in the host. Upon sensing a virus, RIG -1/ melanoma differentiation-associated protein 5 (MDA-5) recruits downstream adaptor proteins, such as mitochondrial antiviral signaling protein (MAVS), where a surface receptor TOM70 is a crucial receptor. Subsequently, the activated factors initiate the transcription of type I interferon (IFN), type III interferon, and other proinflammatory cytokines, which lead to the host immune response (Bowie and Unterholzner 2008, Han et al. 2021). There are reports that suggest the cause of intracellular infection by SARS-CoV-2. In the presence of SARS-CoV-2, its intracellular viral protein Orf9b affects the interaction between TOM70 and natural ligands by blocking TOM70 (Jiang et al. 2020).
IFNs are pleiotropic signaling proteins that act as the first line of defense against viruses (Nan et al. 2014). Their activities include blocking viral transcription, degrading viral RNA, and inhibiting viral translation (Sadler and Williams 2008). Numerous studies and clinical trials have demonstrated antagonizing properties of interferons on SARS-CoV-2 (Bosi et al. 2020; Monfared et al. 2020). Such as efficacy of IFN-beta 1a and 1b was checked, where IFN-beta 1a has been found suitable for an effective add on therapy (Rahmani et al. 2020; Darazam et al. 2021). Trials with IFN-alpha 2b also showed efficacy for the clearance of viral load from infected lungs (Zhou et al. 2020). However, recent studies suggest the development of an evasion mechanism that enables SARS-CoV-2 to escape from the interferon-based innate response by impeding the IFN pathways (Oh and Shin 2021). This viral evasion mechanism is exhibited by the expression and localization of the alternative SARS-CoV-2 open reading frame (Orf)9b on the mitochondrial membrane, suppressing IFN pathways by interacting with TOM70 (Jiang et al. 2020).
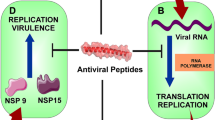
In this COVID-19 pandemic situation, antiviral peptides (AVP) are also being explored to fight against SARS-CoV-2 (Huan et al. 2020; Su et al. 2022; Madhavan et al. 2021), such as ricin-based peptide BRIP from Hordeum vulgare inhibits the Mpro (main protease) of SARS-CoV-2 (Kashyap et al. 2022). This study aimed to bring attention to the antiviral peptides as novel therapeutics to address threat of intracellular infection caused by Orf9b protein of SARS-CoV-2. Here, we performed a high-throughput virtual screening of a library of 267 antiviral peptides from the AVPdb, which is a manually curated, comprehensive database of antiviral peptides targeting many medically important viruses (Qureshi et al. 2014), to find small peptides that can competitively bind to Orf9b. The interaction between AVPs and TOM70 leaving TOM70 disengaged for further MAVS (Mitochondrial antiviral signaling pathway) signal transduction to initiate interferon response pathways (Fig. 1). Ultimately, it suggests the potential of AVPs to be used as prospective therapeutics to treat intracellular infection related to the host’s immunity.

IFN response pathways are hindered by the Orf9b TOM70 complex (A), and Orf9b is bound to small antimicrobial peptides, promoting innate immunity (B). Panel A: This panel likely illustrates how the Orf9b TOM70 complex suppresses the interferon (IFN) response pathways, a vital component of the innate immune system. This suggests Orf9b, when bound to TOM70, might act as an immune antagonist. Panel B: Conversely, Panel B portrays Orf9b bound to small antimicrobial peptides. This interaction might enhance innate immunity, potentially signifying a role for Orf9b as an immune activator when complexed with these specific peptides
Materials and methods
To investigate the potential inhibitory effect of SARS-CoV-2 Orf9b on TOM70 binding, crystal structures of the unbound Orf9b (PDB ID: 6Z4U) (Weeks et al. 2020) and the Orf9b–TOM70 complex (PDB ID: 7KDT) (QCRG Structural Biology Consortium 2020) were retrieved from the Protein Data Bank (PDB) (Berman et al. 2000). Alongside, potential inhibitors for Orf9b were also investigated. A library of antiviral peptides for virtual screening was generated by retrieving the list of antiviral peptides from the AVPdb database and modeling the structure of all the small peptides having more than 90% identity and e-Value 0 to its template homologs.
Antiviral peptides (AVPs) structure prediction and model generation
The structural prediction of these peptides was carried out using Modeller 10.3 and HHpred (Fiser and Sali 2003, Söding et al. 2005), which are widely used homology modeling-based structural prediction tools. A hex command line interface, Hex 8.0, was utilized to virtually screen the peptide library for potential antiviral peptides that have more affinity for Orf9b than its natural ligand, TOM70 (Ritchie and Kemp 2000). For modeling the sequences of AVPDB, we used the modeller command line tool. The process was to be performed for almost 2000 AVPDB sequences, so the process was automated with the help of Perl programming language. In the first step, the search for structures related to peptides was performed. Then the template was selected. In this step, templates having e-Val = 0 and identity > = 95% were selected for the next step; in case the model did not have these features, the peptides were kept aside and continued with the accepted templates. The selected templates were sent to next for comparison among them. The templates having low resolution and good coverage were selected for the next step. After that, aligning the peptide with the template was done. Rest, all other parameters were kept default.
Next was to build the model. Over here we were asked for the number of conformations which we wanted from the template to be built, which was set to 100. After which, based on dope score, the best among all the models was to be selected. The best model was selected comparing the DOPE score and GA341 assessment score. For the present modeling, we considered the DOPE score, the lowest DOPE score indicated the best model. These scores are the internal scoring functions of the modeller.
Docking
Hex 8.0 was used as a docking tool to carry out molecular docking between AVPs and Orf9b, Orf9b,as and TOM70.
Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA)
The MM-PBSA was done for the two groups, i.e., ligand and the protein group. The protein group contained the Orf9b protein, and the ligand group contained the AVPs and the TOM70 receptor, respectively. All the frames were considered for this analysis, starting from 0 to 100 ns at the interval of 1. Rest all parameters were generated for the three complexes using gmx_mmpbsa create input command.
Analysis of hydrogen bonds
Hydrogen bonds were determined based on cutoffs for the angle hydrogen–donor–acceptor and the distance donor–acceptor. OH and NH groups were regarded as donors, where O is an acceptor always, N is an acceptor by default. Dummy hydrogen atoms were assumed to be connected to the first preceding non-hydrogen atom. The cutoff in our case was set to default (CompChems 2022).
SASA (solvent accessible surface area)
SASA was calculated by following built-in gromacs module.
A group for the surface calculation was considered, which consisted all the non-solvent atoms. To compute the SASA, one group from the protein was selected to compute SASA. Surface protein was calculated using GROMACS SASA. SASA value for the protein was calculated as a result in xvg format (sasa.xvg). The output was passed to Xmgrace, where a plot of the surface area (nm2) as a function of time (ps) was generated (CompChems 2022).
GMX_Cluster
GMX clustering data were accessed by following the gromacs manual. Here, the cutoff was set to 0.5 (https://manual.gromacs.org/current/onlinehelp/gmx-cluster.html#:~:text=gmx%20cluster%20can%20cluster%20structures,define%20the%20distance%20between%20structures.)
The entire process was automated using in-house written Perl scripts. Peptides with a high affinity for Orf9b were further studied using atom-interaction map and molecular dynamic simulations. The interaction maps were generated using Ligplot + , and molecular dynamics studies were carried out using GROMACS 2021.4 (Laskowski and Swindells 2011, Pronk et al. 2013).
MD simulation
For the molecular dynamics (MD) simulation, top two AVPs-Orf9b protein complexes with Orf9b-Tom70 complex were taken for the simulation. For this, GROMACS was used. GROMACS is a full-featured suite of programs to perform MD simulations, i.e., to simulate the behavior of systems with hundreds to millions of particles using Newtonian equations of motion. After this, the simulation graphs were generated using xmgrace/gracebat. Following guidelines based manual was applied for this study. (http://www.mdtutorials.com/gmx/lysozyme/01_pdb2gmx.html).
Results and discussion
The spread of SARS-CoV-2 results from its ability to evade host innate immunity (Oh and Shin 2021; Taefehshokr et al. 2020). In this regard, competitive inhibition of Orf9b–TOM70 binding is seen as a promising strategy for fighting COVID-19 (Han et al. 2021; Jiang et al. 2020). Our screening indicated that two AVPs, AVP1896 and AVP1442, have a higher affinity of binding with Orf9b than the general interaction between Orf9b and TOM70 (Table 1).
The AVP1896 heptad is a repeat-derived peptide, which has shown an inhibitory effect on SARS-CoV virus (Bosch et al. 2004), whereas AVP1442 is a viral fusion inhibitory peptide derived from the HIV-1 gp41 protein and was reported to restrict the transmission of HIV to uninfected cells (U.S patent and Trademark office, 2006). Our molecular interaction study of Orf9b-AVP1896 (Supplementary Fig. 1) and Orf9b-AVP1442 (Supplementary Fig. 2) shows that both peptides were able to form strong hydrogen bonds with the Orf9b protein. Specifically, AVP1896 hydrogen bonded with Orf9b’s threonine at location 84 with a 2.95 Å bond distance, whereas AVP1442 bound to Orf9b’s leucine at position 15 with a 3.05 Å bond distance (Fig. 2). Other crucial interactions were also analyzed.

Multiple sequence alignment of Orf9b with its homologs. The blue box shows the conserved binding site for AVP1896, and the red box depicts the conserved binding site for AVP1442 on Orf9b. The conservation in this region suggests that this binding site is essential for AVP1896 and AVP1442 function and likely remains consistent across these Orf9b homologs. Each row in the alignment represents a different protein sequence, and the columns represent corresponding amino acid positions across the sequences. Matching amino acids across sequences are shown with identical letters, while mismatched positions are shown with different letters
The molecular dynamics simulation (MDS) for 100 ns on these peptide-Orf9b complexes are consistent with the docking studies. There are 10 states observed after an interval of 10 ns from 0 to 100 ns (Fig. 4). This interaction depicts the hydrogen bonding within the complex formation. Overall interaction between amino acid residues within the formed complexes between AVPs and Orf9b is similar to the amino acid residue sites of interaction between TOM70 and Orf9b, and other additional sites reported in the literature (Bouhaddou et al. 2020). The calculated hydrogen bonds, SASA, RMSF show optimum results in MDS (Supplementary Data, Sect. 9 and Sect. 10). RMSD values for both complexes, AVP1442–Orf9b and AVP1896–Orf9b, were observed up to 1.2 nm similar to standard complex TOM70–Orf9b. It shows that the formed complexes were similar to standard structure in the time frame of 1 to 100 ns. Radius of gyration (Fig. 3) further supported this data, where the radius of gyration values in the plot fluctuate around a central value suggests that the structures of the molecules in these simulations are relatively stable over time. The radius of gyration values for all three simulations appear to be relatively constant throughout the simulation time. This suggests that the overall size and shape of the molecules in these simulations are not changing significantly over time. Ultimately, the radius of gyration (Fig. 3) and RMSD (Fig. 4a) values from the MDS indicated that both the peptide complexes, AVP1442–Orf9b and AVP1896–Orf9b, are relatively compatible, and highly compact than the counterpart reference complex, i.e., TOM70–Orf9b complex. In addition, the in silico analysis of these peptides using the ToxinPred server elucidates the non-toxic nature of these peptides (Gupta et al. 2013).

The radius of gyration trajectories of antiviral peptide–protein complexes over 100 ns. Complexes are indicated in blue (Orf9b–TOM70), orange (Orf9b-AVP1896), and green (Orf9b-AVP1442). Rg reflects compactness (low) or spread (high). All complexes show stable Rg trajectories, suggesting minimal peptide-induced conformational changes within the simulated timeframe

RMSD trajectories of antiviral peptide–protein complexes (a) up to 100 ns of MD simulation (b,c,d). The figure shows root mean square deviation (RMSD) trajectories of several antiviral peptide–protein complexes over a 100 ns molecular dynamics (MD) simulation. The RMSD trajectories of all the complexes (b, c, and d) appear to reach a plateau after a short initial increase, which suggests that the peptide–protein complexes achieve structural stability during the 100 ns simulation timeframe
In addition, several parameters were analyzed through MM-PBSA. Here, complex energy, ligand energy, receptor energy, and delta energy were calculated with respect to the other factors, such as angle and bond, in the complex formation (Table 2). Overall, end state energy was found to be suitable in AVPs-Orf9b interaction than the Orf9b–TOM70 complex. RMSD cluster index was also plotted for the same (Fig. 5). SASA (solvent accessible surface area) shows suitability of complexes structure in respective to the solvent (Supplementary Fig. F20).

Comparison of RMSD cluster index between natural blocked complex in infection (a) and AVPs facilitated blocking (b and c). The figure shows that the Orf9b–TOM70 complex (a) has the highest RMSD cluster index, followed by the AVP1896–Orf9b complex (c) and the AVP1442–Orf9b complex (b). This suggests that the Orf9b–TOM70 complex is the most structurally variable of the three complexes, while the AVP1442–Orf9b complex is the least structurally variable
In an attempt to find novel strategies against SARS-CoV-2, several in silico studies have been carried out in the past (Chowdhury et al. 2020a, Jang et al. 2021, and Hosseini et al. 2021). This pursuit has resulted in several candidate small molecules/peptides that are currently under further trials. However, the majority of these efforts revolved around restricting the entry of SAR-CoV-2 into the host cell by targeting spike proteins.
In this study, all the antiviral peptides (AVPs) were docked with protein Orf9b. The binding region was specified through random docking and as per predicted active site of Orf9b protein by CASTp server. The amino acid residue regions of active site were detected in both chain A and chain B. There were 11 active sites present in the chain A amino acid residues, such as Ala(Alanine)57, Lys(Lysine)67, Phe(Phenylalanine)69, Leu(Leucine)71, respectively, with the atoms, whereas in chain B, there were 25 active sites in amino acid residues, such as Asp(Aspartic acid)2, Pro(Proline)3, Lys4, Ile(Isoleucine)5, Met(Methionine)8, Leu12, Ile45, Arg(Arginine)47, Thr(Threonine)84, Leu87, Phe91, Val(Valine) 93 with respect to the different atoms within these amino acid residue locations. The overall active site region contains the pocket surface area of 173.315 and volume of 226.262 (SA). The binding affinity of docked complexes was analyzed and compared with the control. Here, we considered Orf9b–TOM70 binding affinity as a standard complex for reference. However, we found binding affinity of Orf9b–TOM70 complex, which was -810 kcal.mol−1. As per our knowledge, there is no report of binding affinity value (in kcal.mol−1) of Orf9b–TOM70 complex up to date. There were 283 antiviral peptides–Orf9b protein docked complexes, out of which only four AVPs showed the close and competitive binding affinity with Orf9b in comparison to the reference complex (shown in supplementary Table T1). Out of those, most favorably interacted AVPs were AVP1896 and AVP1442 with the binding affinity of − 846.3 kcal.mol−1 and − 820 kcal.mol−1, respectively (Table 1). Accordingly, these AVP–Orf9b complexes were carried out for ligplot analysis to determine the amino acid interaction sites.
Protein Orf9b consists of two chains, chain A and chain B. In this investigation, the core interaction between chain A of Orf9b protein and TOM70 receptor amino acid residues was Lys(Lysine)97 and Glu(Glutamic acid)419, respectively. Additional amino acid interacting residues of Orf9b in this complex were Thr95, Lys59, Phe69, and Thr 24 and interacting residues of TOM70 were Leu384, Gln(Glutamine)222, and Lys230, respectively. Between Orf9b Chain B and TOM70 interaction, amino acid residues were His(Histidine)9 and Asp415, Thr79 and Lys233, Thr84 and Gln222, and Asp89 and Gln222. Similarly, here also, other interacting residues for complex formation in Orf9b were Pro10, Glu7, Pro17, Gln18, Gly(Glycine)49, Ser(Serine)50, Pro51, Leu52, Ser53, Asn(Asparagine)55, Ala75, Met(Methionine)78, Leu48, Gln77, Pro73, Pro3, Glu86, Glu85, Leu87, Pro88, Pro43, and Leu14. Other side, interacting amino acids residues of TOM70 receptor were Thr451, Arg447, Tyr450, Lys230, Leu234, Gln416, Thr387, Pro383, Leu384, Gln380, Gln381, Leu414, Lys237, Gln382, Asn453, Met225, Phe219, Asn221, Leu226, and Asn229, respectively. We also found reported interaction regions S50 and S53 of Orf9b interacted with Gln416 and Pro383 of TOM70, respectively.
The amino acid residues of Orf9b chain A involved in the core interaction with AVP1442 were Leu33 and Arg25, and Asn34 and Arg25, respectively. The amino acid residues of Orf9b chain B found to be involved in interaction with AVP1442 were Pro73, Ile74, Ala75, Val76, Met78, Gln77, Asp16, Pro51, Leu52, Leu48, Gly49, Pro10, His9, Ala11, Arg(Arginine)13, Lys80, and Arg58 (for protein Orf9b), whereas AVP1442 interaction sites are Tyr(Tyrosine)28, Glu24, Ile21, Lys25, Gln22, Thr33, Glu17, Glu10, Phe7, Leu8, Asn11, Ala14, Glu18, Leu15, and Ser35. The determined amino acid residues of core interaction between chain B and AVP1442 were Lys25 and Asp16, Gln17 and Leu52, Asn11 and His9, Asn11 and Arg13, Glu18 and Arg13, and Glu18 and Ly80, respectively (Supplementary Tables T5 and T6). These results showed additional interaction possibilities of AVP1442 with Orf9b.
There are other infection-driven phosphorylation sites in Orf9b protein, which are already reported, i.e., Ser50 and Ser53 (Bouhaddou et al. 2020). Two hydrogen bonds are contributed by Ser53 site with Glu477 site in TOM70, during Orf9b–TOM70 complex formation (Brandherm et al. 2021). We also found these interaction sites in the complex formation between Orf9b and TOM70. In addition, recent report does not suggest new sites of interaction other than Glu477. Here, alongside the site Ser50 and Ser53, Leu52 also contributed (a site of interaction behind S53). Leu52 was blocked by Glu17 of AVP1442 (Supplementary data Fig. F2). It suggests the possibility of disrupting the complex formation between Orf9b and TOM70.
AVP1896 has also shown interaction with phosphorylation-driven infection-based region. Amino acid residues found to be involved in the core interaction between protein Orf9b chain A and AVP1896 were Thr84 and Asn35, respectively. Other interacting residues of chain A were Asp2, Met8, Met1, Ala82, Lys4, Thr83, Ile5, Glu85, Pro3, Arg47, Thr 95, Val 96, and Lys 97, whereas AVP1896 interacting residues were Val33, Gln37, Asp41, Lys38, Ile36, Ser32, Asn30, Ile29, and Gly28. Core interaction residues between chain B and AVP1896 were Arg13 and Val21, Arg13 and Asp22, Lys80 and Asp20. Other interaction residues of Or9b were Leu48, Leu52, Gln77, Met78, Pro51, Leu64, Glu65, and Lys67. And AVP1896 interaction residues, respectively, for the mentioned amino acid residues are Leu23, Pro19, Gly24, Asp25, Ile26, Ile29, Thr17, Arg42, Glu39, and Lys38. Supplementary Figure F4, F5, F6, F7, F8 illustrates the core interacting residues of protein Orf9b-AVP1896 complex (Supplementary Table T7 and T8).
These interactions create an evidence of AVPs flexibility for an early interaction with Orf9b (even in its dimer form).
The toxicity analysis of these AVPs was performed by analyzing the subsets of peptides under various parameters, such as SVM score, hydrophobicity, steric hindrance, side bulk, and hydropathicity. A detailed value analysis of these parameters is mentioned in Supplementary Table T3 2.1, for AVP1442, and in Supplementary Table T4 2.2 for AVP1896. Analysis of the peptide fragments under the specified conditions showed no toxicity. This suggests that these peptides have potential as antiviral agents, and further studies could be conducted to assess their overall quality.
Accordingly, with the help of SAVES server, quality factor of antiviral peptides was determined. The Ramachandran plot, according to the plot statistics, of AVP1442 (Supplementary Fig. F23a) and AVP1896 (Supplementary Fig. F23b) showed torsion angles consisting amino acid residues in the allowed regions. In case of AVP1442, there were 33 residues found in the most favored regions (A, B, L) which were non-glycine and non-proline residues. Number of end residues excluding glycine and proline were only two. Therefore, according to plot analysis, the total number of residues involved were 35. In case of AVP1896, residues in the most favored regions (red) (A, B, L) were 46 in number. Residues in the additional allowed region (yellow colored) were four. Accordingly, number of non-glycine and non-proline residues were found to be 50, alongside the number of end residues, glycine residues, and proline residues were two. Therefore, total number of residues were 56. Subsequently, AVP1442 and AVP1896 provided the quality factor of 96.293% and 56.8182%, respectively. This quality factor can be considered suitable for AVPs action.
The interaction and stability of docked complexes were analyzed under MD simulation, which was conducted using GROMACS tool. There were four parameters such as pressure, density, radius of gyration, and RMSD of docked complexes considered for MD simulation. Radius of gyration (Rg) shows compactness of peptide–proteins in a complex (Lobanov et al. 2008). Rg in case of TOM7-Orf9b was found to be ranging from 2.8 to 2.9 nm, whereas AVP1442–Orf9b interaction showed more compactness with 1.8–1.9 nm under 100 ns. AVP1896–Orf9b interaction lies in the region of 2.3–2.4 nm, which is again less than the Rg of standard complex under 100 ns. Accordingly, we found that both AVP1442 and AVP1896 are substantially compact and more stable in complex with Orf9b for up to 100 ns of time duration similar to the standard complex TOM70–Orf9b.
Potential of AVPs as binding inhibitors has been studied since COVID-19 pandemic. AVPs are structurally and functionally versatile due to their simple primary structure (Tonk et al. 2021). Some AVPs are also found against main protease protein and nucleo-protein of SARS-CoV-2 (Mahdi et al. 2022). AVPs are also shown effective against spike protein under in silico analysis based on the structure activity relationship (Chowdhury et al. 2020b). It is evident that AVPs can be efficiently delivered as therapeutics for intracellular infection with nano-delivery system (Essa et al. 2022). In these recent studies, most of the targets were those proteins whose blocking can prevent viral entry, replication or post-translational modifications. Ultimately, therapeutics for preventative approach has been tried instead of focusing on post-infection stage. Intracellular infection causing proteins of SARS-CoV-2 like Orf9b is a novel target to address due to its direct interference with host’s immune system. Also, this protein is capable of fold switching (Porter 2021). As our study suggested both interaction sites of interest for therapeutics development. Hence, flexible peptides with an approach to target different regions close to core interaction regions also become crucial.
Our human body has a complicated and well-developed immune system that is competent enough to prevent pathogenic success. Despite this, SARS-CoV-2 has mastered mechanisms that allow it to escape from our immune system. Therefore, our study seems to be a need-of-the-hour strategy because it attempts to identify peptides that can check and counteract these viral evasion mechanisms. Since these peptides are expected to restrict viral evasion via the host immune response, we propose and intend to check their efficacy in vitro and in vivo as a drug cocktail component in small molecule peptide cocktails and with interferon-based therapeutics. We believe our findings will facilitate the development of the next generation of drugs against the current menace of COVID-19.
Conclusion
Virtual screening of AVPs and their molecular docking with Orf9b of SARS-CoV-2 showed substantial binding affinity to block Orf9b interaction with the TOM70 receptor. The top two AVPs provided the indications to block Orf9b with higher binding affinity in comparison to Orf9b–TOM70 complex. Structural analysis of formed AVP–Orf9b complexes showed that interaction sites occur at conserved regions and other crucial regions of interaction mentioned in the recent studies, which suggests the appropriate binding potential of AVPs. MD simulations of docked AVPs with Orf9b were suitable in terms of different parameters such as pressure, density, radius of gyration, and RMSD along with the respective timing of 10–100 ns. Further, the potential AVPs were found to be non-toxic, with a good quality factor of 50–90%. The findings of present study indicate that AVP1442 and AVP1896 can be propounded for prospective in vitro experiments for their adequate validation against Orf9b of SARS-CoV-2. Prospective studies can support the development of novel therapeutics to maintain interceptive MAVS pathway for interferon production.
Data availability
Not applicable.
Code availability
Not applicable.
References
Armstrong K (2021) Covid-19 and the investigator pipeline. N Engl J Med 385:7–9. https://doi.org/10.1056/NEJMp2100086
Batista C, Shoham S, Ergonul O, Hotez P, Bottazzi ME, Figueroa JP, Gilbert S, Gursel M, Hassanain M, Kang G, Kaslow D, Kim JH, Lall B, Larson H, Naniche D, Sheahan T, Smith AW, Sow SO, Yadav P, Wourgaft NS (2021) Urgent needs to accelerate the race for COVID-19 therapeutics. E Clin Med 36:100911. https://doi.org/10.1016/j.eclinm.2021.100911
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucl Acids Res 28(1):235–242. https://doi.org/10.1093/nar/28.1.235
Bolognesi DP (2006) Fusion proteins comprising DP-178 and other viral fusion inhibitor peptides useful for treating aids. U.S. Patent and Trademark Office. https://patents.google.com/patent/US7122190B2/en. Accessed 17 November 2022
Bosch BJ, Martina BEE, Zee RVD, Lepault J, Haijema BJ, Versluis C, Heck AJR, Groot RD, Osterhaus ADME, Rottier PJM (2004) Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc Natl Acad Sci USA 101(22):8455–8460. https://doi.org/10.1073/pnas.0400576101
Bosi E, Bosi C, Querini PR, Mancini N, Calori G, Ruggeri A, Canzonieri C, Callegaro L, Clementi M, Cobelli FD, Filippi M, Bregni M (2020) Interferon β-1a (IFNβ-1a) in COVID-19 patients (INTERCOP): study protocol for a randomized controlled trial. Trials 21:939. https://doi.org/10.1186/s13063-020-04864-4
Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Marrero MC, Polacco BJ, Melnyk JE, Ulferts S, Kaake RM, Batra JBR, Mullins RD, Fischer ER, Kochs G, Grosse R, Garcia-Sastre A, Vignuzzi M, Johnson JR, Shokat KM, Swaney DL, Beltrao P, Krogan NJ (2020) The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182:685-712.e19. https://doi.org/10.1016/j.cell.2020.06.034
Bowie AG, Unterholzner L (2008) Viral evasion and subversion of pattern-recognition receptor signaling. Nat Rev Immunol 8:911–922. https://doi.org/10.1038/nri2436
Brandherm L, Kobaš AM, Klöhn M, Brüggemann Y, Pfaender S, Rassow J, Kreimendahl S (2021) Phosphorylation of SARS-CoV-2 Orf9b regulates its targeting to two binding sites in TOM70 and recruitment of Hsp90. Int J Mol Sci 22:9233. https://doi.org/10.3390/ijms22179233
Chowdhury SM, Talukder SA, Khan AM, Afrin N, Ali MA, Islam R, Parves R, Mamun AA, Sufian MA, Hossain MD, Hossain MA, Halim MA (2020a) Antiviral peptides as promising therapeutics against SARS-CoV-2. J Phys Chem B 124(44):9785–9792. https://doi.org/10.1021/acs.jpcb.0c05621
Chowdhury SM, Talukder SA, Khan AM, Afrin N, Ali MA, Islam R, Parves R, Mamun AA, Sufian MA, Hossain CompChems (2020b) How to compute the Solvent Accessible Surface Areas (SASA) with GROMACS. https://www.compchems.com/how-to-compute-the-solvent-accessible-surface-areas-sasa-with-gromacs/#solvent-accessible-surface-area-sasa. Accessed 17 November 2022
CompChems (2022) How to study Hydrogen bonds using GROMACS. https://www.compchems.com/how-to-study-hydrogen-bonds-using-gromacs/#list-of-atoms-involved-in-hydrogen-bonds. Accessed 17 Nov 2022.
Darazam IA, Shokouhi S, Pourhoseingholi MA, Irvani SSN, Mokhtari M, Shabani M, Amirdosara M, Torabinavid P, Golmohammadi M, Hashemi SP, Azimi A, Maivan MHJ, Rezaei O, Zali A, Hajiesmaeili M, Dehbsneh HS, Kusha AH, Shoushtari MT, Khalili N, Soleymaninia A, Gachkar L, Khoshkar A (2021) Role of interferon therapy in severe COVID-19: the COVIFERON randomized controlled trial. Sci Rep 11:8059. https://doi.org/10.1038/s41598-021-86859-y
Essa RZ, Wu YS, Batumalaie K, Sekar M, Poh CL (2022) Antiviral peptides against SARS-CoV-2: therapeutic targets, mechanistic antiviral activity, and efficient delivery. Pharmacol Rep 74(6):1166–1181. https://doi.org/10.1007/s43440-022-00432-6
Fiser A, Sali A (2003) Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol 374:461–491. https://doi.org/10.1016/S0076-6879(03)74020-8
Gordon DE, Jang GM, Bouhaddou MXu et al (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583:459–468. https://doi.org/10.1038/s41586-020-2286-9
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Consortium OSDD, Raghava GPS (2013) In silico approach for predicting toxicity of peptides and proteins. PLOS ONE 8(9):e73957. https://doi.org/10.1371/journal.pone.0073957
Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, Zhang XJ, Gao C, Wang PH (2021) SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J Med Virol 93(9):5376–5389. https://doi.org/10.1002/jmv.27050
Hossain MN, Halim MA (2020) Antiviral peptides as promising therapeutics against SARS-CoV-2. J Phys Chem Biol Syst Biomol 124(44):9785–9792. https://doi.org/10.1021/acs.jpcb.0c05621
Hosseini M, Chen W, Xiao D, Wang C (2021) Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. Precision Clin Med 4(1):1–16. https://doi.org/10.1093/pcmedi/pbab001
Huan Y, Kong Q, Mou H, Yi H (2020) Antimicrobial peptides: classification, design, application and research progress in multiple fields. Front Microbiol 11:582779. https://doi.org/10.3389/fmicb.2020.582779
Jang WD, Jeon S, Kim S, Lee SY (2021) Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc Natl Acad Sci USA 118(30):e2024302118. https://doi.org/10.1073/pnas.2024302118
Jiang HW, Zhang HN, Meng QF, Xie J, Li Y, Chen H, Zheng YX, Wang X, Qi H, Zhang J, Wang PH, Han ZG, Tao SC (2020) SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol 17:998–1000. https://doi.org/10.1038/s41423-020-0514-8
Kalil AC, Mehta AK, Patterson TF, Beigel JH... on behalf of the ACTT-3 study group members (2021) Efficacy of interferon beta-1a plus remdesivir compared with remdesivir alone in hospitalised adults with COVID-19: a double-bind, randomised, placebo-controlled, phase 3 trial. The Lancet Respiratory Medicine 9(12):1365-1376. https://doi.org/10.1016/S2213-2600(21)00384-2
Kashyap P, Bhardwaj VK, Chauhan M, Chauhan V, Kumar A, Purohit R, Kumar A, Kumar S (2022) A ricin-based peptide BRIP from Hordeum vulgare inhibits Mpro of SARS-CoV 2. Sci Rep 12:12802. https://doi.org/10.1038/s41598-022-15977-y
Kumaravel SK, Subramani RK, Jayaraj Sivakumar TK, Elavarasan RM, Vetrichelvan AM, Annam A, Subramaniam U (2020) Investigation on the impacts of COVID-19 quarantine on society and environment: Preventive measures and supportive technologies. 3 Biotech 10, 393. https://doi.org/10.1007/s13205-020-02382-3
Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inform Model 51(10):2778–2786. https://doi.org/10.1021/ci200227u
Lobanov MIu, Bogatyreva NS, Galzitskaia OV (2008) [Radius of gyration is indicator of compactness of protein structure]. Mol Biol (Mosk) 42(4):701–706
Madhavan M, AlOmair LA, KS D, Mustafa S, (2021) Exploring peptide studies related to SARS-CoV to accelerate the development of novel therapeutic and prophylactic solutions against COVID-19. J Infect Publ Health 14(8):1106–1119. https://doi.org/10.1016/j.jiph.2021.06.017
Mahase E (2021) Covid-19: How many variants are there, and what do we know about them? BMJ 374:n1971. https://doi.org/10.1136/bmj.n1971
Mahdi I, Yeasmin H, Hossain I, Badhan RM, Ali MA, Kaium MA, Islam R, Sufian MA, Halim MA (2022) Potential antiviral peptides against the nucleoprotein of SARS-CoV-2. Chem Pap. https://doi.org/10.1007/s11696-022-02514-4
Mahendran ASK, Lim YS, Fang C-M, Loh H-S, Le CF (2020) The potential of antiviral peptides as COVID-19 therapeutics. Front Pharmacol 11:575444. https://doi.org/10.3389/fphar.2020.575444
Mahmud S, Biswas S, Paul GK, Mita MA, Afrose S, Hasan MR, Shimu MSS, Uddin MAR, Uddin MS, Zaman S, Kibria KMK, Khan MA, Emran TB, Saleh MA (2021) Antiviral peptides against the main protease of SARS-CoV-2: A molecular docking and dynamics study. Arab J Chem 14(9)103315:1878–5352. https://doi.org/10.1016/j.arabjc.2021.103315
Mahmud S, Paul GK, Biswas S, Afrose S, Mita MA, Hasan MR, Shimu MSS, Hossain A, Promi MM, Ema FK, Chidambaram K, Chandrasek aran B, Alqahtani AM, Emran TB and Saleh MA (2021) Prospective Role of Peptide-Based Antiviral Therapy Against the Main Protease of SARS-CoV-2. Front. Mol. Biosci. 8:628585. https://doi.org/10.3389/fmolb.2021.628585
Monfared ED, Rahmani H, Khalili H, Hajiabdolbaghi M, Salehi M, Abbasian L, Kazemzadeh H, Yekaninejad MS (2020) A randomized clinical trial of the efficacy and safety of interferon β-1a in treatment of severe COVID-19. Antimicrob Agents Chemother 64(9):e01061-e1120. https://doi.org/10.1128/AAC.01061-20
Nan Y, Nan G, Zhang Y (2014) Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses 6(12):4999–5027. https://doi.org/10.3390/v6124999
Oh SJ, Shin OS (2021) SARS-CoV-2 nucleocapsid protein targets RIG-I-like receptor pathways to inhibit the induction of interferon response. Cells 10(3):530. https://doi.org/10.3390/cells10030530
Porter LL (2021) Predictable fold switching by the SARS-CoV-2 protein ORF9b. Protein Sci 30:1723–1729. https://doi.org/10.1002/pro.4097
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, Spoel DVD, Hess B, Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29(7):845–854. https://doi.org/10.1093/bioinformatics/btt055
QCRG Structural Biology Consortium (2020) Human Tom70 in complex with SARS CoV2 Orf9b. RCSB-PDB. https://doi.org/10.2210/pdb7kdt/pdb
Qureshi A, Thakur N, Tandon H, Kumar M (2014) AVPdb: a database of experimentally validated antiviral peptides targeting medically important viruses. Nucl Acids Res 42(D1):D1147–D1153. https://doi.org/10.1093/nar/gkt1191
Rahmani H, Monfared ED, Nourian A, Khalili H, Hajizadeh N, Jalalabadi NZ, Fazeli MR, Ghazaeian M, Yekaninejad MS (2020) Interferon β-1b in treatment of severe COVID-19: A randomized clinical trial. Int Immunopharmacol 88:106903. https://doi.org/10.1016/j.intimp.2020.106903
Ratre YK, Kahar N, Bhaskar LVKS, Bhattacharya A, Verma HK (2021) Molecular mechanism, diagnosis, and potential treatment for novel coronavirus (COVID-19): a current literature review and perspective. 3 Biotech 11: 94. https://doi.org/10.1007/s13205-021-02657-3
Rishi P, Thakur K, Vij S, Rishi L, Singh A, Kaur IP, Patel SKS, Lee JK, Kalia VC (2020) Diet, gut microbiota and COVID-19. Indian J Microbiol 60:420–429. https://doi.org/10.1007/s12088-020-00908-0
Ritchie DW, Kemp GJ (2000) Protein docking using spherical polar Fourier correlations. Proteins 39(2):178–194. https://doi.org/10.1002/(SICI)1097-0134(20000501)39:2%3c178::AID-PROT8%3e3.0.CO;2-6
Sadler AJ, Williams BRG (2008) Interferon-inducible antiviral effectors. Nat Rev Immunol 8:559–568. https://doi.org/10.1038/nri2314
Söding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucl Acids Res 33(2):W244–W248. https://doi.org/10.1093/nar/gki408
Su P, Zhai D, Wong AHC, Liu F (2022) Development of a novel peptide to prevent entry of SARS-CoV-2 into lung and olfactory bulb cells of hACE2 expressing mice. Mol Brain 15(71). https://doi.org/10.1186/s13041-022-00956-1
Taefehshokr N, Taefehshokr S, Hemmat N, Heit B (2020) Covid-19: perspectives on innate immune evasion. Front Immunol 11:580641. https://doi.org/10.3389/fimmu.2020.580641
Thakur V, Bhola S, Thakur P, Patel SKS, Kulshrestha S, Ratho RK, Kumar P (2022a) Waves and variants of SARS-CoV-2: understanding the causes and effect of the COVID-19 catastrophe. Infection 50:309–325. https://doi.org/10.1007/s15010-021-01734-2
Thakur P, Thakur V, Kumar P, Singh Patel SK (2022) Emergence of novel omicron hybrid variants: BA(x), XE, XD, XF more than just alphabets. Int J Surg (104):106727. https://doi.org/10.1016/j.ijsu.2022.106727
Tonk M, Růžek D, Vilcinskas A (2021) Compelling evidence for the activity of antiviral peptides against SARS-CoV-2. Viruses 13(5):912. https://doi.org/10.3390/v13050912
Weeks SD, De Graef S, Munawar A (2020) X-ray Crystallographic Structure of Orf9b from SARS-CoV-2. RCSB-PDB. https://doi.org/10.2210/pdb6Z4U/pdb
Zhou Q, MacArthur MR, He X, Wei X, Zarin P, Hanna BS, Wang ZH, Xiang X, Fish EN (2020) Interferon-α2b treatment for COVID-19. Viruses 13(1):44. https://doi.org/10.3390/v13010044
Funding
There is no funding source for this study.
Author information
Authors and Affiliations
Contributions
Conceptualization, data generation, data collection, data analysis, manuscript preparation, writing, and proofreading, GS; data generation and analysis. PP; manuscript writing and editing, PK; data collection, manuscript writing and editing, AD; data collection, manuscript writing, review, and editing; PK; research work, manuscript preparation, reviewing, and editing, VKG; research work, manuscript preparation, reviewing, and editing, SK
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest in the publication.
Declarations
The authors declare that none of the work reported in this paper could have been influenced by any known competing financial interests or personal relationships.
Conflicts of interest
The authors declare that they have no conflict of interest in the publication.
Ethical statement
This article does not have any studies with human participants or animals performed by any of the authors.
Consent to participate
Not applicable.
Consent for publication
All the authors mutually agreed that the work should be published in the 3 Biotech.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sharma, G., Paul, P., Dviwedi, A. et al. In silico screening and evaluation of antiviral peptides as inhibitors against ORF9b protein of SARS-CoV-2. 3 Biotech 14, 192 (2024). https://doi.org/10.1007/s13205-024-04032-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-024-04032-4