
Abstract
Immunity and inflammation play critical roles in the pathogenesis of acute ischemic stroke. Therefore, immune intervention, as a new therapeutic strategy, is worthy of exploration. Here, we tested the inflammation modulator, vinpocetine, for its effect on the outcomes of stroke. For this multi-center study, we recruited 60 patients with anterior cerebral circulation occlusion and onset of stroke that had exceeded 4.5 h but lasted less than 48 h. These patients, after random division into two groups, received either standard management alone (controls) or standard management plus vinpocetine (30 mg per day intravenously for 14 consecutive days, Gedeon Richter Plc., Hungary). Vinpocetine treatment did not change the lymphocyte count; however, nuclear factor kappa-light-chain-enhancer of activated B cell activation was inhibited as seen not only by the increased transcription of IκBα mRNA but also by the impeded phosphorylation and degradation of IκBα and subsequent induction of pro-inflammatory mediators. These effects led to significantly reduced secondary lesion enlargement and an attenuated inflammation reaction. Compared to controls, patients treated with vinpocetine had a better recovery of neurological function and improved clinical outcomes during the acute phase and at 3-month follow-up. These findings identify vinpocetine as an inflammation modulator that could improve clinical outcomes after acute ischemic stroke. This study also indicated the important role of immunity and inflammation in the pathogenesis of acute ischemic stroke and the significance of immunomodulatory treatment. Clinical Trial Registration Information: www.clinicaltrials.gov. Identifier: NCT02878772
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke is the second leading cause of death worldwide and is associated with serious disability for the vast majority of its victims [1, 2]. The overwhelming burden of mortality and morbidity imposed by stroke makes exploration of its pathogenesis and identification of new therapeutic resources absolutely essential.
Immune responses have recently emerged as important elements contributing to the pathogenesis of stroke, acting as major players during its onset and progression [3, 4]. Cerebrovascular occlusion induces a massive infiltration of peripheral immune cells into ischemic brain tissue through the damaged blood–brain barrier. The associated activation of microglia and astrocytes promotes local inflammation by producing abundant inflammatory mediators [5, 6]. Post-ischemic inflammation incites brain swelling, which leads to brain cell death and the exacerbation of neurological deficits [7]. Studies of experimental stroke have indicated that interceding to block inflammatory signaling can reduce acute brain parenchymal destruction; one such strategy is based on regulation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), mTOR pathway [8,9,10]. Prophylactic antibiotics did not improve functional outcomes at 3 months or mortality at 14 or 90 days and could not be recommended for prevention of post-stroke infection. These findings could imply that post-stroke pneumonia can be viewed as a post-stroke respiratory syndrome, rather than solely a bacterial infection [11,12,13]. Recently, increased understanding of the role played by post-ischemic inflammation prompted us to inquire whether intervention at the level of brain inflammation might provide an alternative treatment for acute ischemic stroke patients, thereby alleviating their poor status when standard treatment is delayed. In fact, our three recent clinical trials revealed that the immunomodulator fingolimod (FTY720, Gilenya, Novartis) could limit the secondary tissue injury of ischemic and hemorrhagic stroke, decrease microvascular permeability, attenuate neurological deficits, and promote clinical recovery by reducing circulating blood lymphocytes and subsequently blocking these inflammatory cells from homing to the brain [14,15,16,17]. This potential caused us to seek an anti-inflammatory agent that could modulate brain inflammation without significant numerical alteration of peripheral immune cells.
Vinpocetine is an alkaloid extracted from the periwinkle plant and easily passes the blood–brain barrier [18,19,20,21]. As a potent inhibitor of the voltage-dependent Na+ channels and Ca2+/calmodulin-dependent phosphodiesterase-1, vinpocetine has long been used in ischemic stroke patients [22,23,24,25,26]. As recently demonstrated, vinpocetine possesses an anti-inflammatory property that suppresses the activation of NF-κB in a variety of cell types and in a rat model of ischemic stroke [27,28,29]. Compared to traditional steroids and non-steroidal anti-inflammatory drugs, vinpocetine has no known significant side effects and can pass the blood–brain barrier effectively [30], thus making it an attractive alternative anti-inflammatory agent for use in patients with acute ischemic stroke. Brain ischemia orchestrates a series of inflammatory responses, including (1) activation of innate brain immune cell microglia [31, 32], which subsequently leads to the disruption of blood–brain barrier and massive influx of peripheral immune cells [33], and (2) activation of endothelial cells to a pro-inflammatory and pro-thrombogenic type [34], followed by the formation of inflammatory thrombosis, exacerbating tissue ischemia. On the other hand, vinpocetine could inhibit the inflammatory pathway in multiple cell types including endothelial cells, muscle cells, and macrophage, etc. Despite its long use in the clinic for treatment of ischemic stroke known as improving the cerebral blood flow and perfusion, its function of anti-inflammatory role in this type disease needs to be clarified, especially when regarding the multiple type cells’ activation after brain ischemia. Therefore, we designed the multi-center, randomized, and evaluator-blinded study to investigate the impact of vinpocetine as an anti-inflammatory agent for the treatment of patients with acute ischemic stroke.
Methods
Study Population
During open enrollment, a total of 350 patients with acute ischemic stroke were screened. Sixty patients who met the criteria were recruited into this trial at Tianjin Medical University General Hospital, Tianjin Huanhu Hospital, and Tianjin First Central Hospital, Tianjin, China. A number of inclusion and exclusion criteria were adopted. Inclusions were (1) >18 years of age, (2) anterior circulation ischemic stroke: all patients had symptoms of focal neurological deficits and simultaneous radiological evidence (MRI) of an ischemic brain lesion, (3) measurable neurological deficit (National Institutes of Health Stroke Scale (NIHSS) ≥5), and (4) interval between symptom onset and admission more than 4.5 h and less than 48 h. That is, all patients we recruited were beyond the 4.5 h of symptom onset and, therefore, past the accepted time window for thrombolytic therapy. Exclusion criteria were (1) hemorrhagic stroke and severe hemorrhage in other organs, (2) other diseases of the central nervous system (CNS), (3) diabetes mellitus, (4) tumor or hematological systemic diseases, (5) any infection before acute ischemic stroke, (6) concomitant use of antineoplastic or immune modulating therapies, and (7) contraindication to MRI.
This project was designed as a multi-center, randomized, and evaluator-blinded study. The trial protocol and supporting documentation were approved by the institutional review boards of each participating center. Informed consent was obtained at enrollment from all patients or legally acceptable surrogates.
Trial Design
Sixty patients diagnosed with acute ischemic stroke were randomly placed into two groups: a control group (standard treatment designated by current American Heart Association guidelines) and a vinpocetine group (standard treatment plus vinpocetine, Gedeon Richter Plc., Hungary). Each patient in the vinpocetine group received 30 mg of the drug by intravenous infusion once daily for 14 consecutive days beginning within 1 h after the baseline MRI and no later than 48 h after the onset of symptoms. All recruited patients were randomly allocated in a 1:1 ratio to the vinpocetine or control groups. Randomization was computer generated and stratified by center. Allocation concealment was achieved using a centralized web-based randomization system in which the participant identifier was entered before the allocation was revealed (Y. Y. and X. M.). The treating clinicians were aware of the treatment assignments (C. W., S. L., and Z. G.), but the evaluators did not know this (J. H. and F. Z.). We also recruited 20 age-matched and gender-matched healthy volunteers for the comparative study in the real-time PCR and western blot part.
Clinical Assessments
Clinical status was based on each patient’s enrollment (baseline) and on days 3, 7, 14, 30, and 90 afterwards in an evaluator-blinded fashion. The extent of neurologic deficit was determined using the NIHSS. Higher scores indicate more serious neurologic impairment. Global outcomes were assessed with the modified Rankin Scale (mRS). The scale runs from 0 to 6, running from perfect health without symptoms to death (0: no symptoms; 1: no significant disability, able to carry out all usual activities, despite some symptoms; 2: slight disability, able to look after own affairs without assistance, but unable to carry out all previous activities; 3: moderate disability, requires some help, but able to walk unassisted; 4: moderately severe disability, unable to attend to own bodily needs without assistance and unable to walk unassisted; 5: severe disability, requires constant nursing care and attention, bedridden, and incontinent; 6: dead). Limitation of the ability to perform activities of daily living was calculated with the modified Barthel Index (mBI). The total score is 100, and the higher score means the better independent daily living ability.
Imaging Protocol
For MRI, 3 Tesla GE scanners were used at admission, 3 and 7 days later, with a comprehensive acute stroke MRI protocol, including diffusion-weighted imaging (DWI), T2 FLuid Attenuated Inversion Recovery (T2FLAIR), as well as magnetic resonance spectroscopy (MRS). Lesion volumes were measured on DWI (baseline) and T2FLAIR (7 days). 1H MRS value for brain myo-inositol (MI) was the marker of brain inflammation (7 days) [35]. Acute lesions were identified from diffusion-weighted images. Lesions on follow-up were identified from FLAIR images. MI and creatine (Cr) were obtained from MRS. Typical imaging parameters were as follows: DWI: b = 0, 1000 s/mm2, repetition time (TR)/echo time (TE) = 6000/72 ms; FLAIR: TR/TE = 9000/140 ms; inversion time (TI) = 2200 ms; field of view (FOV) = 22 cm; matrix = 256 × 128 × 40; number of acquisitions (NEX) = 1; resolution = 0.85 × 1.7 × 3.5 [14, 36, 37]; MRS: TR/TE = 3000/30 ms, using stimulated echo acquisition mode, single-voxel spectroscopy. The 1 × 1 × 1 cm3 voxel was placed midsagittally in the infarct core and peripheral area. Proper repositioning of volumes of interest (VOIs) for repeat MRS was ascertained by matching VOI positions on sagittal T1 MPRAGE and axial T2-weighted images [38,39,40]. TR indicates repetition time; TE, echo time; TI, inversion time; FOV, field of view; and NEX, number of acquisitions.
Measurements were done independently and blindly by two neuroradiologists using MIPAV software. Lesion volume was manually outlined on the DWI and FLAIR slices and then automatically calculated for each slice from the measured area and corresponding slice thickness. We also found the third radiologist and used a semiautomated technique (Cheshire; Perceptive Informatics) to measure ischemic lesion volumes for validation. Metabolite quantification was carried out using the LCModel software [41] relative total Cr concentration. This means of internal referencing is often used in clinical spectroscopy due to the relative stability of the Cr peak [42, 43]. Metabolite areas were converted to metabolite ratios MI/Cr to correct for image and localization method differences and uncontrollable experimental conditions such as gain instabilities, and further avoided the need to correct for different contributions of CSF to the analyzed MRS volumes. All processed spectra were visually inspected for quality and artifacts.
Isolation of Mononuclear Cells from Human Peripheral Blood
Peripheral blood anticoagulated by ethylene diamine tetraacetic acid (EDTA) was obtained, first from all patients to be included in the vinpocetine treatment group at baseline (<48 h), which preceded the first dose, and subsequently at days 3 and 7 after the first dose for comparison with control patients at the same time points. Human peripheral blood mononuclear cells (PBMCs) were isolated with Ficoll-Hypaque gradients, and blood plasma was also collected at this step.
RNA Extraction and Real-Time PCR
Total RNAs were extracted from PBMCs of ischemic stroke patients by using TRIzol (Invitrogen, USA) following the manufacturer’s instructions. RNA quantity and quality were assessed using the NanoDrop ND-100 Spectrophotometer (NanoDrop Technologies, USA) and the 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit, Germany), with a 260:280 ratio of ≥1.5. For the RT reaction, SYBR Green RT reagents (Bio-Rad) were used. In brief, the RT reaction was performed for 60 min at 37 °C, followed by 60 min at 42 °C, using oligo (dT) and random hexamers. For PCR amplifications, a SYBR Green Universal Master Mix was used to yield duplicate reactions containing 2× Universal Master Mix, 1 μL of template cDNA, and 100 nM primers in a final volume of 12.5 μL, followed by analysis in a 96-well optical reaction plate (Bio-Rad). The relative quantities of mRNAs were obtained by using the comparative Ct method.
Western Blot Analysis
PBMCs of ischemic stroke patients were lysed, and equal amounts of extract were added to electrophoresis sample buffer (Invitrogen). After boiling, the extracts were loaded on 10% SDS-PAGE gels and electrophoretically separated. Proteins were visualized by the usage of primary antibodies specific for IκBα, p-IκBα (1–2 μg/mL, Abcam, MA, USA), and then incubated with goat anti-rabbit and goat anti-rat horseradish peroxidase-conjugated (Abcam) secondary antibody, respectively. The protein-specific signals were detected using Bio-Rad 721BR08844.
ELISA
For analysis of inflammatory mediators in the blood plasma, human TNF-α, IL-6, IL-8, IL-10, IL-17, IL-1β, IFN-γ, TGF-β1, MCP-1, VCAM-1, ICAM-1, and C-reactive protein (CRP) multi-analyte ELISArray kits were purchased from BioLegend for five vinpocetine-treated and five control patients. From these 12 inflammatory mediators, we chose six trend clear factors, TNF-α, IL-6, MCP-1, ICAM-1, VCAM-1, and CRP, for further analysis using the single-factor ELISA kit. The ELISA was performed according to the manufacturer’s protocol.
Outcomes
The primary outcomes of this study were changes in lesion volume from baseline (DWI) to day 7 (FLAIR), the brain inflammatory level (MI, MI/Cr) at day 7, and the extent of clinical improvement at days 7 and 14, as measured by the changes on the NIHSS score from baseline to days 7 and 14. Secondary outcomes were probability of excellent recovery at day 90 (defined as a score of 0 or 1 on the mRS).
Statistical Analysis
SPSS for Windows version 17.0 software (SPSS, Inc., Chicago, IL, USA) was used for the analyses. For continuous variables, such as lesion volumes, descriptive statistics were calculated and reported as means ± SE. Non-normally distributed or discontinuous variables were reported as median (range) and compared as groups using a Mann–Whitney test. All continuous variables were compared for the vinpocetine treatment group versus the control group using the t test for independent samples. Categorical variables were compared for the vinpocetine treatment group versus controls using the χ 2 test. Infarct lesion volume for the vinpocetine treatment group and controls was analyzed with a two-way ANOVA and post hoc analysis. Statistical significance is defined as P < 0.05.
Results
Baseline Characteristics
During open enrollment, 350 patients with acute ischemic stroke were screened. Sixty patients (17%) met the inclusion criteria and were recruited into this trial (Fig. 1). These patients were subsequently randomized into either the treatment (vinpocetine) or control groups, as described in the “Methods” section. Patients with internal carotid artery occlusion, basilar artery occlusion, diabetes mellitus, infection before stroke, or a contraindication to MRI were excluded from the study. No patient died, dropped out, or was lost to follow-up during the course of this study. The time from disease onset to vinpocetine treatment was between 4.5 and 48 h in all patients. Demographic, clinical, and radiological characteristics are shown in Table 1. At baseline, there were no significant differences with regard to age, stroke etiology, NIHSS score, lesion volume, or lesion location between the two groups.

Effects of vinpocetine in patients with acute ischemic stroke: trial profile. Sixty patients with acute ischemic stroke, who exceeded the therapeutic window for tPA upon enrollment, were randomly assigned into one of two groups. All were treated with standard stroke management, and half (n = 30) also received vinpocetine (Gedeon Richter Plc, 30 mg intravenously once daily for 14 consecutive days) at the indicated time points. Clinical assessments (NIHSS, mRS, and mBI) were conducted at the indicated time points. Infarct volume and MI/Cr were measured by MRI at the indicated time points. Percentages of circulating lymphocyte subsets were monitored by flow cytometry. The expression of IκBα mRNA was assessed by real-time PCR at baseline and at days 3 and 7 after stroke onset. Western blotting analysis was carried out to evaluate the levels of phosphorylated IκBα, total IκBα, and β-actin at baseline and post-stroke days 3 and 7. The inflammatory factors TNF-α, IL-6, MCP-1, VCAM-1, ICAM-1, and CRP were measured by ELISA
Vinpocetine Treatment Improves Clinical Outcomes of Acute Ischemic Stroke Patients
Baseline and follow-up clinical assessments are shown in Fig. 2. The vinpocetine recipients exhibited mild neurological deficits, most of which improved during the first 2 weeks after treatment. Specific comparison of values during the first 2 weeks revealed that control patients registered a mean decrease in their NIHSS score from 7 to 6. In contrast, patients treated with vinpocetine had a mean decrease in their NIHSS score from 7 to 4. The mean difference in the NIHSS score change (NIHSS change = baseline − 14 days) between the two groups was statistically significant (1.8 ± 1.4 vs. 3.3 ± 1.5, P = 0.002) (Fig. 2a, b). Additionally, the mBI scores were significantly higher in the vinpocetine group than in the control group at post-stroke day 90 (94 ± 13.6 vs. 83.1 ± 17.5, P = 0.044) (Fig. 2c). The mRS 0–1 at post-stroke day 90 was 53% in the control group compared with 70% in the vinpocetine treatment group (P < 0.05); this implies that vinpocetine promoted rehabilitation in patients with acute ischemic stroke (Fig. 2d).

Clinical outcomes in control and vinpocetine-treated stroke groups. a Trends of NIHSS scores from control and vinpocetine-treated stroke patients at the indicated time points. b Sharp contrasts are clear in changes of NIHSS scores for control and vinpocetine-treated subjects in the first week (NIHSS change = baseline − 14 days). c Comparison of mBI between groups. d Distribution of the degree of disability at day 90; comparisons were performed with the chi-squared test. mRS modified Rankin Scale. Data represent means ± SE. *P < 0.05 versus control at same time point
Vinpocetine Reduces Secondary Lesion Enlargement in Patients with Acute Ischemic Stroke
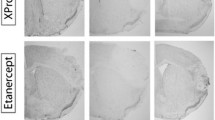
The local inflammatory response after stroke may contribute to secondary lesion extension, cerebral edema, and worsened clinical outcome. Therefore, all patients in this study were assessed for infarct volume at baseline and at 7 days (Fig. 3a).

Impact of vinpocetine on lesion volume in patients and representative MRIs. a Representative MRI scans showed acute left hemisphere infarct in control (upper panel) and vinpocetine-treated patient (lower panel). Lesion volumes were measured on DWI (baseline) and T2FLAIR (7 days). b Lesion volumes compared between the two groups. Lesion volumes of the two groups changed with time to a significant extent (lesion volume growth = 7-day lesion volume − baseline lesion volume). Values are means ± SE; comparisons were performed with independent t tests. **P < 0.01 versus control at same time point
Before treatment, lesion volumes did not differ significantly between the prospective vinpocetine recipients and controls at baseline. Subsequently, 7 days after treatment, the lesion volume on FLAIR of the vinpocetine group was smaller than that of the control group, but this effect was not statistically significant (P = 0.37). However, the lesions (7-day infarct volume–baseline infarct volume) were significantly smaller in the vinpocetine-treated group compared to the control group (0.56 ± 0.37 vs. 3.62 ± 0.35, P < 0.01) (Fig. 3b). Since these data indicate the likelihood that vinpocetine was responsible for decreasing the extension of lesion volume in acute ischemic stroke patients from the onset of symptoms to day 7, vinpocetine evidently limited the progression of these patients’ secondary injury.
Vinpocetine Inhibits Intracerebral Inflammation Response
Previous observations demonstrated that increased regional microglia activation, as a consequence of stroke, can be ascertained by using MRS to detect the local level of MI metabolites, which are synthesized mainly by activated microglial cells [44, 45]. Since the level of Cr in the brain is relatively more stable [46, 47], we used the MI and MI/Cr ratio at post-stroke day 7 in this study to measure the activation level of microglia, thus reflecting one aspect of intracerebral inflammation (Fig. 4a). As shown in Fig. 4b, c, no significant difference was present in MI or MI/Cr at the infarction core between the vinpocetine and control groups. However, within the peri-infarct region, the MI and MI/Cr of controls were much larger than that in the vinpocetine group (76.0 ± 16.0 vs. 61.0 ± 13.0, P = 0.042; 0.70 ± 0.07 vs. 0.41 ± 0.07, P = 0.039).

Impact of vinpocetine on intensity of intracerebral inflammation. a Single-voxel volume-selective water-suppressed proton MRS was performed by using a stimulated-echo acquisition mode. MI and MI/Cr ratios measured the activation level of microglia, thus reflecting the intracerebral inflammation. b, c No significant difference was observed in MI or MI/Cr of the infarction core between the vinpocetine-treated and control groups. However, the MI and MI/Cr of the peri-infarct region in controls much exceeded those in the vinpocetine group. Values are means ± SE; comparisons were performed with independent t tests. *P < 0.05 versus the control at the time point
Vinpocetine Increases Total IκBα Levels by Promoting Transcription of IκBα mRNA and Inhibiting IκBα Phosphorylation
In a previous study, vinpocetine inhibited NF-κB-dependent inflammatory responses by directly inhibiting IKK and subsequent IκBα phosphorylation/degeneration in vitro and in experimental animals [22,23,24]. We thus tested vinpocetine’s ability to influence the transcription of IκBα mRNA. Although the transcription levels of IκBα mRNA in vinpocetine-treated patients were similar to those of controls at baseline, IκBα mRNA expression increased steadily until a statistically significant difference emerged between vinpocetine-treated patients and controls at day 7 after symptom onset (1.46 ± 0.05 vs. 0.96 ± 0.23, P = 0.022) (Fig. 5a). Purportedly, vinpocetine inhibited the NF-κB-dependent inflammatory response by increasing the expression levels of IκBα mRNA.

Vinpocetine reduces NF-κB activation not only by inhibiting phosphorylation and degradation of IκBα but also by increasing transcription of IκBα mRNA. a The transcription of IκBα mRNA in PBMCs (RT-PCR). Vinpocetine increased the transcription of IκBα mRNA in acute ischemic stroke patients. b PBMCs from patients with acute ischemic stroke treated with or without vinpocetine were analyzed by western blotting for phospho-IκBα and total IκBα. The relative protein levels were quantified by densitometry and normalized to β-actin. c, d The quantity of total IκBα decreased in the vinpocetine and control groups along with increases of phospho-IκBα when compared with normal people at baseline, but not to a level of statistically significant difference. The total IκBα level decreased and phospho-IκBα increased gradually with time in the control group. However, the total IκBα and phospho-IκBα content, after treatment with vinpocetine, had no obvious increase or decrease over time. Data represent means ± SE. *P < 0.05 versus control at the same time point. **P < 0.01 versus control at the same time point
We also measured total and phosphorylated IκBα protein by western blot analyses (Fig. 5b). As shown in Fig. 5c, d, the quantity of total IκBα decreased in the vinpocetine and control groups along with increases of phospho-IκBα when compared with healthy volunteers at baseline, but not to the level of statistical significance. Thereafter, in controls, total IκBα levels decreased and phospho-IκBα increased gradually. However, after treatment with vinpocetine, neither total IκBα nor phospho-IκBα concentrations obviously increased or decreased over time. These results demonstrate that vinpocetine also inhibits NF-κB activation by preventing IκBα phosphorylation and degradation, upstream of IκBα.
Vinpocetine Decreases the Levels of Inflammatory Mediators and CRP
Since vinpocetine could inhibit NF-κB activation by preventing IκBα phosphorylation and degradation, we next determined if vinpocetine inhibits pro-inflammatory mediators. As the ELISA denotes (Fig. 6a–f), vinpocetine potently inhibited the upregulation of TNF-α, IL-6, MCP-1, ICAM-1, VCAM-1, and CRP in blood plasma, especially at post-stroke day 7.

Vinpocetine inhibits expression of inflammatory factors, chemokines, and CRP in blood plasma. a–f The levels of TNF-α, IL-6, MCP-1, VCAM-1, ICAM-1, and CRP were measured by ELISA. Vinpocetine treatment potently decreased the levels of TNF-α, IL-6, MCP-1, ICAM-1, VCAM-1, and CRP in patients’ plasma, especially at 7 days after stroke onset compared to controls. Data represent means ± SE. *P < 0.05 versus controls at the same time point
Safety
Six control patients became infected after stroke: four with severe pulmonary infections, one with an upper respiratory tract infection, and one with a urinary tract infection. Five of these patients had temperatures higher than 38 °C and received antibiotic therapy. In the vinpocetine group, three patients had infections, all in the upper respiratory tract. Interestingly, none of the latter group had a fever or antibiotic therapy (Supplementary Table 1).
Discussion
In this study, we evaluated the impact of modulating inflammatory reactions on the outcome of acute ischemic stroke. For this purpose, we monitored patients who had exceeded the accepted timing for tPA treatment but then received treatment to reduce inflammation via regulation of the NF-κB pathway. This treatment involved the intravenous administration of vinpocetine, 30 mg, given daily for 14 consecutive days between 4.5 and 48 h after the onset of acute ischemic stroke. Clearly, the results indicated that vinpocetine attenuated inflammatory responses at the peri-infarct area, prevented secondary brain injury, and substantially improved clinical outcomes. Notably, vinpocetine decreased the incidence of post-stroke infection, an outcome that duplicates our prior results from a clinical retrospective trial (Supplemental Fig. 1). Recent studies showed that stroke-associated pneumonia might be partly a pneumonitis and harbor a greater non-infective inflammatory component than previously thought [48]. Therefore, apart from antibiotics and standard measures to prevent pneumonia, novel strategies should be pursued. Since not only dysphagia but also stroke-induced immune disorders are important risk factors, the use of targeted immune modulators might provide a more powerful approach for its prevention [49]. Vinpocetine’s elimination half-life (t1/2) is 4.83 ± 1.29 h. Because of the low effective drug concentration and good drug safety, vinpocetine was ministered 30 mg once daily. From the discovery of vinpocetine to now, the targets of vinpocetine have been discovered, including Ca2+/calmodulin-stimulated cyclical nucleotide phosphodiesterase-1 (PDE-1) and voltage-dependent Na+ channels and Ca2+ channels. The effects of vinpocetine are composed of improvement of cerebral blood flow and enhancement of neuronal ATP production via increasing the uptake of glucose and oxygen from blood. So, vinpocetine was always ministered for 14 consecutive days. Given that vinpocetine has been proven safe for long-term use, our findings suggest that vinpocetine has substantial potential for use as anti-inflammatory therapy for patients with acute ischemic stroke.
Brain lesions on day 7 mixed the ischemia injury with the secondary inflammation injury that is the main target of vinpocetine, which we want to detect. Thus, we deem the change of lesion size from day 1 to day 7 as a primary outcome for determining the efficacy of vinpocetine. For that reason, we quantified the changes of lesion size from baseline to day 7, which we deem to be a primary outcome. A remarkable restriction in lesion growth between baseline and day 7 was observed in patients who received the combination of vinpocetine and antiplatelet drugs, as compared to that in patients who received antiplatelet drugs only.
Microglia, the brain-resident innate immune cells [50], can release neutral proteinases, produce oxidative radicals, and secrete immunoregulatory factors that influence lymphoid cells as well as the glial cells themselves after being activated [51]. Thus, microglia can be considered as a specialized subtype of tissue macrophage in the CNS and can be monitored to indicate a central inflammatory response. MRS to assess MI, the concentration of which reflects the relative level of activated glial cells, is a useful glia-specific marker of inflammatory responses in the brain [44, 45, 52]. In this study, we found that vinpocetine could decrease the MI and MI/Cr ratio in the peri-infarct region. Thus, we speculated that vinpocetine alleviated the inflammatory response at local areas of infarction in the CNS. Inflammation is impactful at all stages of the ischemic cascade, from the early damaging events triggered by arterial occlusion to the late regenerative processes [6, 53]. NF-κB, as a key transcriptional factor, plays a critical role in modulating inflammatory responses via regulating the expression of pro-inflammatory mediators. When inactive, NF-κB resides in the cytoplasm and forms a multi-protein complex with an inhibitory subunit, inhibitor of NF-κB (IκB). When activated by external stimuli, IκB phosphorylation, and degradation, the liberated NF-κB then enters the nucleus and activates transcription of multiple inflammatory response genes by interacting with κB elements in the promoter region [54]. Thus, increased NF-κB activation is considered as an important pathogenic factor in many inflammatory disorders. In this study, vinpocetine inhibited NF-κB activation not only by increasing the transcription of IκBα mRNA but also by inhibiting the phosphorylation and degradation of IκBα, thus reducing the subsequent induction of pro-inflammatory mediators. Further, unlike conventional immunomodulators that affect the percentage and number of lymphocytes (Supplemental Fig. 2), vinpocetine had no such impact but, instead, inhibited the release of inflammatory cytokines and lowered the magnitude of brain inflammation after acute ischemic stroke. Vinpocetine’s additional benefits were decreases in secondary lesion enlargement.
Limitations of this study include the small sample size and the lack of a parallel arm of patients treated with vinpocetine only beyond 4.5 h of disease onset to differentiate the effect of vinpocetine alone or in combination with antiplatelet agents. However, it is unethical to administer vinpocetine alone to patients with acute ischemic stroke who are eligible for antiplatelet drugs. Unfortunately, we are unable to figure out the relationship between inhibiting inflammation and good outcome in the current study, because using relative blockers to make sure that was the case was unethical. However, based on the data of immune intervention for stroke from patients, we believe that, at least partially, the beneficial outcome of vinpocetine-treated patients was due to its anti-inflammatory function. Cr has higher concentrations in white matter than in neurons. Therefore, the use of MI/Cr ratios reflecting inflammatory levels may have errors. In order to make up for this, we also calculated the absolute value of MI. We think that MI and MI/Cr complement each other well to reflect inflammatory levels. We did not do the multiple comparisons for the inflammatory mediators in the blood plasma. Although the small sample size presented here precludes definite conclusions, our results suggest that vinpocetine as an effective anti-inflammatory agent without causing significant immunosuppression may alleviate the outcome of acute ischemic stroke. This study’s results also indicate that the modulation of inflammatory reactions produces an appreciably more favorable disease prognosis, which is consistent with our recent results of clinical trials [14,15,16,17] and encourages further investigation of inflammatory modulators and manipulation of immune reactions as a new avenue for managing patients with acute ischemic stroke by large-scale clinical trials.
References
Lapchak PA, Zhang JH. The high cost of stroke and stroke cytoprotection research. Transl Stroke Res. 2016; doi:10.1007/s12975-016-0518-y.
Cassidy JM, Cramer SC. Spontaneous and therapeutic-induced mechanisms of functional recovery after stroke. Transl Stroke Res. 2016; doi:10.1007/s12975-016-0467-5.
Liesz A, Kleinschnitz C. Regulatory T cells in post-stroke immune homeostasis. Transl Stroke Res. 2016;7(4):313–21. doi:10.1007/s12975-016-0465-7.
Xia Y, Cai W, Thomson AW, Hu X. Regulatory T cell therapy for ischemic stroke: how far from clinical translation? Transl Stroke Res. 2016;7(5):415–9. doi:10.1007/s12975-016-0476-4.
Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–65. doi:10.1056/NEJMra0910283.
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. doi:10.1038/nm.2399.
Jin R, Liu L, Zhang S, Nanda A, Li G. Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res. 2013;6(5):834–51. doi:10.1007/s12265-013-9508-6.
Tulsulkar J, Glueck B, Hinds TD Jr, Shah ZA. Ginkgo biloba Extract prevents female mice from ischemic brain damage and the mechanism is independent of the HO1/Wnt pathway. Transl Stroke Res. 2016;7(2):120–31. doi:10.1007/s12975-015-0433-7.
Hasegawa Y, Suzuki H, Altay O, Rolland W, Zhang JH. Role of the sphingosine metabolism pathway on neurons against experimental cerebral ischemia in rats. Transl Stroke Res. 2013;4(5):524–32. doi:10.1007/s12975-013-0260-7.
Kim EJ, Raval AP, Hirsch N, Perez-Pinzon MA. Ischemic preconditioning mediates cyclooxygenase-2 expression via nuclear factor-kappa B activation in mixed cortical neuronal cultures. Transl Stroke Res. 2010;1(1):40–7.
Kalra L, Irshad S, Hodsoll J, Simpson M, Gulliford M, Smithard D, et al. Prophylactic antibiotics after acute stroke for reducing pneumonia in patients with dysphagia (STROKE-INF): a prospective, cluster-randomised, open-label, masked endpoint, controlled clinical trial. Lancet. 2015; doi:10.1016/s0140-6736(15)00126-9.
Westendorp WF, Vermeij JD, Zock E, Hooijenga IJ, Kruyt ND, Bosboom HJ, et al. The Preventive Antibiotics in Stroke Study (PASS): a pragmatic randomised open-label masked endpoint clinical trial. Lancet. 2015;385(9977):1519–26. doi:10.1016/S0140-6736(14)62456-9.
Meisel A, Smith CJ. Prevention of stroke-associated pneumonia: where next? Lancet. 2015; doi:10.1016/s0140-6736(15)00127-0.
Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A. 2014;111(51):18315–20. doi:10.1073/pnas.1416166111.
Fu Y, Hao J, Zhang N, Ren L, Sun N, Li YJ, et al. Fingolimod for the treatment of intracerebral hemorrhage: a 2-arm proof-of-concept study. JAMA Neurol. 2014;71(9):1092–101. doi:10.1001/jamaneurol.2014.1065.
Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, et al. Combination of an immune modulator fingolimod with alteplase in acute ischemic stroke: a pilot trial. Circulation. 2015; doi:10.1161/circulationaha.115.016371.
Fu Y, Liu Q, Anrather J, Shi FD. Immune interventions in stroke. Nat Rev Neurol. 2015;11(9):524–35. doi:10.1038/nrneurol.2015.144.
Vas A, Gulyas B, Szabo Z, Bonoczk P, Csiba L, Kiss B, et al. Clinical and non-clinical investigations using positron emission tomography, near infrared spectroscopy and transcranial Doppler methods on the neuroprotective drug vinpocetine: a summary of evidences. J Neurol Sci. 2002;203-204:259–62.
Gulyas B, Halldin C, Karlsson P, Chou YH, Swahn CG, Bonoczk P, et al. Cerebral uptake and metabolism of (11C) vinpocetine in monkeys: PET studies. Orv Hetil. 1999;140(30):1687–91.
Gulyas B, Toth M, Schain M, Airaksinen A, Vas A, Kostulas K, et al. Evolution of microglial activation in ischaemic core and peri-infarct regions after stroke: a PET study with the TSPO molecular imaging biomarker [((11))C]vinpocetine. J Neurol Sci. 2012;320(1–2):110–7. doi:10.1016/j.jns.2012.06.026.
Vas A, Shchukin Y, Karrenbauer VD, Cselenyi Z, Kostulas K, Hillert J, et al. Functional neuroimaging in multiple sclerosis with radiolabelled glia markers: preliminary comparative PET studies with [11C]vinpocetine and [11C]PK11195 in patients. J Neurol Sci. 2008;264(1–2):9–17. doi:10.1016/j.jns.2007.07.018.
Szobor A, Klein M. Ethyl apovincaminate therapy in neurovascular diseases. Arzneimittelforschung. 1976;26(10a):1984–9.
Bonoczk P, Gulyas B, Adam-Vizi V, Nemes A, Karpati E, Kiss B, et al. Role of sodium channel inhibition in neuroprotection: effect of vinpocetine. Brain Res Bull. 2000;53(3):245–54.
Feigin VL, Doronin BM, Popova TF, Gribatcheva EV, Tchervov DV. Vinpocetine treatment in acute ischaemic stroke: a pilot single-blind randomized clinical trial. Eur J Neurol. 2001;8(1):81–5.
Patyar S, Prakash A, Modi M, Medhi B. Role of vinpocetine in cerebrovascular diseases. Pharmacol Rep: PR. 2011;63(3):618–28.
Bagoly E, Feher G, Szapary L. The role of vinpocetine in the treatment of cerebrovascular diseases based in human studies. Orv Hetil. 2007;148(29):1353–8. doi:10.1556/oh.2007.28115.
Jeon KI, Xu X, Aizawa T, Lim JH, Jono H, Kwon DS, et al. Vinpocetine inhibits NF-kappaB-dependent inflammation via an IKK-dependent but PDE-independent mechanism. Proc Natl Acad Sci U S A. 2010;107(21):9795–800. doi:10.1073/pnas.0914414107.
Medina AE. Vinpocetine as a potent antiinflammatory agent. Proc Natl Acad Sci U S A. 2010;107(22):9921–2. doi:10.1073/pnas.1005138107.
Wang H, Zhang K, Zhao L, Tang J, Gao L, Wei Z. Anti-inflammatory effects of vinpocetine on the functional expression of nuclear factor-kappa B and tumor necrosis factor-alpha in a rat model of cerebral ischemia-reperfusion injury. Neurosci Lett. 2014;566:247–51. doi:10.1016/j.neulet.2014.02.045.
Balestreri R, Fontana L, Astengo F. A double-blind placebo controlled evaluation of the safety and efficacy of vinpocetine in the treatment of patients with chronic vascular senile cerebral dysfunction. J Am Geriatr Soc. 1987;35(5):425–30.
Savman K, Heyes MP, Svedin P, Karlsson A. Microglia/macrophage-derived inflammatory mediators galectin-3 and quinolinic acid are elevated in cerebrospinal fluid from newborn infants after birth asphyxia. Transl Stroke Res. 2013;4(2):228–35. doi:10.1007/s12975-012-0216-3.
Wu LJ. Microglial voltage-gated proton channel Hv1 in ischemic stroke. Transl Stroke Res. 2014;5(1):99–108. doi:10.1007/s12975-013-0289-7.
Shi Y, Leak RK, Keep RF, Chen J. Translational stroke research on blood-brain barrier damage: challenges, perspectives, and goals. Transl Stroke Res. 2016;7(2):89–92. doi:10.1007/s12975-016-0447-9.
Reuter B, Rodemer C, Grudzenski S, Meairs S, Bugert P, Hennerici MG, et al. Effect of simvastatin on MMPs and TIMPs in human brain endothelial cells and experimental stroke. Transl Stroke Res. 2015;6(2):156–9. doi:10.1007/s12975-014-0381-7.
Garg M, Gupta RK, Husain M, Chawla S, Chawla J, Kumar R, et al. Brain abscesses: etiologic categorization with in vivo proton MR spectroscopy. Radiology. 2004;230(2):519–27. doi:10.1148/radiol.2302021317.
Gaudinski MR, Henning EC, Miracle A, Luby M, Warach S, Latour LL. Establishing final infarct volume: stroke lesion evolution past 30 days is insignificant. Stroke. 2008;39(10):2765–8. doi:10.1161/strokeaha.107.512269.
Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, et al. Combination of the immune modulator fingolimod with Alteplase in acute ischemic stroke: a pilot trial. Circulation. 2015;132(12):1104–12. doi:10.1161/CIRCULATIONAHA.115.016371.
Young AC, Yiannoutsos CT, Hegde M, Lee E, Peterson J, Walter R, et al. Cerebral metabolite changes prior to and after antiretroviral therapy in primary HIV infection. Neurology. 2014;83(18):1592–600. doi:10.1212/wnl.0000000000000932.
Voevodskaya O, Sundgren PC, Strandberg O, Zetterberg H, Minthon L, Blennow K, et al. Myo-inositol changes precede amyloid pathology and relate to APOE genotype in Alzheimer disease. Neurology. 2016;86(19):1754–61. doi:10.1212/wnl.0000000000002672.
Cousins JP. Clinical MR spectroscopy: fundamentals, current applications, and future potential. AJR Am J Roentgenol. 1995;164(6):1337–47. doi:10.2214/ajr.164.6.7754871.
Provencher SW. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 2001;14(4):260–4.
Valenzuela MJ, Sachdev P. Magnetic resonance spectroscopy in AD. Neurology. 2001;56(5):592–8.
Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. 2012;206(2):275–82. doi:10.1093/infdis/jis326.
Valero IP, Baeza AG, Hernandez-Tamames JA, Monge S, Arnalich F, Arribas JR. Cerebral volumes, neuronal integrity and brain inflammation measured by MRI in patients receiving PI monotherapy or triple therapy. J Int AIDS Soc. 2014;17(4 Suppl 3):19578. doi:10.7448/ias.17.4.19578.
Ciccarelli O, Barkhof F, Bodini B, De Stefano N, Golay X, Nicolay K, et al. Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. Lancet Neurol. 2014;13(8):807–22. doi:10.1016/s1474-4422(14)70101-2.
Roricht S, Meyer BU, Grafin von Einsiedel H, Sander B. A solitary toxoplasmosis focus simulating a brain tumor as the first manifestation of AIDS. RoFo: Fortschritte auf dem Gebiete der Rontgenstrahlen und der Nuklearmedizin. 1997;167(2):201–3. doi:10.1055/s-2007-1015517.
Luyten PR, Marien AJ, Heindel W, van Gerwen PH, Herholz K, den Hollander JA, et al. Metabolic imaging of patients with intracranial tumors: H-1 MR spectroscopic imaging and PET. Radiology. 1990;176(3):791–9. doi:10.1148/radiology.176.3.2389038.
Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665–71. doi:10.1056/nejm200103013440908.
Meisel C, Meisel A. Suppressing immunosuppression after stroke. N Engl J Med. 2011;365(22):2134–6. doi:10.1056/NEJMcibr1112454.
Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends Neurosci. 1988;11(6):273–7.
Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183–93. doi:10.1016/s1474-4422(14)70256-x.
Fernando KT, McLean MA, Chard DT, MacManus DG, Dalton CM, Miszkiel KA, et al. Elevated white matter myo-inositol in clinically isolated syndromes suggestive of multiple sclerosis. Brain. 2004;127(Pt 6):1361–9. doi:10.1093/brain/awh153.
Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8(7):401–10. doi:10.1038/nrneurol.2012.98.
Perez JM, Chirieleison SM, Abbott DW. An IkappaB kinase-regulated feedforward circuit prolongs inflammation. Cell Rep. 2015; doi:10.1016/j.celrep.2015.06.050.
Acknowledgements
We thank our patients for participating this study and to Yao YR and Lu HY for facilitating recruitment of the patients; we also thank Shi HL for technical support. This work was financially supported by the National Basic Research Program of China (2013CB966900 to FDS), the National Natural Science Foundation of China (81571600, 81322018, 81273287, and 81100887 to JWH), the Youth Top-Notch Talent Support Program, and the National Key Clinical Specialty Construction Project of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Funding
This work was financially supported by the National Basic Research Program of China (2013CB966900 to FDS), the National Natural Science Foundation of China (81571600, 81322018, 81273287, and 81100887 to JWH), the Youth Top-Notch Talent Support Program, and the National Key Clinical Specialty Construction Project of China.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Electronic Supplementary Material
Supplemental Figure 1
Comparison of infection rates in recipients of vinpocetine or standard treatment. In another retrospective study of infection associated with stroke, we collected records from 200 patients with acute ischemic stroke who received only standard treatment, then compared the scores with those from 200 age-, gender- and infarct volume-matched patients with acute ischemic stroke (control vs vinpocetine: Female, n, 74 vs 79; age, year, 61.3 ± 16.7 vs 65.1 ± 19.2) who received standard treatment and vinpocetine (30 mg per day intravenously for 14 consecutive days). The inclusion and exclusion criteria were as follows: Inclusions: (1) >18 years of age; (2) Anterior-circulation ischemic stroke: All patients had symptoms of focal neurological deficits and radiological evidence (nuclear magnetic resonance, NMR) of a concurrent ischemic brain lesion; (3) measurable neurological deficit (NIHSS ≥3). Exclusion criteria were (1) evidence of other diseases of the central nervous system (CNS); (2) diabetes mellitus; (3) tumor and hematological system diseases; (4) any infection before acute ischemic stroke; (5) concomitant use of antineoplastic or immune modulating therapies. (A) The infection rate of vinpocetine group (13%) was lower than that of the control group (28%). Statistically significant differences were found between these groups (P = 0.014 Chi-square text). (B) Among the infected patients of vinpocetine group, 88% were respiratory infection and the last 12% were urinary infection. In the control group, respiratory infections account for 74%, urinary infections account for 20%, oral and ocula infections account for 3% respectively. (DOCX 48.1 kb).
Supplemental Figure 2
Dynamics of lymphocyte subsets during vinpocetine treatment. The dynamic changes of lymphocyte subsets were monitored at baseline (<48 h after stroke onset) in whole-blood samples from all patients to receive vinpocetine, preceding the first dose, and then at days 3 and 7 after the first vinpocetine dose. These values were compared with those from control patients (no vinpocetine) at the same time points. Peripheral blood mononuclear cells were isolated from the whole-blood specimens and stained with antibodies to CD4-FITC, CD8-PE, CD3-PerCP, CD19-APC, CD11c-FITC and CD56-APC (BD Biosciences, Franklin Lakes, NJ, USA). Data were acquired using a FACS Calibur (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) and analyzed with Flow Jo software (Tree Star, Ashland, OR, USA). At baseline, day 3 and day 7 after symptom onset, the lymphocyte counts and percentages of CD4+ T, CD8+ T, CD19+ B, CD56+ NK and CD11c+ DC cells in the vinpocetine group were similar to those of controls. These results suggest that vinpocetine exerts anti-inflammatory effects but not by decreasing the number of lymphocytes. Data represent means ± SE (DOCX 68.9 kb).
Supplementary Figure 3
Baseline characteristics. (a) NIHSS score and (b) lesion volume of two goups at baseline. Values are means ± SE. (DOCX 67.3 kb).
Supplementary Figure 4
Lesion volumes compared between two groups. Lesion volumes did not differ significantly between the vinpocetine-treated group and control group at baseline and day 7. Values are means ± SE. (DOCX 46.4 kb).
Supplemental Table 1
(DOCX 30 kb).
Rights and permissions
About this article

Cite this article
Zhang, F., Yan, C., Wei, C. et al. Vinpocetine Inhibits NF-κB-Dependent Inflammation in Acute Ischemic Stroke Patients. Transl. Stroke Res. 9, 174–184 (2018). https://doi.org/10.1007/s12975-017-0549-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-017-0549-z