
Abstract
The cardioprotective activity of rosuvastatin (R) is yet to be known. The objective of this study was to research whether R perfusion before global ischemia can mitigate myocardial ischemia-reperfusion damage, considering the metabolic condition in which these effects occur, and to contemplate potential mitochondrial benefits. Protein kinase B (Akt)/glycogen synthase kinase-3β (GSK-3β) and mitochondrial permeability transition pore (MPTP) are key elements in myocardial injury produced by ischemia-reperfusion. Isolated rat hearts were subjected to 25-min ischemia and 1-h reperfusion in the presence or absence of R, with or without Wortmannin (W), a phosphatidylinositol 3-kinase (PI3K)/Akt inhibitor. Akt and GSK-3β were measured by Western blot analysis; lactate, glycogen, and G6PDH were determined; and Ca2+-induced MPTP opening was evaluated using a spectrophotometric method. Contractility was assessed by left ventricular developed pressure (LVDP), and rate-pressure product (RPP), peak rate of contraction and peak rate of relaxation (± dP/dt), and left ventricular end-diastolic pressure (LVEDP) were determined. Tissue samples were extracted to evaluate mitochondrial damage by electron microscopy and to assess infarct size. Statistical analysis employed ANOVA (n = 6/per group). Myocardial infarct size was significantly reduced by R, which also improved cardiac function. MPTP opening was delayed to 300 μM CaCl2, while use of W resulted in MPTP opening at 200 μM CaCl2. Electron microscopy showed better mitochondrial preservation with R, which reduced lactic acid production, increased glycogen consumption and G6PDH activity, as well as phosphorylation of Akt and GSK-3β. R before ischemia is cardioprotective against ischemic and reperfusion damage, activating Akt and regulating GSK-3β negatively and attenuating the MPTP opening.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases, especially acute myocardial infarction, have been a major health problem and the leading cause of death in many countries for several decades. The World Health Organization (WHO) predicts that by 2020, ischemic heart disease will be responsible for 11.1 million deaths, so delving into the potential endogenous mechanisms involved in this process is of vital importance [36].
Two medical strategies are currently being used against ischemic heart disease: restoration of oxygen supply through percutaneous coronary intervention and use of therapies that decrease oxygen consumption by the myocardium. In both strategies, the goal is to reduce the myocardial tissue damage that occurs as a result of the so-called “ischemia-reperfusion injury” associated with the indispensable restoration of coronary flow. Although the coronary percutaneous intervention is relatively safe, the procedure-related complications are associated with local inflammation that can trigger thrombotic events [17].
Statins are reversible inhibitors of the 3-hydroxy-3-methylglutaryl coenzyme A reductase, interfering in the synthesis of mevalonate, a rate-limiting step in the synthesis of cholesterol [16]. Throughout all these years since their implementation as a hypocholesterolemic, a relationship between the administration of statins and the reduction of cardiovascular morbidity and mortality has been reported in a significant number of clinical trials [23].
In the last decade, the prophylactic use of statins in patients before cardiac surgery or during percutaneous coronary interventions (PCI) has increased due to their pleiotropic actions, independent of cholesterol reduction [37]. Moreover, perioperative statin therapy is related to less severe myocardial injury and lower incidence of atrial fibrillation after a cardiovascular procedure like surgery or PCI. Some studies showed that statins improved endothelial function because of their anti-inflammatory and antioxidant effects. Among other pleiotropic actions of statins, antithrombotic and anti-inflammatory properties were described, which could stabilize atherosclerotic platelet reducing ischemic cardiovascular events [19, 21].
Whether the cardioprotection offered by statins is due to the recruitment of the same cardiac endogenous responses involved in myocardial preconditioning, the most powerful procedure to improve myocardial post-ischemic recovery, still remains unclear. Among all statins currently prescribed, rosuvastatin (R) therapy can achieve effective lipid level goals, showing greater effectiveness than other statins [35].
Henning et al. demonstrated that protein kinase B or Akt is involved in the limitation of apoptosis and necrosis in cultures of myocytes subject to hypoxia [14]. In this regard, Akt has a recognized ability to protect the heart from injury by ischemia-reperfusion. In the same way, some authors have postulated that Akt could promote post-ischemic recovery in preconditioned hearts acting at the mitochondrial level, modulating the opening of the mitochondrial permeability transition pore (MPTP) [27].
Likewise, the enzyme glycogen synthase kinase 3 (GSK-3) was initially described in the context of glycogen synthesis, but it is currently known to be involved in the regulation of numerous cellular processes. There are two isoforms of this enzyme, α and β, which are inactivated when they are phosphorylated in Ser 9 by other regulatory proteins, such as Akt [33]. In the context of cardiac injury, the GSK-3β isoform would be related to the development of pro-apoptotic signals. In this regard, some publications reported an increase in P-GSK-3β in mitochondria induced by agonists of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, such as insulin or erythropoietin [25], suggesting that Akt may regulate the activity of this enzyme in mitochondria. The mechanism by which GSK-3β inhibition would mediate the protective effects exerted by Akt in mitochondria is not clear yet but suggests an inhibition of the MPTP opening as a consequence of a reduction in the Ca2+ overload caused by reduced hydrolysis of adenosine triphosphate (ATP) [18, 32, 38].
Given the cardioprotective role of Akt as a component of reperfusion injury salvage kinase (RISK) and the amount of evidence on the beneficial effects of treatment with R on periprocedural cardiac interventions, the present study proposed to deepen the investigation into elucidating the metabolic, energetical, functional, and structural changes that take place in the treatment with R in hearts subjected to ischemia-reperfusion.
Methods
Experimental protocol
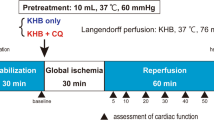
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and Argentine Law No. 14346 concerning animal protection. The experimental protocols were approved by the Institutional Animal Care and Use Committee of the School of Pharmacy and Biochemistry, University of Buenos Aires (EXP-FYB No. 0031654/14). Fifty-eight female Wistar rats fed ad libitum, weighing 250–350 g and maintained on a 12-h dark/light cycle, were used. Rats were anesthetized with diethyl ether and heparin (250 IU) injected into the jugular vein. The hearts were quickly excised and cooled in ice-cold saline until contractions stopped. They were subsequently mounted on a modified Langendorff apparatus (Hugo Sachs Elektronik) and perfused isovolumetrically at a constant pressure of 70 mmHg with a non-recirculating Krebs-Ringer bicarbonate solution of the following composition (mM): NaCl 120, NaHCO3 25, KCl 4.8, MgSO4 1.33, KPO4H2 1.2, CaCl2 1.6, Na2EDTA 0.02, and glucose 10. The perfusate was gassed with 95% O2, 5% CO2 (pH 7.4) and kept at a constant temperature of 37 °C. After instrumentation, hearts were randomly assigned to one of the following groups (n = 6/group), shown in Fig. 1:
Control (C): 25-min stabilization + 25-min global ischemia (IS) (started by shutting off the perfusate flow) + 60-min reperfusion (RP).
Control DMSO (DMSO): 10-min stabilization + 15 min of 0.01% dimethyl sulfoxide (DMSO) + 10 min of R + 25-min global IS + 60-min RP.
Rosuvastatin (R): 15-min stabilization + 10 min of R (added to the perfusion medium in a concentration of 3 μM, Roemmers, Argentine) + 25-min global IS + 60-min RP.
Wortmannin (W): 10-min stabilization + 15 min of W (added to the perfusion medium in a concentration of 100 nM dissolved in dimethyl sulfoxide (DMSO), Sigma, USA) + 25-min global IS + 60-min RP.
R + W: 10-min stabilization + 5 min of W + 10 min of R and W + 25-min global IS + 60-min RP.

Scheme of the experimental protocols. C: control hearts; W: hearts treated with wortmannin at minute 10; R: hearts treated with rosuvastatin at minute 15; R + W: hearts treated with wortmannin at minute 10 during 5 min, and then with rosuvastatin and wortmannin during the next 10 min until the ischemic period; OX: hearts treated with oxfenicine at minute 10; OX+R: hearts treated with oxfenicine at minute 10, and then with rosuvastatin and oxfenicine during the next 10 min until the ischemic period
In order to study the use of fatty acids by R, Oxfenicine (OX) was used to inhibit the enzyme CPT-1: 2 mM (Sigma, St Louis, MO, USA) was added to the perfusion medium to investigate if the use of fatty acids is involved in the beneficial effects of R. The groups to study this hypothesis were as follows:
Oxfenicine (OX): 10-min stabilization + 15 min of OX + 25-min global IS + 60-min RP.
OX + R: 10-min stabilization + 5 min of OX + 10 min of R and OX + 25-min global IS + 60-min RP
Based on bibliographic data, differences in gender and species and in experimental models [11, 20], experiments were performed with four different concentrations of rosuvastatin: 1, 3, 10, and 30 μM. We chose the lowest one that demonstrated cardioprotective effects (3 μM).
Measurement of heart function
In order to measure left ventricular pressure, the left atrium was removed, and a latex balloon connected to a pressure transducer was inserted into the left ventricle through the mitral valve. The volume of the balloon was adjusted to obtain left ventricular end-diastolic pressure (LVEDP) of 10 mmHg. Values for left ventricular developed pressure (LVDP), peak rate of contraction (+dP/dt), and peak rate of relaxation (−dP/dt) were obtained using a digital data acquisition system (Unkel Scope Configuration Program for the PCLabCard Data Acquisition Boards from Advantec, USA; this program was adapted and modified by the technical assistant). Heart rate (HR) was measured with a counter triggered by the LVDP pulse. Rate-pressure product (RPP) was determined by multiplying HR by LVDP. Only hearts with LVDP > 60 mmHg and HR > 200 bpm at the end of the equilibration period were included in the study.
Creatine kinase release
Spectrophotometric measurement of creatine kinase (CK) released (IU/g wet weight (gww)) in the coronary effluent during the first 10 min of RP was used as an indicator of cardiac injury. Since measurement of CK in the effluent is subject to changes in coronary flow, it is worth noting that there were no differences in effluent volume among groups during the first 10 min of RP.
Infarct size
Hearts were removed at the end of the reperfusion period, frozen, and then cut into six to eight slices (approximately 0.8–1 mm). Following defrosting, slices were incubated at room temperature with 1% triphenyltetrazolium chloride in phosphate buffer (100 mmol/L, pH 7.4) for 90 min and then fixed in 10% formaldehyde solution to clearly distinguish stained viable tissue from unstained necrotic tissue. The areas of viable tissue were determined by densitometry using Image J software. Infarct size was calculated as a percentage of the risk area. Because the hearts had been exposed to global ischemia, the risk area was the total ventricular area minus cavities.
Tissue lactate content
At the end of the 25-min ischemic period, hearts were removed and immediately frozen between two blocks of cold ice for determination of tissue lactate. Lactate was extracted from 100 mg of frozen tissue into 6% ice-cold perchloric acid and measured enzymatically at 340 nm. A sample of 60 mg of tissue was used to determine the dry weight in order to express lactate values as μmol/g dry weight (gdw) [28].
Tissue glycogen content
Glycogen was determined in 10–20-mg samples of frozen ventricular tissue. Glycogen extraction was carried out by incubating the tissue at 70 °C for 30 min in a 30% KOH solution, and then precipitated with 6% Na2SO4 and absolute ethanol. The treatment with anthrone in sulfuric acid enabled determination of hexoses released from the reserve polysaccharide at 620 nm [10]. Glycogen content was expressed as μg/100 mg dry weight (mgdw).
Glucose-6-phosphate dehydrogenase activity
Frozen heart tissue was homogenized in 50-mmol/L cold phosphate buffer (pH 7.4). The activity of G6PDH was measured using spectrophotometry at 340 nm [12].
Enzyme activity was expressed as units/g protein. The amount of protein was determined by the method of Lowry [24] with BSA as the standard.
Mitochondrial isolation
At the end of reperfusion, the ventricles were rapidly removed from the hearts, weighed, and homogenized in ice cold sucrose buffer solution (300 mmol/L sucrose, 10 mmol/L Tris-Cl, 2 mmol/L EGTA, 5 mg/mL bovine serum albumin (BSA), pH 7.4). The homogenate was centrifuged at 2000g for 2 min to remove cell debris and the supernatant was centrifuged at 10,000g for 5 min to sediment the mitochondria. The mitochondrial pellet was then washed three times in sucrose isolation buffer solution lacking BSA. Cardiac mitochondria prepared using this procedure have been shown to be metabolically active with respiratory control ratios of 3.5–5.0 with succinate and of 8.0–10.0 with glutamate/malate and corresponding ADP/O ratios of 1.5–1.7 and 2.5–2.7 [31].
Mitochondrial swelling assay
Fresh mitochondria were used for each experiment. The MPTP opening was assessed spectrophotometrically following changes in mitochondrial volume by monitoring the classic decrease in absorbance at 540 nm [22] for up to 8 min at 25 °C. Isolated mitochondria (0.5 mg) were added to 1 mL of buffer (200 mM sucrose, 5 mM Tris, 10 mM Mops, 10 M EGTA, 5 mM KH2PO4, 4 μM rotenone, 0.2 μg/mL antimycin, 8 mM succinate). After a basal line was established, Ca2+ (100–500 μM) was added.
Since cyclosporin A (CsA) is considered to be a potent direct inhibitor of the MPTP, mitochondria incubated in the presence of CsA 1 μM were used as negative controls.
Measurement of mitochondrial ATP synthesis
Since it is well documented that complex I of the respiratory chain is the most sensitive to ischemia-reperfusion injury [30], mitochondrial ATP synthesis was measured in the presence of the complex I substrates pyruvate and malate. Mitochondria (1 mg protein/mL) were incubated for 5 min in a metabolic shaker at 37 °C, in a medium containing (mM): KCl 125, Mops 20, Tris 10, EGTA 0.5, KH2PO4 2.4, MgCl2 2.5, malate 2.5, and pyruvate 2.5, pH 7.4. ATP synthesis was then initiated by adding 2.5 mM ADP, which is the amount of ADP that corresponds to the physiological concentration found in myocytes. Aliquots of 100 μL were taken from the incubation mixture for the assay and ATP synthesis was measured by luciferin-luciferase luminometry (Sigma bioluminescent assay kit) every 5 s over a 1-min period. Mitochondrial protein concentration was determined by the method of Lowry using BSA as the standard, and the rate of mitochondrial ATP synthesis was calculated and expressed as nmol per gram of mitochondrial protein per minute [24].
Western blotting
Hearts were homogenized in lysis buffer (18.2 mM HEPES, 1 mM EDTA, 0.28 M sucrose, 2 mM dithiothreitol, pH 7.4, 2 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail from Thermo Scientific), an aliquot was saved to measure proteins by the method of Lowry [24], and the remaining sample was subjected to SDS-PAGE.
Equal amounts of protein were mixed with LAEMMLI sample buffer (BioRad), boiled for 5 min, and then resolved on 12% SDS-PAGE gels at 120 V. Proteins were afterwards transferred to polyvinylidene difluoride membranes at 100 V for 90 min and the membranes were then blocked for 1 h with 5% non-fat powdered milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T). Membranes were then washed with TBS-T and incubated overnight at 4 °C with polyclonal rabbit anti total Akt (Cell Signaling) 1:1000, anti-phosphorylated Akt (Ser 473) (Cell Signaling) 1:1000, polyclonal rabbit anti GSK-3β (Santa Cruz Technologies) 1:1000, polyclonal rabbit anti-phosphorylated GSK-3β (Santa Cruz Technologies) 1:1000, and polyclonal rabbit anti-β-actin antibody (Thermo Scientific) at 1:2000 dilution. Then, HRP-conjugated donkey anti-rabbit antibody (Thermo Scientific) at 1:2500 dilution was incubated 1 h at room temperature. Following TBS-T washes, bands were detected with Bio-Lumina detection reagent (Kalium Technologies) and exposed to an autoradiography film (Carestream). For band quantification, the film was scanned using a Hewlett-Packard scanner and the intensity of the immunoblot bands was analyzed by densitometry using Image J software and normalized to β-actin. Results were expressed in arbitrary units.
Electron micrographs
After 60 min of RP, heart tissue samples (C, R, W, and R + W) (1 mm thick) were fixed with 2.5% glutaraldehyde in phosphate buffer (pH 7.4) for 4 h and then postfixed in 1% osmium tetroxide for 1 h. After this, block staining with 2% uranyl acetate, dehydration in a graded series of ethanol, and embedding in Durcupan resin were performed. Thin sections were prepared with a diamond knife and stained with lead citrate. Grids were examined under a Zeiss 109 electron microscope (National Laboratory of Research and Services in Electronic Microscopy, University of Buenos Aires).
Statistical analysis
Values are mean ± SEM. Changes in ventricular contractile function were compared statistically using a three-factor ANOVA for repeated measures on one factor followed by Tukey’s test. All other experimental data were evaluated statistically using two-way ANOVA followed by Tukey’s test. Statistical analysis was performed using statistical software SPSS Statistics 19.0. p values ≤ 0.05 were considered statistically significant.
Results
Measurement of heart function
Toward the end of the stabilization period, none of the experimental groups exhibited any modifications in the development of HR, RPP, +dP/dt, and −dP/dt (data not shown). Exposure to 25 min of total ischemia prompted complete cessation of spontaneous contractions and, over the 60 min of RP, HR slowly reestablished its pre-ischemic values (pre-ischemic: C 233 ± 14, R 222 ± 12; 60-min RP: C 240 ± 13, R 232 ± 12 beats/min). W did not exert any impact on HR in either C or R hearts (pre-ischemic: W 227 ± 11, R + W 225 ± 13; 60-min RP: C + W 228 ± 14, R + W 237 ± 13 thumps/min). Recuperation of RPP, +dP/dt, and −dP/dt was improved by R (5-min RPP (%): C 6.8 ± 2.0; W 10 ± 3.0; R 24.3 ± 6.0*; R + W 8.5 ± 3.0; E 24.11 ± 3.0*; E + W 11.0 ± 2.7, *p < 0.05 vs. C, W, and R + W; n = 6/group) (Fig. 2a), and abundancy of LVEDP during the earlier period of RP was essentially decreased (5-min RP LVEDP (%): C 20.0 ± 3.0; R 6.0 ± 1.0*, *p < 0.05). These useful impacts were canceled by W (5-min RP LVEDP (%): W 18.0 ± 3.0; R + W 16 ± 1.0 (*p < 0.05 vs. C, W and R + W) (Fig. 2b). In no case did W change RPP, +dP/dt, −dP/dt, or LVEDP in C hearts (Fig. 2c, d). DMSO did not by itself show any effects in a separate experimental test (data not shown).

Effects of rosuvastatin (R) on: a rate-pressure product (RPP); b left ventricular end-diastolic pressure (LVEDP); c peak rate of contraction (+dP/dt); d peak rate of relaxation (−dP/dt) due to ischemia-reperfusion in control (C) and rosuvastatin (R) hearts. Values expressed as a percentage of the respective basal value at the end of the 60-min stabilization period. Squares: C hearts; Rhombuses: R hearts; Filled symbols: hearts perfused with wortmannin (W). Values are mean ± SEM (n = 6)
On the other hand, OX did not exhibit any effect on the baseline values of HR, RPP, +dP/dt, and −dP/dt (data not shown). The exposure to 25 min of global ischemia led to complete cessation of spontaneous contractions and during the 60-min RP, HR returned progressively to pre-ischemic values, with the same extent in both conditions (pre-ischemic: R 230 ± 5 and OX 243 ± 10; 30-min RP: R 221 ± 18; and OX 229 ± 14 expressed as beats/min). Neither R nor OX exerted any effect on HR in the ischemic-RP hearts (data not shown). The use of OX did not significantly affect recovery in the post-ischemic R group, no differences were found between controls (RPP (%) 5-min RP: OX 8.4 ± 2.0; R + OX 23.5 ± 3.0*; *p < 0.05). The same pattern of results was obtained when the speeds of contraction and relaxation were determined (5-min of RP: +dP/dT (%): OX 38.0 ± 3.8, R + OX 57.7 ± 4.8*; −dP/dT (%): OX 50.0 ± 5.1, R + OX 69.5 ± 4.5*; *p < 0.05). R prevented the increase of LVEDP as R + OX, which indeed occurred upon RP in OX rat heart, whereas it markedly increased the LVEDP during early RP (LVEDP (%) in the first 5 min of RP: OX 17.0 ± 3.0; R + OX 7.0 ± 3*; *p < 0.05).
Creatine kinase release
As shown in Fig. 3a, concomitant with the improvement in contractile function, R attenuated the ischemia-induced CK release during the first 10 min of reperfusion (CK IU/g wet weight (gww): C 45.4 ± 6.4; R 32.0 ± 3.0*; *p < 0.05). This effect was abolished by W (CK IU/gww: R + W 50.0 ± 8.0), and no effects were observed in W hearts (CK UI/gww: W 45.4 ± 5.6).

Effect of rosuvastatin (R) on creatine kinase (CK) release during the first 10 min of reperfusion (a) and infarct size (b). CK release is expressed as IU/g wet weight and infarct size as a percentage of the total left ventricle area. Values are mean ± SEM (n = 6). *p < 0.05 vs. C, W, and R + W; **p < 0.01 vs. C, W, and R + W
The reduced release of CK produced in the presence of R was not modified by the presence of OX. In the same way, the increased release of CK observed in the C group remained unchanged in the presence of OX (CK(IU/gww): OX 55 ± 4, R + OX 30 ± 4*; *p < 0.05).
Infarct size
Infarct size follows the same pattern as CK release. R-treated hearts had a smaller infarct size than the C group (infarct size (%) C 62.0 ± 5.1; R 41.6 ± 3.8**; **p < 0.01). The use of W avoided the changes generated by R (infarct size (%) W 68.2 ± 10.8; R + W 65.0 ± 6.1) (Fig. 3b). OX did not change the marked decrease in the percentage of infarct size generated by R (OX 55.0 ± 2.3, R + OX 42.0 ± 2.0*, *p < 0.05).
DMSO did not show effects by itself on infarct size in a separate experimental test (data not shown).
Tissue lactate content
Lactate production at the end of the stabilization period did not present significant differences among experimental groups (μmol/gwwC: 21.9 ± 4.7; R: 13.8 ± 2.6; W: 17.9 ± 1.9; R + W: 13.8 ± 2.3), while tissue samples from hearts treated with R markedly decreased the production of fixed acids at the end of ischemia (μmol/gww C: 146.3 ± 6.6#; R: 56.7 ± 6.8; W: 65.3 ± 5.2; R + W: 61.4 ± 4.1, #p < 0.01 vs. R, W and R + W; n = 6/each group) (Fig. 4a).

Lactate and glycogen content (a, b, respectively), glucose-6-phosphate dehydrogenase (G6PDH) activity (c) of hearts at the end of the stabilization period (pre-ischemic, black columns) and at the end of ischemia (ischemic, gray columns). Values are mean ± SEM (n = 6 for each group). **p < 0.01 vs. wortmannin (W) and rosuvastatin and wortmannin (R + W) from ischemic group; #p < 0.01 vs. rosuvastatin (R), W and R + W from ischemic group; @p < 0.05 vs. control (C), W and R + W from pre-ischemic group; ##p < 0.01 vs. C, W, and R + W from ischemic group
Tissue glycogen content
The glycogen content (μg/100 mg dw dry weight) at the end of the stabilization period did not present significant differences among the different groups (μg/100 mg dry weight (mgdw) C: 323.5 ± 22.4**; R: 351.9 ± 33.8**; W: 251.1 ± 29.27; R + W: 365.9 ± 55.4).
The results obtained showed similar glycogen content in both R- and C-treated hearts at the end of ischemia (μg/100 mg dw C: 247.1 ± 47.0**; R: 120.7 ± 25.5**; W: 253.4 ± 26.2; R + W: 324.6 ± 34.8, **p < 0.01 vs. W and R + W; n = 6/group) (Fig. 4b).
Glucose-6-phosphate dehydrogenase activity
The treatment with R generated an increase in the activity of G6PDH at the end of the stabilization period (IU/g protein C: 1.19 ± 0.3; R: 2.2 ± 0.2@; W: 1.87 ± 0.2; R + W: 1.31 ± 0.2, @p < 0.05 vs. C, W and R + W; n = 6/group), and the same profile was maintained at the end of ischemia but with even higher values (IU/g proteins C: 1.76 ± 0.3; R: 3.08 ± 0.2##; W: 1.39 ± 0.2; R + W: 1.53 ± 0.1; ##p < 0.01 vs. C, W, and R + W; n = 6/group) (Fig. 4c).
Mitochondrial swelling assay
The sensitivity of the MPTP opening to calcium in mitochondria obtained from ischemia-reperfused hearts was assessed evaluating the changes in the suspension absorbance at 540 nm in a sucrose-based medium. Calcium-induced mitochondrial swelling was further increased in mitochondria from C, W, and R + W hearts, compared to R-treated hearts (300 μM [Ca2+] vs. 200 μM [Ca2+]), suggesting that mitochondria from R hearts were less susceptible to the MPTP opening than those from the other groups. Despite the fact that W essentially did not have any impact against [Ca2+]-activated MPTP opening, it abolished the useful impacts of R (Table 1). CsA abolished the decrease in absorbance prompted by 500 μM [Ca2+].
Western blotting
We examined whether reperfusion activated Akt in hearts treated with R. Akt contains a critical activating residue at serine 473 (Ser 473). Phosphorylation of this residue is used as an indicator of the activation state of this kinase. In this context, at the end of reperfusion we explored the ratio of phosphorylated Akt in Ser 473 (Akt-P) to total enzyme Akt (Akt-T) by Western blot analysis. The results showed a significant increase in the Akt-P/Akt-T ratio. W, the major non-specific Akt inhibitor used in this model, prevented activation of Akt thus canceling the differences observed between R and C groups (Fig. 5).

Western blot showing phosphorylated Akt (Akt-P) and total enzyme Akt (Akt-T) expression in samples from rat hearts, and phosphorylated GSK-3β in Ser 9 (GSK-3β-P) and total enzyme GSK-3β (GSK-3β-T) at the end of reperfusion. Results are expressed as arbitrary units (ARU) for the Akt-P to Akt-T ratio (Akt-P/Akt-T) and phosphorylated GSK-3β-P to total GSK-3β ratio (GSK-3β-P/GSK-3β-T). Values are mean ± SEM (n = 4 per group) **p < 0.01 vs. control (C), wortmannin (W), rosuvastatin, and wortmannin (R + W); ##p < 0.01 vs. C, W, and R + W
To determine whether the activation of Akt that had been observed was related to a decrease in the MPTP opening in mitochondria from R-treated hearts, we investigated the GSK-3β activation profile. The activity of GSK-3β is regulated by phosphorylation at serine 9 (Ser 9). The phosphorylation in its residue reduces GSK-3β activity. We explored the ratio of phosphorylated GSK-3β in Ser 9 (GSK-3β-P) to total enzyme GSK-3β (GSK-3β-T).
Western blot analysis showed increased activation of Akt and inactivation of GSK-3βB at the end of reperfusion in R-treated hearts (arbitrary units of Akt-P/Akt-T): C 0.51 ± 0.08; W 0.40 ± 0.10; R 0.83 ± 0.05**; R + W 0.35 ± 0.02 (**p < 0.01 vs. C, W, and R + W; n = 4/group). Moreover, with regard to the above, we observed greater levels of phosphorylation of GSK-3β, indicating less activity from this kinase (arbitrary units of GSK-3β-P/GSK-3β-T): C 1.0 ± 0.23; W 0.88 ± 0.12; R 1.99 ± 0.17##; R + W 0.70 ± 0.14##p < 0.01 vs. C, W, and R + W; n = 4/group).
Electron micrographs
As represented in Fig. 6b, electron micrographs showed that R maintained mitochondrial ultrastructure toward the end of RP, while the rest of the experimental groups showed degenerative changes with loss of cristae membranes and matrix swelling. R mitochondria had fewer cristae disruptions and fewer amounts of amorphous density formation.

Mitochondrial ATP synthesis (a) of hearts at the end of the stabilization period (pre-ischemic, black columns) and at the end of ischemia (ischemic, gray columns). Values are mean ± SEM (n = 6 for each group). & p < 0.01 vs. control (C), wortmannin (W), rosuvastatin and wortmannin (R + W). Electron microscopies of mitochondria from each group of hearts are shown in b. Arrows indicate separation between peaks, clearance from mitochondrial matrix, and cristae disruptions (n = 4 per group)
Measurement of mitochondrial ATP synthesis
To analyze whether the conservation of the mitochondrial structure produced by R was directly related to its ability to produce ATP, the rate of ATP synthesis by isolated mitochondria towards the end of RP was assessed. The mitochondria of hearts treated with R had a greater capacity to synthesize ATP than the rest of the experimental groups: C, W and R + W (nmoles/g protein/min) C 46.4 ± 6.7; W 45.3 ± 3.8; R, 77.0 ± 8.7&; R + W 41.1 ± 5.9 (&p < 0.01 vs. C, W and R + W) (Fig. 6a).
Discussion
In the present study, we evaluated the cardioprotective effect of R on hearts subjected to ischemia-reperfusion.
Statins, which produce an inhibition of 3-hydroxy-3-methylglutaryl-coenzymeA reductase, present a wide range of pharmacological functions independent of their hypocholesterolemic action. This places them among the most widely used drugs on the planet.
In this sense, rosuvastatin has proven to be the most effective drug for lowering cholesterol levels, producing increased levels of high density lipoprotein while reducing levels of low density lipoproteins and triglycerides. Therefore, it has become the most commonly used statin in diabetic patients with or without metabolic syndrome [2].
The aim of the present study was to investigate the role of Akt in R-mediated cardioprotection in hearts subjected to total global ischemia and subsequent reperfusion.
This experimental protocol is about how hearts treated with R before a period of ischemia can handle the first minutes of reperfusion, resembling people who have an acute myocardial infarction and are also treated with R. Since gender differences have been demonstrated, not only in terms of the clinical manifestation of ischemic heart disease, but also in the therapeutic approach, and considering that male animal models are the most commonly used as a reference to study cardiovascular diseases, it is absolutely necessary to deepen the approximation of cardiovascular diseases in women. For all above, we decided to evaluate the effects of rosuvastatin in female rat hearts subjected to ischemia-reperfusion.
Our results demonstrate that the cardioprotection exerted by R is partially due to the activation of Akt since W abolished the beneficial effects of R on post-ischemic functional recovery. W, a drug that easily permeates the cell membrane, is an irreversible inhibitor of the PI3K family of kinases and the drug of choice to inhibit Akt in the Langendorff perfusion model. These findings are consistent with multiple reports showing that Akt could be involved in cardioprotective pathways.
In relation to the metabolic aspects involved in the treatment with R, we observed that it reduced the production of lactic acid during ischemia. Paradoxically, glycogen levels were similar in the C and R groups. In contrast to the low production of lactate, we were interested in observing the participation of G6PDH in glucose utilization. In this sense, we were able to determine that the pentose pathway was activated in the hearts treated with R prior to sustained ischemia. The use of endogenous glucose content in said pathway is very important since it is responsible for the production of a reductive environment required to reject the reactive oxygen species in play during ischemia and subsequent heart reperfusion. In this regard, different researchers have shown the participation of R in the reduction of reactive oxygen species (ROS) production after subjecting the heart to ischemia-reperfusion [6, 29]. Wang L et al. observed that R protected primary myocardial cells against ischemia-reperfusion injury by increasing the activity of superoxide dismutase (SOD) and decreasing ROS production [34].
On the other hand, the use of W decreased the cleavage of glycogen during ischemia, thus maintaining lactate production levels similar to those of R. These effects were expected given that W inhibits certain enzymes responsible for the glycolytic process. The same profile was observed in hearts treated with R in the presence of W [1, 15].
We also analyzed whether R promoted the use of fatty acids. It is well known that under aerobic metabolism, most of the heart’s energetical supply derives from fatty acid oxidation (60–90%), about 10–40% from glucose and lactate oxidation in the tricarboxylic acid (TCA) cycle through pyruvate dehydrogenase (PDH), and less than 2% from glycolysis [7, 8, 25]. The greatest part of the oxidation of long-chain fatty acids (LCFAs) to acetyl-CoA takes place in the mitochondrial matrix. LCFAs are first transformed into their CoA esters by acyl-CoA synthase, which is ATP-dependent, and then, they are converted to acylcarnitine derivatives by the enzyme carnitine palmitoyl transferase (CPT-1), which allows them to be translocated from the cytosol to the mitochondrial matrix. Once inside, acylcarnitines are reconverted to acyl-CoAs by the enzyme carnitine palmitoyl transferase 2 (CPT-2). The use of unsaturated fats by means of beta-oxidation is not controlled in isolation, but is instead regulated in response to changes in contractile work, levels of contending substrates (i.e., glucose, lactate, ketones, aminoacids), changes in hormonal conditions, or a decrease in oxygen supply.
We showed that the use of oxfenicine(OX) (CPT-1 inhibitor) did not modify the functional response of R and that OX was not involved in any way in the contractile recovery of the hearts subjected to ischemia-reperfusion. Furthermore, no changes were observed in the beneficial effects produced by rosuvastatin in the presence of OX. These results contrast with those found by Bhuiyan and Seccombe who highlighted that treatment with statins promoted CPT-1 activity [4]. These differences may be due to the use of a different statin (lovastatin) supplied in the feed for 16 weeks and to the use of rabbits instead of rats as an animal model.
Preservation of the contractile function was not only accompanied by a special metabolic environment, but it was also possible to determine a higher mitochondrial ATP synthesis capacity in the hearts treated with R. On the other hand, electron microscopy examination revealed that mitochondria from hearts subjected to R showed preservation of their structure while other groups showed greater separation between peaks and clearance [9]. Furthermore, it has been reported that the increase in exogenous glucose utilization is correlated with an increase in the efficiency of myocardial metabolism enhancing production of ATP for each mole of oxygen used, which may represent a response to R administration [5]. Under these experimental conditions, R preserved structural integrity of mitochondria at the end of reperfusion.
The heart depends almost entirely on mitochondrial supply of ATP for the maintenance of its function. The ease to produce mitochondrial ATP could relate directly to the regulation of the opening of the MPTP. In this sense, we were able to verify that acute administration of R attenuated the opening of the MPTP [3, 26].
Furthermore, we were able to demonstrate that Akt is mostly phosphorylated in its active form compared to treatment with R. Likewise, GSK-3β is also predominantly phosphorylated in its inactive form. The data accumulated so far suggests that R would be regulating the expression of Akt, with GSK-3β as the probable responsible for stabilization of the closed form of the MPTP. Other authors have been able to demonstrate that in cell cultures, GSK-3β translocates to the mitochondrial membrane, interacts with a voltage-dependent anion-selective channel (VDAC), and promotes the open state of the MPTP [18, 32]. This interaction could be dependent on the isoform involved, with a voltage-dependent anion-selective channel 2 (VDAC2) potentially involved, and the production of reactive oxygen species. If so, the mitochondrion would preserve its form, functionality, and structure to sustain the integrity of the membranes that are part of it [13].
The present study brings to light the possible participation of PI3K/Akt/GSK-3β in the direct cardioprotective effects of R, basing part of its explanation on the metabolic changes it induces during ischemia which promote the conservation and maintenance of the energy generating centers at the cellular level, i.e., the mitochondria. Mitochondria undertake multiple critical functions in a cell: they supply all the necessary biological energy, regulate the cellular redox state, produce most of the cellular ROS, buffer cellular Ca2+, and initiate cellular apoptosis. In response to cellular environment, mitochondria can send death signals and decide the cell’s fate.
Evaluating all the results obtained, we can state that the changes that are initiated by R tend to improve the cardiac function in addition to preserving mitochondrial function. This confirms that mitochondria would be the main target of R, and therefore, it would be related to the effects of R over the cardiac energetic metabolism. Much remains to be investigated on the possible cardioprotective mechanisms promoted by acute administration of R.
References
Beckner ME, Gobbel GT, Abounader R, Burovic F, Agostino NR, Laterra J, Pollack IF (2005) Glycolytic glioma cells with active glycogen synthase are sensitive to PTEN and inhibitors of PI3K and gluconeogenesis. Lab Investig 85:1457–1470. https://doi.org/10.1038/labinvest.3700355
Bener A, Dogan M, Barakat L, Al-Hamaq AOAA (2014) Comparison of cost-effectiveness, safety, and efficacy of Rosuvastatin versus atorvastatin, pravastatin, and simvastatin in Dyslipidemic diabetic patients with or without metabolic syndrome. J Prim Care Community Health 5:180–187. https://doi.org/10.1177/2150131914520991
Bernardi P, Rasola A, Forte M, Lippe G (2015) The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev 95:1111–1155. https://doi.org/10.1152/physrev.00001.2015
Bhuiyan J, Seccombe DW (1996) The effects of 3-hydroxy-3-methylglutaryl-CoA reductase inhibition on tissue levels of carnitine and carnitine acyltransferase activity in the rabbit. Lipids 31:867–870
Burkhoff D, Weiss RG, Schulman SP, Kalil-Filho R, Wannenburg T, Gerstenblith G (1991) Influence of metabolic substrate on rat heart function and metabolism at different coronary flows. Am J Phys 261:H741–H750. https://doi.org/10.1152/ajpheart.1991.261.3.H741
Burma O, Onat E, Uysal A, Ilhan N, Erol D, Ozcan M, Sahna E (2014) Effects of rosuvastatin on ADMA, rhokinase, NADPH oxidase, caveolin-1, hsp 90 and NFkB levels in a rat model of myocardial ischaemia-reperfusion. Cardiovasc J Afr 25(5):212–216. https://doi.org/10.5830/CVJA-2014-038
Chiu H-C, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE (2005) Transgenic expression of fatty acid transport protein 1 in the heart causes Lipotoxic cardiomyopathy. Circ Res 96(2):225–233. https://doi.org/10.1161/01.RES.0000154079.20681.B9
Christe M, Rodgers RL (1994) Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J Mol Cell Cardiol 26:1371–1375. https://doi.org/10.1006/jmcc.1994.1155
Daghistani HM, Rajab BS, Kitmitto A (2018) Three-dimensional electron microscopy techniques for unravelling mitochondrial dysfunction in heart failure and identification of new pharmacological targets. Br J Pharmacol. https://doi.org/10.1111/bph.14499
Danchenko EO, Chirkin AA (2010) A new approach to the determination of glycogen concentration in various tissues and comments on the interpretation of its results. Sud Med Ekspert 53:25–28
D'Annunzio V, Donato M, Erni L, Miksztowicz V, Buchholz B, Carrión CL, Schreier L, Wikinski R, Gelpi RJ, Berg G, Basso NJ (2009) Rosuvastatin given during reperfusion decreases infarct size and inhibits matrix metalloproteinase-2 activity in normocholesterolemic and hypercholesterolemic rabbits. Cardiovasc Pharmacol 53(2):137–144. https://doi.org/10.1097/FJC.0b013e318197c5e9
Echler G (1983) Determination of glucose-6-phosphate dehydrogenase levels in red cell preparations. Am J Med Technol 49:259–262
González Arbeláez LF, Ciocci Pardo A, Fantinelli JC, Mosca SM (2016) Cyclosporine-a mimicked the ischemic pre- and postconditioning-mediated cardioprotection in hypertensive rats: role of PKCε. Exp Mol Pathol 100:266–275. https://doi.org/10.1016/j.yexmp.2016.01.009
Henning RJ, Dennis S, Sawmiller D, Hunter L, Sanberg P, Miller L (2012) Human umbilical cord blood mononuclear cells activate the survival protein Akt in cardiac myocytes and endothelial cells that limits apoptosis and necrosis during hypoxia. Transl Res 159:497–506. https://doi.org/10.1016/j.trsl.2012.02.004
Huisamen B, Genade S, Lochner A (2008) Signalling pathways activated by glucagon-like peptide-1 (7-36) amide in the rat heart and their role in protection against ischaemia. Cardiovasc J Afr 19(2):77–83
Istvan ES, Deisenhofer J (2001) Structural mechanism for statin inhibition of HMG-CoA reductase. Science 292:1160–1164. https://doi.org/10.1126/science.1059344
Jiang F, Yang J, Zhang L, Li R, Zhuo L, Sun L, Zhao Q (2014) Rosuvastatin reduces ischemia-reperfusion injury in patients with acute coronary syndrome treated with percutaneous coronary intervention. Clin Cardiol 37(9):530–535. https://doi.org/10.1002/clc.22292
Jovanović A (2018) Cardioprotective signalling: past, present and future. Eur J Pharmacol 833:314–319. https://doi.org/10.1016/j.ejphar.2018.06.029
Kavalipati N, Shah J, Ramakrishan A, Vasnawala H (2015) Pleiotropic effects of statins. Indian J Endocrinol Metab 19:554–562. https://doi.org/10.4103/2230-8210.163106
Kelle I, Akkoç H, Uyar E, Erdinç M, Evliyaoğlu O, Sarıbaş S, Tunik S, Özoğul C (2015) The combined effect of rosuvastatin and ischemic pre- or post-conditioning on myocardial ischemia-reperfusion injury in rat heart. Eur Rev Med Pharmacol Sci 19(13):2468–2476
Kinlay S, Schwartz GG, Olsson AG, Rifai N, Sasiela WJ, Szarek M, Ganz P, Libby P (2004) Effect of atorvastatin on risk of recurrent cardiovascular events after an acute coronary syndrome associated with high soluble CD40 ligand in the myocardial ischemia reduction with aggressive cholesterol lowering (MIRACL) study. Circulation 110(4):386–391. https://doi.org/10.1161/01.CIR.0000136588.62638.5E
Li W, Zhang C, Sun X (2018) Mitochondrial Ca2+ retention capacity assay and Ca2+-triggered mitochondrial swelling assay. J Vis Exp 135:e56236. https://doi.org/10.3791/56236
Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group (1998) Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 339:1349–1357. https://doi.org/10.1056/NEJM199811053391902
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Miyamoto S, Murphy AN, Brown JH (2009) Akt mediated mitochondrial protection in the heart: metabolic and survival pathways to the rescue. J Bioenerg Biomembr 41(2):169–180. https://doi.org/10.1007/s10863-009-9205-y
Nesci S, Trombetti F, Ventrella V, Pagliarani A (2018) From the Ca 2+ −activated F 1 F O -ATPase to the mitochondrial permeability transition pore: an overview. Biochimie 152:85–93. https://doi.org/10.1016/j.biochi.2018.06.022
Ong S-B, Hall AR, Dongworth RK, Kalkhoran S, Pyakurel A, Scorrano L, Hausenloy DJ (2015) Akt protects the heart against ischaemia-reperfusion injury by modulating mitochondrial morphology. ThrombHaemost 113:513–521. https://doi.org/10.1160/TH14-07-0592
Shimojo N, Fujino K, Kitahashi S, Nakao M, Naka K, Okuda K (1991) Lactate analyzer with continuous blood sampling for monitoring blood lactate during physical exercise. Clin Chem 37:1978–1980
Sicard P, Lauzier B, Oudot A, Busseuil D, Collin B, Duvillard L, Moreau D, Vergely C, Rochette L (2005) A treatment with rosuvastatin induced a reduction of arterial pressure and a decrease of oxidative stress in spontaneously hypertensive rats. Arch Mal Coeur Vaiss 98(7–8):804–808
Solaini G, Harris DA (2005) Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem J 390:377–394. https://doi.org/10.1042/BJ20042006
Solem LE, Wallace KB (1993) Selective activation of the sodium-independent, cyclosporin A-sensitive calcium pore of cardiac mitochondria by doxorubicin. Toxicol Appl Pharmacol 121:50–57
Tanno M, Kuno A, Ishikawa S, Miki T, Kouzu H, Yano T, Murase H, Tobisawa T, Ogasawara M, Horio Y, Miura T (2014) Translocation of glycogen synthase Kinase-3® (GSK-3®), a trigger of permeability transition, is kinase activity-dependent and mediated by interaction with voltage-dependent Anion Channel 2 (VDAC2). J Biol Chem 289(42):29285–29296. https://doi.org/10.1074/jbc.M114.563924
Tian M, Xie Y, Meng Y, Ma W, Tong Z, Yang X, Lai S, Zhou Y, He M, Liao Z (2019) Resveratrol protects cardiomyocytes against anoxia/reoxygenation via dephosphorylation of VDAC1 by Akt-GSK3 β pathway. Eur J Pharmacol 843:80–87. https://doi.org/10.1016/j.ejphar.2018.11.016
Wang L, Lin R, Guo L, Hong M (2018) Rosuvastatin relieves myocardial ischemia/reperfusion injury by upregulating PPAR-γ and UCP2. Mol Med Rep 18(1):789–798. https://doi.org/10.3892/mmr.2018.9062
Welty FK, Lewis SJ, Friday KE, Cain VA, Anzalone DA (2016) A comparison of statin therapies in hypercholesterolemia in women: a subgroup analysis of the STELLAR study. J Women's Health (Larchmt) 25(1):50–56. https://doi.org/10.1089/jwh.2015.5271
WHO | World Health Organization. https://www.who.int/. Accessed 9 Jan 2019
Zheng Z, Jayaram R, Jiang L, Emberson J, Zhao Y, Li Q, Du J, Guarguagli S, Hill M, Chen Z, Collins R, Casadei B (2016) Preoperative rosuvastatin in cardiac surgery. N Engl J Med 375:901–903. https://doi.org/10.1056/NEJMoa1507750
Acknowledgments
The authors thank Roemmers for the donation of Rosuvastatin. We also express our gratitude to Federico Reznik for his invaluable support in this investigation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
Hearts subjected to ischemia-reperfusion treated with rosuvastatin:
• Improved post-ischemic functional recovery and decreased infarct size.
• Decreased lactic acid production and increased the activity of G6PDH.
• Preserved mitochondrial structure, increasing the capacity of ATP synthesis.
• Activated Akt and negatively regulated GSK-3β, attenuating opening of the MPTP.
Rights and permissions
About this article

Cite this article
Vélez, D.E., Mestre-Cordero, V.E., Hermann, R. et al. Rosuvastatin protects isolated hearts against ischemia-reperfusion injury: role of Akt-GSK-3β, metabolic environment, and mitochondrial permeability transition pore. J Physiol Biochem 76, 85–98 (2020). https://doi.org/10.1007/s13105-019-00718-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-019-00718-z