
Abstract
The water resources in the Guanahacabibes Peninsula are distributed in two areas. The northeastern area is characterized by swamps, wetlands and lagoons, with a low contribution of seawater, whereas the area in the southwestern plain shows a considerable development of the karst structures that limits the existence of superficial waters but permits the ingression of the surrounding seawater. In this latter area, the groundwater showed a marked increase in salinity with the depth. In particular, groundwater with a seawater fraction of 0.1 had the lowest Ca-(Mg)-carbonates saturation indexes calculated by modeling the mixing between freshwater and seawater using different software, thermodynamic databases and equations for activity coefficients. Generally, seawater and groundwaters with an added seawater fraction above 0.60–0.65 showed similar oversaturated indexes in high-Mg calcites and pure Ca-carbonates (calcite and aragonite). Differently, in the groundwater that showed carbonates undersaturation (generally with a seawater fraction between 0.02 and 0.60), the saturation indexes in high-Mg calcites were 0.2 lower than pure Ca-carbonates. Locally, the bacterial reduction of the dissolved sulfate enhanced the dissolution of the limestone, contributing to the increased development of the karst structures and the seawater intrusion. Finally, the presence near the coastline of fresh Ca- and Na-bicarbonate waters was in accordance with the upward flow of the shallow freshwater during the formation of the saline wedge. However, the oxygen and hydrogen stable isotope composition of the waters showed a probable contribution to the area from a deep aquifer that is recharged in the highest reliefs of the province (Cordillera de Guaniguanico).
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Cuban land territory encompasses 110,992 km2, which is 66 % karstified rocks, plus 67,831 km2 of the shallow-water shelf karst (Iturralde-Vinent and Gutiérrez Domech 1999). Carbonate karst aquifers are the primary source of freshwater and are therefore essential to the future needs of the country (Seale et al. 2004). Unfortunately, coastal plains are often contaminated by seawater intrusion, and the vulnerability to salinization is the most common and widespread problem for an aquifer. In the Guanahacabibes Peninsula of western Cuba, the contamination of the fresh groundwater by the saline seawater may be intensified by different, concomitant natural and anthropogenic factors: the rain regime, sea tides, the conditions of recharge of the aquifer(s), excessive aquifer exploitation by wells for human consumption and agricultural activities (Fagundo Castillo and González Hernandez 1999; Hernández et al. 2013; Molerio León and Parise 2009). Therefore, the implementation of suitable monitoring and protective actions is fundamental for the preservation of these aquifers and for assuring the future use of this resource. In this paper, the first results of a study to characterize the groundwaters of Guanahacabibes based on their major chemical and isotopic composition were presented. Specifically, the two main objectives are the following: (a) the calculation of the seawater fraction that mixes with the groundwater together with the influence of this fraction on karst development in terms of the carbonates saturation index (SI) of the waters; and (b) the discrimination between the seawater ingression and the isotope effects due to tropical climate and topographic relief on evaporation and rain recharge, respectively. Concerning geochemical mixing modeling, new topics of wide interest were treated. In particular, the errors on the pure calcite, Mg-calcite and aragonite saturation indexes due to the incomplete chemistry of the end-members, to the use of different thermodynamic databases and the related activity coefficient equations are discussed.
Mixing modeling: the state of the art
When seawater and fresh groundwater mix, the addition of ions, such as Na+ and Cl−, to a bicarbonate water generally decreases the activity of Ca2+ and HCO3 −, among other ions, and so increases the solubility of calcium carbonates. This result is known as the ionic-strength effect, but is sometimes termed the foreign-ion effect or, simply, the salt effect. Generally, when two waters are mixed, the species concentrations in the mixture are volume-weighted averages of the two concentrations in the end-member solutions. However, the thermodynamic activities of the ionic species that control mineral–water reactions in the mixture are not linear functions of mixing, because of the non-linear variation with ionic strength in the activity coefficients of individual ions. Two end-member solutions could, therefore, be saturated with respect to a given mineral, but the mixed solution could be either undersaturated or supersaturated (Runnells 1969). Mean surface seawater is supersaturated in calcite and aragonite, with SIcalcite = 0.8 and SIaragonite = 0.6, and the North Atlantic and North Pacific oceans are slightly above and below these values, respectively (Zeebe and Wolf-Gladrow 2001). Because of global warming and the increasing transport of carbon dioxide from the land to atmosphere and then into seawater, the aqueous CO2 species in seawater increases and the pH of the water decreases slightly, resulting in a decrease in the degree of carbonate supersaturation of coastal zone waters (Mackenzie and Ver 2010). However, note that because of inhibition effects, mainly from dissolved magnesium but also from organic acids and phosphates, calcite, low-Mg calcite (<5 % mol Mg) and dolomite cannot precipitate from surface seawater, whereas aragonite and high-Mg calcite (>5 % mol Mg) are formed (e.g., Appelo and Postma 2007). Therefore, most authors prefer to define the surface seawater calcite supersaturation as “apparent” (Cooke and Kepkay 1984) and use the laboratory solubility product experimentally determined in seawater (Emerson and Hedges 2003).
Using the WATEQ code and the Truesdell-Jones approach for activity calculation, Plummer (1975) showed that mixtures of freshwater with a high percentage of seawater were supersaturated with respect to calcite mainly because of the seawater supersaturation, whereas at lower seawater percentages, the mixtures were undersaturated. Seawater ingression at the Yucatan Peninsula (Mexico) was extensively studied by Back et al. (1986) and Stoessell et al. (1989) with the software PHREEQE and REACT software, which used a modified (Davies or Truesdell-Jones) or extended Debye-Hückel approach, respectively, for activity coefficients calculation. Certain minor differences between the modeling results were imputed to the diversity in activity estimation and to the different end-members compositions (Stoessell et al. 1989). Later, Stoessel (1992) compared an updated mixing model with the real water samples from Barbados and discovered that minimal sulfate reduction (<50 % of sulfate removed as sulfide) enhanced the dissolution of calcite in brackish to saline mixing-zone fluids, whereas considerable sulfate reduction (>50 % of sulfate removed as sulfide) enhanced precipitation in all mixing-zone fluid compositions (Stoessel 1992).
Other studies followed, and different software were used; for example, PHREEQE and its later versions (Lin et al. 2013; Price and Herman 1991; Sanford and Konikow 1989) or EQ3/6 (Liu and Chen 1996). Some of these models were further improved with a solute transport model, but despite this, most of authors do not specify: (a) what equation was employed in the activity coefficients calculation, thus ignoring the error of the saturation indexes and; (b) the inhibition effect of Mg on the saturation indexes of carbonates.
Geological and hydrogeological settings
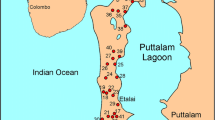
Cuba is a tectonically complex island and its geological history spans the Mesozoic and Cenozoic (e.g., Seale et al. 2004). In western Cuba, carbonate rocks range in age from recent to Upper Jurassic. Cretaceous and Jurassic limestones constitute part of the Cordillera de Guaniguanico (Allochthonous Terrane), which is mainly formed by the belts of Guajaibon–Sierra Azul, Northern Rosario, La Esperanza (westernmost sector of the Northern Rosario, also named Alturas de Pizarras del Norte), Southern Rosario and the Sierra de Los Organos belt that has been subdivided into the Mogotes and the Pizarras del Sur Alturas (Pardo 2009; Fig. 1). These carbonates are telogenetic because they have experienced significant diagenetic alteration, including burial and uplift (Choquette and Pray 1970). In contrast with the tightly faulted, imbricated stack of south-dipping plates in central Cuba, the western belts are less intensely tectonized, north-dipping, superimposed thrust sheets (Pardo 2009). Miocene through recent limestones is expansive in Cuba and is separated from the Guaniguanico Terrane by the Pinar Fault (Fig. 1). These limestones are eogenetic carbonates because they are in situ and have experienced little burial and tectonics (Choquette and Pray 1970). Eogenetic and telogenetic carbonate rocks differ with respect to permeability and porosity. Eogenetic carbonate rocks retain significant matrix permeability and porosity (Budd and Vacher 2004), and may be up to 50,000 times more permeable than telogenetic carbonates (Florea and Vacher 2006). This difference in matrix permeability has important implications when considering groundwater flow, conduit morphology and aquifer response (Florea and Vacher 2006). The carbonate aquifer of the Cordillera de Guaniguanico is confined by the Quaternary sediments, thus representing the hydrological feeding zone for the southwestern area of the Guanahacabibes Peninsula. This peninsula constitutes an example of the recent geologic and geomorphologic development with wide presence of Pliocene and Quaternary carbonate rocks that were modified mainly by karst processes. The southwestern portion of this area, managed as a national park, is formed by two flat peninsulas with playas and coastal cliffs, characterized by the typical diente de perro (dog’s tooth) karst surface, whereas the northeastern portion is characterized by swamps, wetlands and lagoons. The karstification of this area is intense and characterized by dissolution and collapse forms represented by the development of dolines, cenotes, extensive karrenfield and caves (Díaz Guanche et al. 2013).

Geologic maps of Cuba. a allochtonous terrane of Western Cuba and belts subdivision of the Cordillera de Guaniguanico (modified from Pardo 2009). b distribution of the sampling sites in the Guanahacabibes Peninsula. For the sake of simplicity and considering the lithological similarity between Vedado, Jaimanitas and Cayo Piedra, only the Guane and Vedado formations are represented. Symbols represent the chemical classification. See text and Fig. 2 for details
The investigated area is subdivided into four geologic formations (Cabrera and Peñalver 2001; Denis and Díaz Guanche 1993; Díaz Guanche et al. 2013; Hernández et al. 2013): (a) Vedado Formation, consisting of cream-colored to white massive marly limestone of biogenic origin (reef), constituting the main nucleus of the emerged area of the peninsula and dating from the Upper Pliocene until the lower Pleistocene. (b) Jaimanitas Formation, consisting of limestone and biodetritic coral reef, lying on the training Vedado, occupying the first marine terrace and dating from the Middle Pleistocene age. (c) Cayo Piedras Formation, consisting of the middle biocalcarenitic of the retro-reef environment, also corresponding with the Versalles-Guanabo (carbonatic) and Villaroja (terrigenous) Formations, and dating from the Middle Pleistocene age. (d) Guane Formation, consisting of conglomerates, gravels, sands and sandy clays, weakly cemented by clays, showing indefinite lenticular and more rarely inclined stratification deposited in a sea with abundant alluvial contribution, that could be estuarine, measures up to 50 m in thickness and can be interdigitated with the Vedado Formation, and dates from the Upper Pliocene-Lower Pleistocene age. Considering the lithological similarity and their distinctness only at local scale, the Vedado, Jaimanitas and Cayo Piedra formations were unified in Fig. 1.
Two main hydrogeological units are recognized in the area (Peláez García and González Cabrera 2013). The first unit is called the Neogene–Quaternary complex and corresponds to the Guane aquifer of Cabrera Bermúdez et al. (2004). The complex extends broadly through the whole area in association with the carbonate formations (Vedado, Jaimanitas), and it can reach a total thickness of 750 m and be strongly karstified in the upper 300 m. The underground waters of this aquifer are fundamentally of the chloride–sodium type, due to the marine intrusion with a salinity that oscillates between 1 and 5 g/l and chlorides that may vary from 500 to 800 mg/l. At isolated points of the peninsula, lenses of Ca-bicarbonate freshwater with a salinity of 0.6–0.8 g/l and chlorides between 90 and 290 mg/l overlie on the salty waters. The lagoon and mangrove deposits, located along the north coastal belt and in some inner spots as the case of Los Remates Swamp, control the regime of groundwater flow in the northeastern part of the aquifer (Hernández et al. 2013). The second hydrogeological unit is the Upper Jurassic–Lower Cretaceous “Guasasa complex”. It is composed by the gray karstified and cracked limestones of the Upper Jurassic that forms the karst denuded low mountains relief in the Sierra de Los Organos where the hydrographic Cuyaguateje Basin is located. It includes the Guasasa Formation (micritic limestones, calcareous sandstones and flint lens) and the Guaniguanico Basin–Los Organos Sub-Basin of Cabrera Bermúdez et al. (2004). There are no outcrops of this unit in the studied area because they were buried. However, it seems that waters emerge as fresh submarine discharge in the southwestern part of the Guanahacabibes Peninsula; therefore, a relation with the large karst springs located in the Sierra de Los Organos has been proposed (Peláez García and González Cabrera 2013).
Water sampling, analysis, and calculations
Samples’ description
From 22 June 2013 to 27 August 2013, 62 water samples were collected at the locations shown in Fig. 1b. The samples included seawaters from four locations (Los Cayuelos, slc; Bahia Guadiana, sbg; Maria La Gorda, sml; Bahia Cortes, sbc), four waters from karst caves (Quintana, cq; Roncali, cr; Lázaro, cl; Julia, cj), four surface waters from swamps (Ciénaga of Los Remates, LR1 and LR2), lagoons (Laguna El Pesquero, LP) and river (Cuyaguateje, rc). The remaining samples were gathered from 25 water wells, all distributed in the Sandino municipality. Of these, three to five samples were collected from the #18, #33 and Pacheco (p) wells at certain depths with respect to the elevation of the piezometric level (meters with respect to the mean sea level).
Field and laboratory methods
At the sampling site, temperature, electrical conductivity (at 25 °C), pH (at 20 °C), Eh (referred to the standard hydrogen electrode SHE) and CO2 measurements were performed using a multiparameter meter. In the laboratory, carbonate alkalinity (cAlk as HCO3) was determined by titration using the HCl and dye indicator method. The concentrations of major ions (Ca, Mg, Na, K, Cl, NO3, NH4 and SO4) and certain trace constituents (Sr, Li, F, and NO2) were determined by ion chromatography using a DIONEX ICS-1000 instrument with a DIONEX AS40 automated sampler. For the determination of cations, the samples were acidified to pH < 3 using concentrated HNO3 (65 % Suprapur®, Merck). In samples with a rotten-egg odor (e.g., at well #33), the level of total reduced sulfur as HS was determined using the photometric method (standard method 4500-S2− D; Eaton et al. 1995). The accuracy of the analyses was verified based on the ionic balance. For the spring water and groundwater, analyses with an ionic balance of ±5 % were considered acceptable.
The oxygen and hydrogen stable isotope composition of the water molecule was analyzed using the water–gas equilibration method. Briefly, water samples were transferred to a Finnigan GLF 1086 automatic equilibration device, online with a Finnigan DELTA-plus mass spectrometer (Laboratorio di Geochimica Isotopica at the University of Parma, Italy). The ratio measurements 2H/1H were obtained on pure hydrogen after the H2(g)–H2O(l) equilibration using a catalyzer (Boschetti et al. 2005), whereas 18O/16O was measured on carbon dioxide after CO2(g)–H2O(l) equilibration (Epstein and Mayeda 1953), both at 18 °C. Both ratios results were recorded in delta per mil (δ ‰) relative to the V-SMOW (Vienna Standard Mean Ocean Water) standard, thus obtaining the δ18O and δ2H values. The analytical precision was within ±0.2 ‰ for oxygen and ±1 ‰ for hydrogen.
Calculations of activity and saturation index
The saturation indexes (SI) of the carbonate minerals calcite, aragonite and high-Mg calcites (HMC) were calculated on the basis of the complete physico-chemical analyses of the sampled water. Furthermore, saturation indexes resulting from the theoretical mixing of Na-chloride seawater with fresh Ca-bicarbonate groundwater were also calculated. During the mixing, the increasing salt concentration of the solutions produces higher ionic strengths (IS), thus altering the activities of the dissolved species and the calculation of SI (e.g., Appelo and Postma 2007; Langmuir 1997). Moreover, different equations for the calculation of the activity coefficients are available, which may produce different results on activity and consequently on SI (Bethke 2008). Therefore, the following software, equations and thermodynamic databases to calculate the activity coefficients and SI were used and compared: (a) PHREEQCI version 3.0.6 (Parkhurst and Appelo 2013) for the Debye–Hückel/Davies, Truesdell-Jones, SIT (Specific Ion interaction Theory) and Pizter approaches to the activity coefficient calculations and the thermodynamic datasets llnl.dat, phreeqc.dat, wateq4f.dat, sit.dat and pitzer.dat and (b) the Geochemist’s Workbench version 7 (Bethke and Yeakel 2008), for the B-Dot equation with the dataset thermo.dat and Pitzer parameters with the datasets and hmw.dat and phrqpitz.dat. In addition, EQ3/6 version 8 (Wolery and Jarek 2003) and the B-Dot equation for activity coefficient calculation supported by the data0.cmp thermodynamic dataset were used to calculate the SI after the formation of an ideal solid solution for CaCO3–MgCO3 resulting from the freshwater–seawater mixing. Differently, in the above described (a) and (b) models, the Mg-calcite’s saturation indexes of solutions resulting from mixing and containing a mole fraction x of Mg-carbonate were calculated as following (Glynn and Reardon 1990):
where IAPMg–calcites is the ion activity product of the solution and K Mg–calcites the solubility constant of the HMCs. For these latter, 12 % < x < 14 % were considered in accordance with the typical values found in tropical carbonates (Agegian and Mackenzie 1989; Garrels and Wollast 1978; Perry et al. 2011), whereas −logK Mg–calcites of 8.42 and 8.25 at 25 °C and 1 bar for synthetic(inorganic) and biogenic were chosen, respectively (Morse and Mackenzie 1990).
Chemical composition
Results and main characteristics
The chemical composition of the sampled waters is shown in the Electronic Annex 1. The mean temperature of the samples, T = 27.3 ± 1.6 °C, agreed with the local air temperature of the summer period (25 °C). Considering the calculated total dissolved solids (TDS; Eaton et al. 1995) and the salinity classification by Robinove et al. (1958) that use this parameter, all of the groundwaters collected from the surface of the water table were fresh because they had TDS <1 g/l, but two slightly saline (1 < TDS < 3 g/l) samples from #66 (TDS = 1.1 g/l) and Mella (me, TDS = 1.7 g/l) wells. Moreover, the TDS increased with the sampling depth in the wells explored at different depths. For example, at the bottom of the wells #18 and #33, the TDS increased toward the highly saline range (10 < TDS < 35 g/l), thus approaching the mean seawater value (35 g/l), whereas the TDS in the Pacheco well remained in the freshwater range. Water from the well at Roncali lighthouse (Faro Roncali, fr) showed the highest TDS value, similar to that of the seawater, both at the surface and the well bottom. Excluding this sample, all of the water sampled at the surface of the water table showed a mean pH of 7.43 ± 0.31, which is similar to the mean value of water from the Cuyaguateje Basin (Fig. 1; 7.00 ± 0.86; Fagundo et al. 1986). Water samples from the wells explored in depth showed values that scattered not only between this values and that of the local seawater (pH = 8.53 ± 011), as expected for seawater intrusion (well #18), but also values lower than 6 (well #33). Therefore, in this latter case, more complex phenomena than the pure mixing must have occurred.
Evaluation of seawater ingression and related effects
After converting the anion and cation concentration data of the major chemical constituents from mg/l to meq/l, the four following compositional facies, listed from the least to the most salty, could be distinguished in the sampled waters (Electronic Annex 1): six Ca-bicarbonate freshwaters (well #3; Rio Cuyaguateje, rc; Laguna El Pesquero, lp; RGB29 well, #29; Ngouabi well, ng; Cueva Roncali, cr); two Ca-chloride freshwaters (well Quintana, q; well #60) and two Na-bicarbonate freshwaters (Pacheco well at the surface of the water table, p; Abasto Maria La Gorda P1, mlg1). All of the TDS of the Ca-bicarbonate and Ca-chloride waters were 0.1 < TDS < 0.5 g/l, which are excellent to good waters for irrigation and drinking purposes in terms of salinity (CASS 2003; WHO 2011), but the water quality deteriorates when approaching to the Na-chloride composition passing through Na-bicarbonate. Because of the swelling effect in clays, a sodium excess in water can produce a decreased permeability with negative repercussion on the normal water supply and aeration required for plant growth. Therefore, the sodium adsorption ratio is another parameter widely used to classify irrigation waters (SAR, Appelo and Postma 2007; Electronic Annex 1). However, using waters classification by this parameter, only 4 samples picked up at water table surface showed SAR value above its critical limit SAR >10, which correspond to a TDS > 1.4 g/l (me, #18, fr wells and Los Remates LR2 swamp). Whereas the Na-chloride is the typical chemical facies resulting from the pure mixing with the seawater in a coastal karst environment, both the Ca-chloride and Na-bicarbonate are other two recurrent facies resulting from cation exchange in this type of environment. Ca-chloride is the result of seawater intrusion in an aquifer with Ca-bearing sediment (Ca2+ released by soil exchanger, Cl− from seawater), whereas the latter occurs when freshwater flushes a salt water aquifer with Na-bearing sediments (Appelo and Postma 2007; Jones et al. 1999). These effects are suitably shown in the diamond of the Piper diagram (Fig. 2), where freshening and intrusion cation exchange effects depart from the mixing line connecting the Ca-bicarbonate freshwater corner and the saline seawater point. A pure mixing effect is shown for well #18 down to a depth of 10 m (Fig. 2b), whereas exchange effects after seawater intrusion can be hypothesized in well #33 down to 20 m (Fig. 2c). The Pacheco well samples are scattered in the freshening area, probably due to a secondary mixing (Fig. 2d). The presence of freshwater very close to the coastline, as occurred for the Na-bicarbonate water at Maria La Gorda and Ca-bicarbonate water at Cabo San Antonio promontories, might represent the effect of a lens of freshwater floating on a deeper salty one according to the typical salt-wedge effect (Custodio 1987; Sáinz et al. 2010). Also, calcite dissolution–precipitation and sulfate reduction could be potentially visualized on the Piper plot (Fig. 2; Appelo and Postma 2007), but the discernment is hard when more effects are combined. Moreover, the interpretation could be complicated further by additional processes (secondary mixing, sulfide and organic matter oxidation). Finally, the sum of the variables that characterize this diagram does not help the purpose. Therefore, the additional effects were searched using different variables and diagrams.

Piper’s diagrams where waters sampled in the Guanahacabibes Peninsula are plotted using major chemical composition (mg/l). The chemical classification of the waters (symbols in legend) was obtained using the sample chemical concentration in meq/l. Arrows depict the paths due to some processes (Appelo and Postma 2007). The transitional compositions Ca-chloride and Na-bicarbonate, which represent the switch from Ca-bicarbonate to Na-chloride, are highlighted by the dashed area. Well #18, #33 and Pacheco explored at certain depths are shown in b, c and d, respectively (numbers: sampling depth in meters with respect to the mean sea level)
The use of the conservative constituents Br and Cl provided further confirmation that mixing between the two end members was the main salinization process because the plotted samples depict a distinct straight line that joins freshwater and seawater (Fig. 3). The only exception concerns the swamp sample LR2 which showed a halogens enrichment, most likely due to the dissolutions of halite in an evaporitic environment (Fig. 3). Chloride can be also used as follows to calculate the theoretical seawater fraction (f) in a mixed sample, assuming conservative mixing between freshwater and seawater (Appelo and Postma 2007):

Bromide vs. chloride (in meq/l) diagram. Symbols and dashed area as in Fig. 2
where Cls, Clfw and Clsw are the chloride content, in mol/l or meq/l, of the sample, the local freshwater and the seawater, respectively. For Clfw and Clsw, the average content in the Cuyagateye Basin (0.621 mol/l; Fagundo et al. 1986) and of the four local seawater sampled in this study (583 mol/l) were used, respectively. Most of the groundwater samples showed 0.00 < f < 0.04, except the coastal part of LR2 (f = 0.28) and fr well (f = 0.86). According to Hernández et al. (2013), also #18, #33 and Pacheco wells have f < 0.04 at depths between 5 and 15 m. The f value gradually increased up to 0.1 at depths of 16–25 m; below this depth range, the f value increased abruptly and quickly toward that of pure seawater (f = 1) estimated at 34–42 m (Fig. 4).

Sampling depth (in meters with respect to the mean sea level) vs. seawater fraction f diagram (see text for details on f calculation). Wells explored at certain depths are plotted
The intrusion and freshening effects in the wells monitored at different depths are also effectively shown on binary diagrams that plot chloride and major cations (Fig. 5). In particular, the samples from well #33 showed Ca enrichment and a Mg, Na and K depletion compared with the curves that represent binary mixing between seawater and freshwater or rainwater on which well #18 samples are clustered. The water from Pacheco well showed a Na-chloride composition at depth, but Na-bicarbonate at the water table and more shifted toward rainwater, probably due to the rain washing of soil’s clay having exchangeable sodium. The greater diversity among the fresh groundwaters and the eastern Cuba (Rodríguez et al. 2004) and Caribbean (Cerón et al. 2005) rainwaters shown in the Cl–Ca plot (Fig. 5a) is explainable by the high variability in the Ca concentration of the Ca-bicarbonate waters coming from the Sierra de Los Organos and Rio Cuyaguateje, whose lower values are quite similar to those of western Cuban rainwater (Molerio León 1992, 2012).

Binary diagrams plotting calcium (a), sodium (b), magnesium (c) and potassium (d) vs. chloride concentration (all in meq/l). Symbols and dashed area as in Fig. 2. Main concentration in the rainwater samples (Western Cuba WC, Molerio León 1992, 2012; Eastern Cuba EC, Rodríguez et al. 2004; Central America CA, Cerón et al. 2005), Cuyaguateje River (light gray area, Fagundo et al. 1986) and Sierra de Los Organos (dark gray area, Fagundo et al. 1986) are also showed for comparison. Binary mixing between local seawater and CA-rain or Cuyaguateje River are represented by dashed lines
The mixing of freshwater and seawater, which are both saturated with calcium carbonate, can result in water that is undersaturated with calcium carbonate; that is, calcite dissolution may occur (Richter and Kreitler 1993). In the Ca vs HCO3 and SO4 vs Cl diagrams (Fig. 6), the calcium excess and sulfate deficit of the well #33 samples could be explained by additional calcium carbonate dissolution occurring in the presence of bacterial sulfate reduction in organic-rich sediments. In fact, the sulfate decline shown by the well #33 occurred at a chloride content that could not be explained by dilution (Fig. 6a). In this scenario, bicarbonate should be increasing according to the following reaction:
and this is confirmed by the presence of dissolved sulfide (up to 1.8 mg/l). However, the bicarbonate unexpectedly decreased most likely because of its conversion to CO2 (up to 504 mg/l) caused by an extremely low pH (down to 5.9), both measured in well #33 at depths down to 30 m. The low pH may have resulted from the oxidation of previously deposited sulfides. Therefore, starting from the groundwater sampled at a shallow depth, which showed high HCO3 most likely because of the presence of organic matter, and continuing to deeper depths, a Ca increase preceded a HCO3 decrease that followed a negative exponential curve fitted to the samples (Fig. 6b). After a steep increase up to 44 meq/l of Ca caused by carbonate dissolution, the Ca concentration of the groundwater decreased at well bottom to resemble seawater, which is consistent with a deep ingression in a manner similar to the previously described “deep regime” intrusion (Back 1966; Custodio 1987). Finally, note that the sulfate-reduction process was also revealed in the neighboring Laguna del Valle de San Juan (Pérez and Pubillones León María 1988).

Binary diagrams plotting sulfate vs. chloride (a) and calcium vs. bicarbonate (b) concentration (all in meq/l). In a symbols as in Fig. 5; moreover, depth sampling in #33 well is highlighted (numbers: sampling depth in meters with respect to the mean sea level). In b dashed lines group samples from the same sampling site. Moreover, calcite and dolomite dissolution (dashed-dotted lines) and seawater mixing following bicarbonate loss after reduction or acidification and CO2-degassing (bold curve with arrow) are shown
Mixing modeling: results and discussion
The results concerning the carbonates saturation indexes (SI) of the samples collected in this study are in the Electronic Annex 2, here discussed and compared with the results of the freshwater–seawater mixing modeling.
Concerning the models, the mean composition of the four local seawater (SW) samples and the Cueva Roncali (FW) sample were used as end-members. The latter was chosen because it has a bicarbonate content similar to that of the surface sample of well #33, but a negligible fraction of seawater typical of the Ca-bicarbonate freshwaters. The discussion and comparison of the results with the real samples in terms of SI follow a technical focus on SI errors, dedicated to the modeler specialists.
Error on saturation index (SI): activity calculation and charge-balance effects
If an error of ±0.2 on SI is considered as acceptable, all of the model’s results were comparable when SIcalcite and SIaragonite were plotted against chloride during FW–SW mixing, except for the Truesdell-Jones model using phreeqc.dat (Fig. 7). Regarding this exception, quite different results on SI were obtained if the input data of the end-members composition included reduced species (e.g., ammonia) without the oxidized counterparts (nitrite, nitrate). In particular, the sampled SWs had a high ammonium content (85.5 mg/l), comparable with that observed in coastal interstitial waters within sediments rich in organic matter (Matisoff et al. 1975). However, in our case, the nitrate data are lacking because they are not measured or below the detection limits. Under these conditions, the error in the calculated SI after mixing is due to the forced charge balance, automatically achieved by the software on both the redox potential (pe or Eh) and the pH. Mass-action and mole-balance equations are written in terms of activity; therefore, the final error in the SI also depends on the equation used for the calculation of the activity coefficients (γ). In fact, the obtained SIs were worst and quite low for the models whose activity coefficients were obtained not only by the Debye-Hückel approaches, as expected, but also for the Truesdell-Jones model (Fig. 7). Differently, the SIs obtained by the SIT modeling were slightly higher than the mean model at approximately 0.2–0.8 of the seawater fraction (Fig. 7). Thus, the results from the different models can be considered always coherent between them when SW fraction ranges from 0.0 up to 0.1. In that range, calcite and aragonite are always undersaturated with SIcalcite = −0.26 ± 0.06, SIaragonite = −0.38 ± 0.10 and SIcalcite = −0.4 ± 0.2, SIaragonite = −0.57 ± 0.07, respectively. Above SW of 0.1, the SIcalcite and SIaragonite show abrupt decrease (up to −3.47 and −3.61 at 0.99 SW, respectively) if activity is calculated also for reduced species, in particular when γ was obtained by Debye-Hückel approach and llnl.dat. This path combines the different effects of the activity coefficient equation form (pure coulombic ion interactions, ion size does not vary with IS, and ions of the same sign do not interact; e.g., Langmuir 1997) with the increased H+ activity due to the forced charge balance. Therefore, it is evident in this case that the equation is inadequate to describe the mixing for seawater fractions above 0.1. In fact, we have IS = 0.083 for SW = 0.1 and IS = 0.153 for SW = 0.2. The Truesdell-Jones equation for the calculation of activity coefficients (Parkhurst and Appelo 2013) was also employed in the mixing models with reduced species (Fig. 7). In this case, calcite and aragonite are again always undersaturated, with results comparable to the other models at SW <0.1, whereas at higher seawater fraction a gentle decrease is shown up to SIcalcite = −0.72 ± 0.12 and SIaragonite = −0.76 ± 0.07 (mean and standard deviation between the employed wateq4f.dat and phreeqc.dat datasets). From a theoretical point of view, the Truesdell-Jones equation should be better than Debye-Hückel for salty solution because the former may explain NaCl solution up to IS = 2. However, SW is more complex in composition than a pure NaCl solution; therefore, the error also increases in this case at higher SW fraction and H+ activity. Therefore, if only the mixing models calculated from complete and consistent end-member composition were considered (i.e., using only the major dissolved components), the obtained mixed waters were oversaturated with respect to calcite or aragonite when the SW fraction was higher than about 0.60 or 0.65 (0.7 if the Truesdell-Jones equation and phreec.dat are used), respectively.

Mixing modeling results in terms of saturation index of calcite (a) and aragonite (b). Results were obtained using different software and thermodynamic databases (see text for details). The physico-chemical composition of the end-members comes from this study: Cueva Roncali for Ca-bicarbonate freshwater (FW) and local mean seawater (SW). The most of the models produced similar results (dark gray area with SI <0.2) when only major composition of the end-member was used in the calculations but Truesdell-Jones with phreeqc.dat. Differently, other models shifted from this “coherence area” when other minor ions (e.g., ammonia), that influenced pH-Eh during the forced charge balance, were considered (dotted-dashed lines)
High-magnesium calcites (HMC) and carbonates saturation index
The local mean SW is oversaturated both in calcite and aragonite, SIcalcite = 1.17 ± 0.04 and SIaragonite = 1.01 ± 0.05, respectively. To overcome the problem of the inhibition effect caused by magnesium on carbonates precipitation, the saturation state of the mixed solutions with respect to HMC was first calculated assuming a pure solid solution between CaCO3 and MgCO3 (EQ3/6, the B-dot model for activity coefficient calculation and the data0.cmp dataset). Using this model, mixed waters start at equilibrium or are slightly undersaturated in HMC (SIHMC = −0.16; x MgCO3 = 5 %). The more seawater fraction is added, the more the HMC saturation state and the MgCO3 fraction increase, reaching oversaturation from approximately 0.35 mixed solutions, up to SW with SIHMC = 1.66 (x MgCO3 = 68 %). Note that the modeled MgCO3 content in seawater is in accordance with the maximum detected content in fish-produced tropical carbonated (Perry et al. 2011). However, the obtained MgCO3 fractions at oversaturation are too high to represent the typical natural HMC, in which the average x MgCO3 content in tropical marine carbonate sediments and marine inorganic cements is approximately 13 % (Perry et al. 2011). In fact, the saturation indexes obtained in the real seawater samples and the oversaturated mixed solutions of the modeling using the logK for synthetic and biogenic HMC at that percentage are similar to pure calcite and aragonite, respectively (Fig. 8). It should be noted that the difference starts to be significant at SW <0.1 in the models (Fig. 8b) and for the undersaturated samples (Fig. 9), the SIHMC of which is lower up to 0.2 in comparison with pure Ca-carbonates. The mean SIs calculated for Ca-bicarbonate (SIcalcite = −0.27 ± 0.28; SIaragonite = −0.43 ± 0.28) and Ca-chloride waters (SIcalcite = −0.19 ± 0.21; SIaragonite = −0.35 ± 0.28) sampled in this study are similar and slightly undersaturated in calcite and more undersaturated in aragonite, whereas the Na-bicarbonate water sample mlg1 is oversaturated in both (Figs. 8, 9). The calculated mean SI of the Na-chloride waters at low SW fractions <0.02 (chloride content <500 mg/l or 0.014 molality) shows that these groundwaters are on average in equilibrium with calcite (SI = −0.09 ± 0.35) but undersaturated in aragonite (SI = −0.25 ± 0.35). Generally, at that low chloride content, the standard deviation of both CaCO3 phases spans between oversaturation and undersaturation, quite similar to the Ca-bicarbonate samples at Cuyaguateje Basin and Sierra Los Organos (Fig. 8b). At SW fractions >0.02, the mixed waters show more extreme variations in the carbonates SI values, in particular the opposite saturation features of wells #18 and #33, which were both sampled at various depths. Especially, at the added SW fraction of 0.1, the groundwater showed oversaturation at well #18 but undersaturation at well #33 (Fig. 8a). For the well #18 groundwater, the carbonates oversaturation was most likely due to the CO2 degassing and/or re-equilibration in the soil environment having a lower CO2 content. In fact, the lowest calcite–aragonite saturation indexes, predicted at SW fraction of 0.1, should occur at pH = 6.85 and logPCO2 = −1.53, whereas the well #18 groundwater at a similar chloride content (−21.5 m sampling depth) had pH = 8.00 and logPCO2 = −2.86. For the well #33 groundwater, and according to Corbella and Ayora (2003), an additional effect such as the organic matter decomposition as in reaction (5), should be invoked to reach the extremely low carbonate saturation indexes (SIcalcite = −2.32; SIaragonite = −2.48). This is confirmed using the REACT tool combined with the thermo.dat dataset of the Geochemist’s Workbench software, which produced a similar calcite saturation index and pH when approximately 0.35 mol of aqueous CH2O was added to the theoretical mixed solution obtained at SW = 0.1.

Results of the freshwater–seawater mixing modeling at the Peninsula of Guanahacabibes in terms of saturation index (SI) of pure calcite (dark gray), pure aragonite (light gray), inorganic Mg-calcite (bricked wall field), biogenic Mg-calcite (squamose field). The continuous curve represents a MgCO3–CaCO3 ideal solid solution as foreseen by EQ3/6 and B-dot activity coefficient calculation. Only calculation models with similar and coherent results were used (dark gray areas in Fig. 7). Globally comparable SIs were obtained between pure calcite and inorganic HMC and between pure aragonite and biogenic HMC; therefore, the SI of the pure Ca-phases of the waters sampled at Peninsula of Guanahacabibes is shown (closed symbols pure calcite, open symbols pure aragonite, symbols shape as in Fig. 2; see also Fig. 9). However, at SW <0.1, the SIHMCs are approximatively 0.15 lower than SI of the pure Ca-carbonates. In (b), the median saturation index for calcite and the median absolute deviation (error bars) in the waters from Sierra de Los Organos (LO, Fagundo et al. 1986) and Cuyaguateje River (CB, Fagundo et al. 1986) are shown for comparison

Comparison between synthetic high-magnesium calcite vs. pure calcite (a) and organic high-magnesium calcite vs. pure aragonite (b) saturation indexes of the waters sampled at Peninsula of Guanahacabibes (data in the electronic annex). The best-fit equations of the samples (bold) and 1:1 lines (dashed) are also shown
Isotope composition
Discriminating between seawater ingression and evaporation
To critically analyze the obtained isotope results (Electronic Annex 1), the sample waters were compared with global (GMWL) and local (LMWL) meteoric water lines (Fig. 10a). For the former, the regression equation of the long-term mean data weighted by the amount of precipitation was given by the following equation (Rozanski et al. 1993):

a hydrogen (δ2H) vs. oxygen (δ18O) water stable isotope composition, both values in ‰ vs. V-SMOW. GMWL global meteoric water line (Rozanski et al. 1993); LMWL local meteoric water line (this study); CMWL cuban meteoric water line (Molerio León 1992, 2012). Mixing and evaporation effect are also shown (dashed-dotted and dotted lines), with slope explicated for these latter (S). Symbols as in Fig. 2, except for waters from kart’s caves (gray). Evaporated (esw) and non-evaporated seawater (nesw) are highlighted. b Chloride (mg/l) vs. hydrogen (δ2H) vs. oxygen (δ18O) isotope composition. Symbols as in (a). c Altitude (meters a.s.l.) vs. oxygen isotope composition (δ18O). In spite of the exponential trend of GNIP’s station data and neighboring karst springs, we obtained a recharge area coherent with the local reliefs using the linear elevation-18O gradient as calculated in this work
For the LMWL, the isotope data for the precipitations were chosen from the following four stations of the Global Network of Isotopes in Precipitation (GNIP; IAEA/WMO 2013): El Salto (37 m asl), Finca Ramirez (170 m asl) in the Pinar del Rio Province; Santa Ana-Santa Cruz (80 m asl), Ciro Redondo (480 m asl) in the near northeastern portion of the Artemisa Province. The least square fit was calculated as follow for all of the unweighted monthly mean data but one outlier (Santa Ana-Santa Cruz from the 15 November 1983):
Other authors obtained different equations using the same data from the same stations (Arellano et al. 1989, 1992). Despite this discrepancy and the fact that fragmentary data were available for these stations (from June to September of the 1983 and from March to November of the 1984), our equation is quite similar to the following one obtainable for Cuba from the weighted monthly mean data of Molerio León (1992) (CMWL in Fig. 10a):
The main isotopic characteristic of the waters sampled in this study was their displacement to the right of the meteoric water lines, with all but one (Cienaga Los Remates P1, LR1) roughly arranged along a straight line with lower slope (Fig. 10a). Considering that wells #18 and #33 were heavily intruded by seawater during sampling, as previously verified by chemical data, and that they were more shifted toward seawater in comparison with most of the other freshwater samples (Fig. 10a), the straight line may represent a mixing effect. However, according to Gonfiantini and Araguas (1988), for Caribbean areas the mixing effect may be confused with evaporation in this kind of graph. In fact, note that the most isotopically enriched samples were represented by the El Pesquero lagoon (LP) and the swamp LR1, which had a negligible seawater contribution. This problem may be solved by combining the water isotope data with data from a conservative element, such as chloride, in solution (Fig. 10b). In these plots, the mixed groundwaters from wells #18 and #33 are readily discriminated from the evaporated ones from LP and LR1 sites. Additionally, the groundwater of the Pacheco well (p) showed an isotopic enrichment at greater depth, but at moderate chloride content (Fig. 10b). This result was most probably due to the ingression of the waters from the neighboring El Pesquero lagoon, as suggested by the shift toward the LP sample in the plot (Fig. 10b). Furthermore, the seawater samples from Guadiana and Cortés bays (evaporated seawaters; esw in Fig. 10a, b) demonstrated an isotope enrichment due to evaporation in comparison with seawater from Cabo San Antonio and Maria La Gorda samples (unevaporated seawaters, nesw in Fig. 10a, b), which showed a mean isotope composition more similar to those of the Caribbean Sea (δ18O = + 0.84 ‰, δ2H = + 6.6 ‰; Gonfiantini and Araguas 1988).
To calculate the slope of a line describing the local evaporation effect, the isotope composition of the local atmospheric moisture (water vapor) is a key parameter that should be known. By default, the isotope composition of the vapor is often estimated by proxy, assuming isotopic equilibrium with the local precipitation. While this approximation may be acceptable during rainy periods in a continental setting, it is unsatisfactory in the vicinity of an evaporative source, such as a lake sea, and not during prolonged dry periods (Gat 2010). Because the isotope composition of local atmospheric moisture is unknown, we attempted to calculate the evaporation slope from our experimental data, that is, with the least square fit between the evaporated and enriched samples and their possible unevaporated analogs, arranged near the water lines, and identifiable as their meteoric recharge. For example, the Laguna El Pesquero is hydrologically connected by channels to the Cuyaguateje Basin, and the obtained slope from the best fit between their isotope data is approximately 5.9. Furthermore, all the four water samples from karst caves are displaced on a straight line, the linearity and slope (5.1) of which were tested for statistical significance by Fisher’s and Student’s test, respectively. Adding to this regression, the data of the LR1 sample, the linearity and slope maintain their significance, with the slope showing a slight decrease to 4.8 but measuring 4.4 when calculating a best fit between LR1 and the Cuyaguateje River sample data. These latter values are more similar to the evaporation slope of 4.6 revealed for Cuba by other authors (Molerio León 1992, 2012; Peralta Vital et al. 2005). However, the obtained slopes of the samples from the karst caves and Cuyaguateje basin agree better with the 5 and 6 theoretical slope range for the water surface evaporation at the mean local relative humidity of 75 % and T = 25 °C (Gonfiantini 1986; Xinping et al. 2005); therefore, the slope values <5 may be attributable to an evaporative effect from soil and/or shallow groundwater (Gat 2010).
Meteoric recharge estimation
All of the water samples that were not located on the seawater mixing or evaporation lines stayed between these lines and showed a mean deuterium excess of 6.4 ± 0.6 (Fig. 10a). This value is less than those of the meteoric water lines, indicating that these waters falling between the aforementioned evaporation lines suffered a moderate evaporation. The highest isotope values of this water group were consistent with those measured in the Cuyaguateje River (δ18O = −3.62 ‰; δ2H = −22.3 ‰), which in turn agreed with the mean values for the rainy period from May to October (δ18O = −3.46 ± 0.62 ‰; δ2H = −19.3 ± 3.8 ‰; Molerio León 1992, 2012). These higher values should also correspond with the meteoric recharge to the well #18, as shown by extrapolating back from the chloride isotope data of this well (Fig. 10b). Therefore, the northwestern sector of the Guanahacabibes Peninsula is most likely fed from a shallow circulation on the karst formation.
In the same manner, note that the lowest water group values coincided with the intercept between the line fitting well #33 and the caves samples and the meteoric water lines, which occurred at approximately δ18O = −5.3 ‰. A similar value was shown also by well #3 (δ18O = −5.34 ‰) and the Abasto Maria la Gorda well (δ18O = −5.31 ‰). To calculate the elevation effect on the meteoric recharge of groundwater, the meter-δ18O equation of Molerio León (1992) could be used, here rewritten as:
This equation partially coincides with that obtained from the local GNIP’s station data as reworked and reevaluated by Arellano et al. (1992; Fig. 10c). Using this equation to calculate the local elevation effect, we obtained a corresponding recharge elevation of approximately 700 m for the Cuyaguateje River and over 1,000 meter for the Maria La Gorda transect samples, which are well over the real and maximum height of the basin and of the Cordillera de Guaniguanico, respectively. In fact, the Cuyaguateje Basin is characterized by low, quite irregular mountains, with average elevations between 350 and 400 m and residual limestone hills named mogotes. By contrast, the highest elevation of the Cordillera lies in the westernmost sector of the Province between the Doming Fault and the Northern Rosario belt (Pico Pan de Guajaibon, 700 m asl). When we tried as an alternative to obtain a better elevational gradient equation using the GNIP’s station data as reevaluated by Arellano et al. (1989) plus some karst springs from the St. Cruz River basin in the Pizarras del Sur and Cangre belts (Arellano et al. 1989, 1992), we obtained the following equation (Fig. 10c):
Note that the raw sample data showed a non-linear, exponential elevation-δ18O path, as revealed in other data from Central and South America (Rozanski and Araguás-Araguás 1995). This pattern may be attributable to the “shadow effect” on the moistures played by the continental Mexican and eastern Cuban reliefs. Despite the poor linear correlation coefficient, the elevation estimated using the equation [10] and relating to the δ18O of the Cuyaguateje River is coherent with the average river elevation. Differently, the extremely low δ18O values measured in the groundwaters of the Maria La Gorda transect could be influenced by the elevation effect on the rains in the Cordillera de Guaniguanico (Fig. 10c). This supposition is consistent with the hypothesized connection between the fresh submarine discharges in the Maria La Gorda area and the large karst springs located in the Cordillera (Peláez García and González Cabrera 2013). However, we cannot exclude the possibility that the observed, quite low isotope values are also related to the rapid infiltration in the deeper endokarstic conduits by abundant precipitations with more negative isotope composition during the periods of heavy rainfall, that are typical for the tropical, monsoonal climate of the area (Florea and McGee 2010). These hypotheses necessitate the collection of isotope data throughout a complete hydrological year for confirmation.
Conclusions
The groundwaters of the Guanahacabibes Peninsula showed the typical chemical compositions related to seawater ingression. The fresh Ca-bicarbonate waters are mainly located in the northeasternmost sector of the peninsula, where the Cuyaguateje River issues into the area. More toward the southwestern sector, the shallow groundwater shows a mainly Na-chloride composition, but maintains a low seawater fraction (0.00 < f < 0.04) up to 20 m in depth and is promptly diluted by local rain. Waters with a Ca-chloride composition were detected in the wells just at south of the Los Remates swamp. This transitional composition, representing seawater intrusion, occurred where the Guane terrigenous fm. displays interdigitation toward the Vedado limestones fm. The groundwater in the southwestern plain of the Guanahacabibes Park area presented an abrupt salinity increase at a depth of 26–30 m, which corresponded to an added seawater fraction of 0.1. This value coincided with the lowest Ca-(Mg)-carbonate saturation index resulting from the mixing modeling. Generally, seawater and groundwaters with an added seawater fraction above 0.60–0.65 showed similar oversaturated indexes in HMC and pure Ca-carbonates (calcite and aragonite). However, in the groundwaters with a seawater fraction between 0.02 and 0.60 and that showed carbonates undersaturation, the saturation indexes in HMC were lower than pure Ca-carbonates of about 0.2. Moreover, the tendency of the waters to dissolve limestone is locally enhanced in organic-rich waters by sulfate reduction, contributing to the subsequent increase in the generation of the karst structures and the intrusion of seawater into the groundwater. This combined effect of Ca-increase and sulfate reduction may be misinterpreted in the Piper plot as cation exchange with clays. Unexpectedly, Ca-bicarbonate and the opposite transitional Na-bicarbonate, representing groundwater freshening, were detected also in the almost open-ocean conditions, respectively, in the south-westernmost Cabo San Antonio and Maria La Gorda promontories. These waters may be easily explained as a lens of freshwaters floating on a deeper salty layer, enhanced by flow inversion from sea tide effects and/or consistent with the upward freshwater flow during the formation of the saline wedge. However, the lower isotope composition indicated that these waters may come from the deep Jurassic aquifer, which is mainly recharged during the rainy period (summer) occurring in the highest reliefs of Cordillera de Guaniguanico. This hypothesis agrees with the extensive presence of freshwater discharge from submarine springs discovered along the coast by other authors.
References
Agegian CR, Mackenzie FT (1989) Calcareous organisms and sediment mineralogy on a mid-depth bank in the Hawaiian Archipelago. Pac Sci 43:55–66
Appelo CAJ, Postma D (2007) Geochemistry, groundwater and pollution, 2nd edn. A. A. Balkema, The Netherlands
Arellano DM, Feitoó R, Seller KP, Stickler W, Rauert W (1989) Estudio isotópico de la llanura costera sur, provincia Pinar del Río, Cuba. Isotope Hydrology Investigations in Latin America, IAEA-TECDOC-502. Austria, Vienna, pp 229–243
Arellano M, Molerio León LF, Suri Hijos (1992) A Efecto de altitud del 18O en zona de articulación de llanura cripto-karsitica con carso de montaña. In: Llanos HJ, Antiguedad I, Morell I, Eraso A (eds) I Taller Internacional sobre Cuencas Experimentales en el Karst, Matanzas, Cuba. Grupo de Trabajo Internacional Cuencas Experimentales Karsticas, Universitat Jaume I de Castelló, pp 29–42
Back W (1966) Hydrochemical facies and ground-water flow patterns in northern part of the Atlantic Coastal Plain. US Geological Survey Professional Paper 498–A
Back W, Hanshaw BB, Herman JS, Van Oriel JN (1986) Differential dissolution of a Pleistocene reef in the ground-water m1 xing zone of coastal Yucatan. Mex Geol 14:137–140
Bethke CM (2008) Geochemical and biogeochemical reaction modeling. Cambridge University Press, New York
Bethke CM, Yeakel S (2008) The Geochemist’s workbench release 7.0—Essentials guide. Hydrogeology program, University of Illinois, Urbana, Illinois
Boschetti T, Venturelli G, Toscani L, Barbieri M, Mucchino C (2005) The Bagni di Lucca thermal waters (Tuscany, Italy): an example of Ca-SO4 waters with high Na/Cl and low Ca/SO4 ratios. J Hydrol 307:270–293
Budd DA, Vacher HL (2004) Matrix permeability of the confined Floridan aquifer Florida, USA. Hydrogeol J 12:531–549
Cabrera Bermúdez J, Guardado Lacaba R, Peláez García R, González Cabrera N (2004) Regionalizacion hidrogeologica de la Provincia de Pinar del Rio er un SIG. Minería y Geología 1–2:24–31
Cabrera M, Peñalver LL (2001) Contribución a la estratigrafía de los depósitos cuaternarios de Cuba. Cuaternario y geomorfología, 15: 37–49
CASS (2003) Phase I Report—Technical appendix A: salinity and total dissolved solids. United States Department of Interior—Bureau of Reclamation, Central Arizona Salinity Study, Lower Colorado Region, Phoenix Area Office. http://www.usbr.gov/lc/phoenix/programs/cass/pdf/Phase1/ATechapdxTDS.pdf
Cerón RM, Cerón JV, Córdova AV, Zavala J, Muriel M (2005) Chemical composition of precipitation at coastal and marine sampling sites in Mexico. Global NEST J 7:212–221
Choquette PW, Pray LC (1970) Geologic nomenclature and classification of porosity in sedimentary carbonates. Am Assoc Pet Geol Bull 54:207–250
Cooke RC, Kepkay PE (1984) Apparent calcite supersaturation at the ocean surface: why the present solubility product of pure calcite in seawater does not predict the correct solubility of the salt in nature. Mar Chem 15:59–69
Corbella M, Ayora C (2003) Role of fluid mixing in deep dissolution of carbonates. Geologica Acta 4:305–313
Custodio E (1987) Hydrogeochemistry and tracers. In: Custodio E (ed) Ground-water problems in coastal areas. Studies and reports in hydrology no. 45. UNESCO, Belgium, pp 213–269
Denis R, Díaz Guanche C (1993) Características geologicas y geomorfologicas de la Península Guanahacabibes. Dirección Provincial de Planificación Física, Pinar del Río, Cuba
Díaz Guanche C, Aldana Vilas C, Farfán González H (2013) Mapping Groundwater Vulnerability in Guanahacabibes National Park, Western of Cuba. In: Farfán González H, Corvea Porras JL, de Bustamante Gutiérrez I, LaMoreaux JW (eds) Management of water resources in protected areas. Springer, Berlin, pp 87–94
Eaton AD, Clesceri LS, Greenberg AE (1995) Standard methods for the examination of water and wastewater—19th edition. American Public Health Association-American Water Works Association-Water Environment Federation (APHA-AWWA-WEF), Washington, (USA)
Emerson S, Hedges J (2003) Sediment diagenesis and benthic flux. In: Elderfield H, Holland HD, Turekian KK (eds) The oceans and marine geochemistry—treatise on geochemistry. Elsevier, New York
Epstein S, Mayeda T (1953) Variations of 18O/16O ratio in natural waters. Geochim Cosmochim Acta 4:213–224
Fagundo Castillo JR, González Hernandez P (1999) Agricultural use and water quality at karstic Cuban western plain. Int J Speleol 28:175-185
Fagundo JR, Valdés JJ, Pajon JM, de la Cruz A (1986) Caracterizacion geoquimica y geomatematica de formaciones geologicas y sedimentos de la cuenca del rio cuyaguateje. Relacion las caracteristicas hidroquimicas de los acuiferos. Voluntad hidráulica 23:43–48
Florea LJ, McGee DK (2010) Stable isotopic and geochemical variability within shallow groundwater beneath a hardwood hammock and surface water in an adjoining slough (Everglades National Park, Florida, USA). Isot Environ Health Stud 46:190–209
Florea LJ, Vacher HL (2006) Morphologic features of conduits and aquifer response in the unconfined floridian aquifer system, west central Florida. Paper presented at the Proceedings of the 12th Symposium on the Geology of the Bahamas and Other Carbonate Regions, Gerace Research Center, San Salvador, Bahamas
Garrels R, Wollast R (1978) Equilibrium criteria for two-component solids reacting with fixed composition in an aqueous phase—example: the magnesian calcite, discussion. Am J Sci 278:1469–1474
Gat JR (2010) Isotope hydrology: a study of the water cycle vol 6. series on environmental science and management. Imperial College Press, London
Glynn PD, Reardon EJ (1990) Solid-solution aqueous-solution equilibria: thermodynamic theory and representation. Am J Sci 290:164–201
Gonfiantini R (1986) Environmental isotopes in lake studies. In: Fritz P, Fontes J-C (eds) Handbook of environmental isotopes geochemistry, vol 2. Elsevier, Amsterdam, pp 113–168
Gonfiantini R, Araguas LA (1988) Los Isotopes Ambientales En El Estudio De La Intrusion Marina. Paper presented at the Tecnología de la Intrusión en Acuíferos Costeros. Almuñécar, Granada
Hernández R, Ramírez R, López-Portilla M, González P, Antigüedad I, Díaz S (2013) Seawater Intrusion in the Coastal Aquifer of Guanahacabibes, Pinar del Río, Cuba. In: Farfán González H, Corvea Porras JL, de Bustamante Gutiérrez I, LaMoreaux JW (eds) Management of water resources in protected areas. Springer, Berlin, pp 301–308
IAEA/WMO (2013) Global network of isotopes in precipitation. The GNIP Database. Accessible at http://www.iaea.org/water
Iturralde-Vinent MA, Gutiérrez Domech MR (1999) Some examples of karst development in Cuba. Boletín Informativo de la Comisión de Geoespeleología, 14:1–4
Jones BF, Vengosh A, Rosenthal E, Yechieli Y (1999) Geochemical Investigations. In: Bear J, Cheng AHD, Sorek S, Ouazar D, Herrera I (eds) Seawater intrusion in coastal aquifers—concepts, methods and practices. Theory and applications of transport in porous media, vol 14. Springer, Dordrecht, pp 51–71
Langmuir D (1997) Aqueous environmental geochemistry. Prentice Hall, New Jersey
Lin CY, Musta B, Abdullah MH (2013) Geochemical processes, evidence and thermodynamic behavior of dissolved and precipitated carbonate minerals in a modern seawater/freshwatermixing zone of a small tropical island. Appl Geochem 29:13–31
Liu CW, Chen JF (1996) The simulation of geochemical reactions in the Heng-Chun limestone formation, Taiwan. Appl Math Model 20:549–558
Mackenzie FT, Ver LM (2010) Land-sea global transfers. In: Steele JH, Thorpe SA, Turekian KK (eds) Marine chemistry & geochemistry: a derivative of encyclopedia of ocean sciences, 2nd edn. Academic Press, London, pp 485–494
Matisoff G, Bricker III OP, Holdren Jr. GR, Kaerk P (1975) Spatial and temporal variations in the interstitial water chemistry of chesapeake bay sediments. In: Church TM (ed) Marine chemistry in the coastal environment. ACS Symposium Series, vol 18. American Chemical Society, Washington, pp 343–363
Molerio León LF (1992) Composición Química e Isotópica de las Aguas de LLuvia de Cuba. Paper presented at the II Congreso Espeleológico de Latinoamérica y el Caribe, Viñales, Pinar del Río, Cuba
Molerio León LF (2012) Hidrología de trazadores en la gestión ambiental de yacimentos de petróleo onshore. Mapping 154:46–76
Molerio León L, Parise M (2009) Managing environmental problems in Cuban karstic aquifers. Environ geol 58: 275–283
Morse JW, Mackenzie FT (1990) Geochemistry of sedimentary carbonates. Developments in sedimentalogy, vol 48. Elsevier, The Netherlands
Pardo (2009) Overview. In : the geology of Cuba. AAPG Studies in Geology Series 58, pp 1–47
Parkhurst DL, Appelo CAJ (2013) Description of input and examples for PHREEQC Version 3—a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. US Geological Survey Techniques and Methods, book 6, chap A43. http://pubs.usgs.gov/tm/06/a43/. p 497
Peláez García R, González Cabrera NA (2013) Underground water of deep circulation in the national park guanahacabibes, pinar del rio province, cuba: another alternative with water supply aims. In: LaMoreaux JW, Farfán González H, Corvea Porras JL, de Bustamante Gutiérrez I (eds) Management of water resources in protected areas. Springer, Berlin, pp 179–186
Peralta Vital JL, Gil Castillo R, Leyva Bombuse D, Moleiro León L, Pin M (2005) Uso de técnicas nucleares en la evaluación de la cuenca Almendares-Vento para la gestión sostenible de sus recursos hídricos. Forum de Ciencia y Técnica. www.forumcyt.cu
Pérez M, Pubillones León María A (1988) Características microbiológicas de una laguna que ocupa una dolina cársica, Laguna del Valle de San Juan, provincia de Pinar del Río. Paper presented at the Taller Internacional sobre Hidrología del Carso en la Región del Caribe, La Habana, pp 4–12
Perry CT, Salter MA, Harborne AR, Crowley SF, Jelks HL, Wilson RW (2011) Fish as major carbonate mud producers and missing components of the tropical carbonate factory. PNAS 10:3865–3869
Plummer LN (1975) Mixing of sea water with calcium carbonate ground water. In: Whitten EHT (ed) Quantitative studies in geological sciences. Geological Society of America Memoir, vol 142, pp 219–236
Price RM, Herman JS (1991) Geochemical investigation of salt-water intrusion into a coastal carbonate aquifer: Mallorca, Spain. Geol Soc Am Bull 103:1270–1279
Richter BC, Kreitler CW (1993) Geochemical techniques for identifying sources of ground-water salinization. CRC Press, Boca Raton
Robinove CJ, Langford RH, Brookhart JW (1958) Saline-water resources of North Dakota. US Geological Survey, USA
Rodríguez R, Bejarano C, Riverón B, Carmenate JA (2004) Composición química de las precipitaciones, deposición de sales y evaluación de la recarga en la región oriental de Cuba. Boletín Geológico y Minero 115:341-356
Rozanski K, Araguás-Araguás L (1995) Spatial and temporal variability of stable isotope composition of precipitation over the south american continent. Bulletin de l’Institut francais d’études Andins 24:379–390
Rozanski K, Araguás-Araguás L, Gonfiantini R (1993) Isotopic patterns in modern global precipitation. In: Swart PK, Lohmann KC, Mckenzie J, Savin S (eds) Climate change in continental isotopic records. Geophysical monograph, vol 78. American Geophysical Union, Washington
Runnells DD (1969) Diagenesis, chemical sediments, and the mixing of natural waters. J Sediment Petrol 39:1188–1201
Sáinz ÁM, Molinero JJ, Saaltink MW (2010) Numerical modelling of coastal aquifer karst processes by means of coupled simulations of density-driven flow and reactive solute transport phenomena. In: Andreo B, Carrasco F, Durán JJ, LaMoreaux JW (eds) Advances in research in karst media. Environmental earth sciences, pp 237–242
Sanford WE, Konikow LF (1989) Simulation of calcite dissolution and porosity changes in saltwater mixing zones in coastal aquifers. Water Resour Res 25:655–667
Seale LD, Soto LR, Florea LJ, Fratesi B (2004) Karst of western Cuba: observations, geomorphology, and diagenesis. In: Davis RL, Gamble DW (eds) Proceedings of the 12th symposium on the geology of the Bahamas and other carbonate terrains. Gerace Research Center, San Salvador, pp 1–9
Stoessel RK (1992) Effects of sulfate reduction on CaCO3 dissolution and precipitation in mixing-zone fluids. J Sediment Petrol 62:873–880
Stoessell RK, Ward WC, Ford BH, Schuffert JD (1989) Water chemistry and CaCO3 dissolution in the saline part of an open-flow mixing zone, coastal Yucatan Peninsula, Mexico. Geol Soc Am Bull 101:159–169
WHO (2011) Guidelines for drinking-water quality, 4th edn. World Health Organisation, Gutenberg
Wolery TW, Jarek RL (2003) EQ3/6, version 8.0—software user’s manual. Civilian radioactive waste management system, Management and Operating Contractor, Sandia National Laboratories, Albuquerque, New Mexico
Xinping Z, Lide T, Jingmiao L (2005) Fractionation mechanism of stable isotope in evaporating water body. J Geog Sci 15:375–384
Zeebe RE, Wolf-Gladrow D (2001) CO2 in seawater: equilibrium, kinetics, isotopes. Elsevier Oceanography Series, vol 65. Elsevier Science, Amsterdam
Acknowledgments
The authors would like to thank the two anonymous reviewers for their suggestions and comments.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Boschetti, T., González-Hernández, P., Hernández-Díaz, R. et al. Seawater intrusion in the Guanahacabibes Peninsula (Pinar del Rio Province, western Cuba): effects on karst development and water isotope composition. Environ Earth Sci 73, 5703–5719 (2015). https://doi.org/10.1007/s12665-014-3825-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12665-014-3825-1