
Abstract
Geochemical processes, occurring in a stable transition zone between saltwater and freshwater, were simulated in a 2D, multi-layer flow chamber experiment. Mixing, calcite dissolution, and oxidative degradation of organic matter were identified as the main controlling factors. The results of the chamber experiment were compared to field data and verified by thermodynamic modeling. Similarity in most ion distributions suggests the general applicability of the experimental method. Differences in the redox conditions between field and experiment were reflected by the oxidants involved in the mineralization of organic carbon; while field data show evidence of sulfate reduction, the presence of oxygen in the laboratory experiment resulted in the reoxidation of sulfides.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Field observations in coastal areas suggest the presence of a mixing zone between freshwater and seawater which moves seaward during periods of recharge and inland during periods of low freshwater heads. In the beginning of the last century, the dynamics of the two water types was investigated and described (e.g. summarized in Dominco and Schwartz 1998). However, the position and the movement of the interface between waters of different salinity can be influenced by a lot of factors, such as climatic stress, tides (Werner and Gallagher 2006), water discharge, no-effective groundwater management (Djabri et al. 2007) or leakage between aquifers (Foyle et al. 2002).
Bridge and Lloyd (2005) discuss diffusion and the effect of increased ion strength on the solubility and diffusion of dissolved ions at the freshwater–saltwater interface. They conclude that the interface and the water quality are mainly influenced by hydraulic gradients, density-induced currents, and additionally the composition of the aquifer material (Djabri et al. 2007). Depending on the regional hydrogeochemical conditions calcite dissolution (Zilberbrand et al. 2001), dolomitisation (Ward and Halley 1985) or the precipitation of carbonates (Magaritz et al. 1980) or gypsum (Gomis-Yagües et al. 2000) is reported.
The position and the movement of the marine intrusion are of importance for any coastal groundwater management. Therefore, modeling the behavior of the interface and the water quality is an important tool. Geochemical interpretation of field date is necessary to verify those concepts (Werner and Gallagher 2006; Dai and Samper 2006).
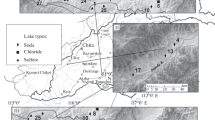
Buried valleys at the European North Sea coast are important groundwater reservoirs. Their use should be based on a good knowledge of the regional hydrogeochemical setting. For the investigation of one of these structures, a coastal aquifer test field (CAT-Field; Kessels et al. 2000) was established. The CAT-Field is located at the German North Sea coast between the Elbe and Weser river estuaries (Fig. 1).

Position of the CAT-Field and its wells. Geological cross-section, to which the set-up of the chamber experiment refers
The geological setting, determining the groundwater quantity, was investigated by geological and geophysical methods (Kirsch et al. 2006; Wiederhold et al. 2005). Groundwater quality depends on the geochemical processes, which are characterized by the present investigations. Most hydrochemical investigations of saltwater–freshwater transition zones focus on field data sometimes compared to numerical simulations. Here, additionally, geochemical processes were simulated by means of a 2D laboratory simulation experiment. Well-defined boundary conditions and intense sampling possibilities are allowing a better understanding of the influences on the water quality.
The 2D flow chamber experiment represents a geological section inland from the coastline (Panteleit et al. 2002). The first phase of the experiment simulated exchanger reactions during the intrusion of saltwater into a sand tank (Panteleit et al. 2003). These processes were modeled using PHREEQC (Parkhurst and Appelo 1999) and PHWAT, which couples a geochemical reaction model with a density-dependent groundwater flow and solute transport model (Mao et al. 2006a, b). After the initial intrusion of the saltwater, the position of the saltwater–freshwater interface stabilized. Diffusion and dispersion processes expanded the initially sharp interface into a zone of transition between both end member water types.
A depletion of magnesium and potassium as well as an excess of calcite, iron, manganese, and lithium could not be caused by exchanger reactions during saltwater intrusion into the sediment (Panteleit et al. 2001b). This indicates the presence of slow geochemical processes, former masked by the exchanger reactions.
For their determination, this paper compares field data collected in the area of investigation with results of the experimental simulation. Differences between field and experimental data are discussed, and the causing processes are identified. Finally, the assumptions are verified by a numerical simulation of the geochemical processes of the field as well as of the laboratory simulation.
Method
Field methods
In several field campaigns, during 2001 and 2002, hydrochemical field samples were collected from a network of 51 screened wells and 26 geological drills between 10 and 120 m in depth in the CAT-Field. Among them also four multi-level well clusters and a fully screened research well with a depth of 120 m (Binot et al. 2002; Panteleit et al. 2006). A brief compilation of the hydrochemical field data by Panteleit and Hammerich (2005) showed the presence of geochemical processes instead of conservative mixing. Now processes resulting in sampled compositions are discussed.
During sampling, a vertical flow-through cell was employed to accommodate probes used for the simultaneous determination of pH, EH, dissolved O2, temperature, and electric conductivity (EC) for all wells. The chamber was flooded from below using a diver pump (Grundfos MP1). Final measurements were recorded after equilibration of the measuring probes. Samples were passed through a 40-μm cellulose filter and either acidified (nitric acid), stored untreated, or immediately titrated for alkalinity (Cook and Miles 1980).
For the analysis of porewater, about 200 g of field fresh sediments from cores, previously stored at 4°C, were centrifuged for 12 min at 2,000 U/min to separate the porewater. EH and pH of these porewater samples were measured potentiometrically. Due to the small sample volume, alkalinity was determined by Gran titration (Grasshoff et al. 1999). The remaining sample volume was acidified with nitric acid for later cation analysis.
Cation analysis were performed by inductive coupled plasma atom emission spectroscopy (ICP-AES; Perkin Elmer, Optima 3000) for Na+, Ca2+, K+, Mg2+, Li+, Sr2+, Ba2+, Mn2+, and Fe2+. ICP-AES was also used to measure the elements Bor and Sulfur which are present in the anions BO3− and SO4 2−. Chloride was determined by high pressure liquid chromatography (HPLC; tsp P100 with a Kratos Spectroflow 773 absorbance spectrometer).
Laboratory simulation
For the laboratory scale simulation of the hydraulic and geochemical processes in the saltwater–freshwater transition zone, a 2D sand tank experiment as shown in Fig. 2 was designed. The sand tank itself consists of a sediment filled flow chamber of length 1.90 m, height 0.50 m, and width 0.05 m. The chamber was covered with a Plexiglas lid to minimize evaporation. To reproduce the geochemical environment encountered in the field, material from different layers of the aquifer was used to fill the flow chamber. These materials were collected from sediment cores during drilling in the investigation area (Binot et al. 2002; Panteleit et al. 2006). The chamber was filled in three layers using different sediment types to simulate the natural conditions (Fig. 2). The uppermost layer (layer 1) with a thickness of 0.15 m consisted of Eemian and Holocene tidal flat and marsh sediments. These are characterized by a high content of calcite and organic compounds. The lithogene component consists of silt and clay with only very small amounts of fine sand. Accordingly the hydraulic conductivity is rather low. The deeper part of the chamber (layer 3) was filled to 0.25 m with glacifluviatile sands from the Drenthe stage. The texture of these sediments ranges from fine sand to fine gravel. Almost no or very low amounts of fine mineral textures, organic or carbonate fractions are present. The intermediate layer (layer 2) is a mixture of the material from layer 1 and 3. It has a thickness of 0.10 m (Table 1).

Experimental set up of the sand tank experiment (200 × 50 × 5 cm); circles indicate locations of sampling points; arrows indicate water flow direction
Each side of the flow chamber is bordered by a water storage chamber. An outflow chamber in the upper part of the saltwater storage chamber avoids the intrusion of mixed water and ensures a constant salt concentration of the intrusion water. A filter fleece installed between the chambers prevented the relocation of sediment while ensuring good hydraulic contact. To control the flow regime, the water level in all four chambers can be adjusted separately through an overflow system. The inflow water on the freshwater side was artificially mixed and saturated with respect to calcite, simulating the groundwater composition from field samples as displayed in Table 2. Saltwater was synthesized according to Grasshoff et al. (1999), diluted to the highest chloride concentration measured at the coastline of the CAT-Field (Table 2), and also calcite saturated. The saltwater was spiked with uranine (disodiumfluoresceine) to allow a quick observation of the salt content of the porewater in the flow chamber.
The flow chamber experiment is divided in three phases. During an initial phase, freshwater from a 200-l storage was circulated through the tank for 3 weeks until the concentrations of the analyzed ions were stable. During the second phase, the saltwater storage chamber of the tank was flooded from the bottom on with saltwater until a hydraulic gradient of 3 mm/m (without density correction) from the left (freshwater) to the right (saltwater). According to the density driven flow field, saltwater intruded into the tank until a hydraulic equilibrium was established. Exchanger sites were occupied related to the changed porewater chemistry (Panteleit et al. 2003; Mao et al. 2006a, b). Also mineral reactions occured during this phase, but were masked by the more intense and quick reactions at the exchanger sites on the mineral surface. After 72 h, the saltwater intrusion was fully established and in equilibrium with the density driven flow field; the system was in a steady state. Ca2+ released from the exchanger sites is flushed out, and Na+ and Mg2+ loss from adsorption is equalized. After 120 h, the slower thermodynamic processes become more important and will control the porewater chemistry in the third phase. During this phase the steady state was held for 9 weeks to establish chemical equilibrium under the changed conditions. The experimental design generated an overall discharge of about 90 ml/h for the three layers. At the end of the experiment, porewater was sampled at 100 locations in the flow chamber. The sampling sites were arranged in six depth profiles, located at different intrusion lengths. Each depth profile had up to 25 sampling depths (Fig. 2). At each point a soil moisture sampler was installed, which consisted of a porous polymer tube (diameter 2.5 mm) with a pore diameter of 1.1 μm. Polymer tubes penetrated the whole sediment and chamber width of 0.05 m, amounting to a relatively high sampling surface of almost 4 cm2 per sampler. Thus, the risk of clogging of the sampling probes was minimized. However, seven of the probes were sealed this way and not available for final sampling. Samples of 4 ml by inducing a vacuum using a 10-ml syringe were taken from top to bottom. Water samples were treated such as the extracted water from the sediment cores.
Thermodynamic modeling
Thermodynamic calculations were performed using the PHREEQC computer program (Parkhurst and Appelo 1999). For a specification of equilibrium states between aqueous and solid phase, saturation indices [SI = log(ion activity in water sample/ion activity in equilibrium)] were determined for the analyzed groundwater compositions. The marine intrusion was simulated by stepwise implementation of the identified reactions (mixing, equilibration with the carbonate system, sulfate reduction, and exchanger reactions). The freshwater analyses of the field sample, respectively of the inflow storage chamber of the sand tank, were taken as starting water composition for the simulations.
Results
Field data
Typical concentrations for different ions analyzed from the field samples are presented in Table 2. The distribution of cation and anion mixing ratios from field samples are shown in Fig. 3 and Table 3. Data vary between typical freshwater and seawater compositions. In freshwater, major cations are Ca2+ and Mg2+; HCO3 − is the major anion, while SO4 2− and Cl− are of minor importance. In a rough classification as indicated in Fig. 3, the freshwaters are of the CaHCO3 type. Water samples of typical seawater composition are of a NaCl type. The transition between these two water types is not linear, but tends toward a NaHCO3 type with Na+ and K+ contributing up to 81% of the total cation equivalent concentration [TEC = Σ(n × Xn+)]. HCO3 − contributes 79% of the total anion equivalent concentration [AEC = Σ(n × Xn−)], while almost no SO4 2− was detected.

Piper plot of field samples (crosses) and experimental data (circles) from a flow chamber experiment. Typical compositions of seawater and freshwater and a rough classification of water types (dashed lines) are shown in the middle diagram
Laboratory experiment
The fraction range of different cations and anions are displayed in Table 3. Depending on the sampling location in the flow chamber, either Ca2+ or Na+ is the dominating cation, while Mg2+ reaches up to 22% only. SO4 2− and Cl− are the predominant species. HCO3 − shares are in principle below 20% and negligible in samples with a high Cl− content.
Mixing ratios of water samples from the laboratory experiment are presented in relation to the field data (Fig. 3), while cations and Cl− show analog fraction ranges in field and laboratory samples. The NaHCO3 water type from the field cannot be reproduced, as the HCO3 − ratios stay well below those in the field. Instead of it SO4 2− becomes much more dominant, resulting in a CaCl water type (“North” shifted in the diamond part of the Piper diagram) in the laboratory samples.
For the interpretation of the data and the identification of the major processes, the spatial distribution of ion concentration (gray scale) and percentage (isolines) are essential. Figure 4a displays the joint distribution of Na+ and Cl−. The saltwater intrusion is indicated by a Cl− concentration exceeding 200 mmol/l. At the bottom of the flow chamber, the Cl− intrusion reaches half of the flow chamber length. At the saltwater inlet, it reaches two-third of the height. Na + shows a similar distribution and so do the mixing ratios of both ions compared to the anion equivalent concentration and the cation equivalent concentration, respectively.

Spatial distribution of different ions in the flow chamber with the mixing zone in a steady state and at a stable position. Gray scale represents effective concentrations in mmol/l for the indicated sampled locations, lines indicate ion shares of the total cation and anion equivalent concentration (interpolated by krigging), respectively
Ca2+ shows a more complex distribution (Fig. 4b). High concentrations in both the saltwater-dominated area and in the upper Pleisto–Holocene layers can be observed. In saltwater-affected Pleisto–Holocene layers, Ca2+ concentrations reach up to a maximum of 45 mmol/l. Lowest concentrations were found in the galcigene sand under freshwater conditions. The observed distribution of the Ca2+ mixing ratio results mainly from changes in the Na+/Ca2+ ratio. The Mg2+ content does not change significantly (Fig. 3); thus, the Ca2+ mixing ratio is equivalent to the inverse Na+ mixing ratio.
The distribution of major anions is more complex than that of the cations. The distribution of HCO3 − is displayed in Fig. 4c. The concentration is highest (up to 15 mmol/l) in the upper layer of the Pleisto–Holocene sediments. In the transition layer, measured HCO3 − concentrations still reach 8 mmol/l, while in the absence of the Pleisto–Holocene sediments a steep concentration decrease down to 1 mmol/l occurs. In the glacigene sands, a slight HCO3 − concentration increase toward the saltwater-dominated side is observed, though values do not rise above 2 mmol/l. The HCO3 −/AEC ratio distribution delivers a different pattern. In the saltwater-dominated part, HCO3 − has a low ratio of 0–5%, while in the freshwater-dominated part ratios up to 18% are reached. In both regions, the derived ratios are higher in the Pleisto–Holocene layer than in the layers containing glacigene sands.
The absolute concentration of SO4 2− is highest (32.0 mmol/l) in the Pleisto–Holocene layers and decreases with depth (Fig. 4d). In the deeper layers, the SO4 2− concentration correlates with the Cl− concentration, resulting in higher values (26.4 mmol/l) in the saltwater-dominated side compared to the freshwater side (2.1 mmol/l). The SO4 2−/AEC ratio is highest close to the surface at the freshwater side, and decreases with depth and toward the saltwater side.
Discussion
First, processes resulting in the observed composition of field samples will be discussed and verified by a stepwise numerical simulation. The processes occurring in the flow chamber are identified and verified subsequently, followed by a discussion of possibilities and limits of the flow chamber experiment.
Field processes
The Piper plot in Fig. 3 shows ion mixing ratios that are characteristic for freshwater and seawater. Field samples from wells located inland match the typical ion ratios for freshwater, while samples from the coastal area resemble seawater. Considering a conservative mixing for the transition from seawater to freshwater, the data should plot on the conservative mixing line in between these two end member water types as indicated in Fig. 5. Ion mixing ratios of field samples from the CAT-Field differ from this conservative mixing line toward higher Na+ and HCO3 − content (Fig. 3). Typical groundwater samples from the CAT-Field (Table 2) are indicated in Fig. 5. Sample 2 originates from the glacigene layers in a depth of 17.5 m, while sample 3 was collected in the Holocene layers (4.5 m depth). Chemical processes that are alterating the hydrochemistry of the water result from conservative mixing, calcite dissolution, sulfate reduction, and exchanger reactions (Werner and Gallagher 2006). These different processes are calculated using PHREEQC (Parkhurst and Appelo 1999); their effects will be discussed separately as follows.

Piper plot showing typical compositions for freshwater and seawater and a rough classification of water types. Numbers indicate typical water samples from the CAT-Field (Table 3). Arrows indicate reaction pathways as discussed in the text
Mixing
The effect of mixing on the hydrochemistry can be derived from the measured Cl− concentration, since Cl− is supposed to behave conservative. This is done in a first step of the thermodynamical calculations (column 2 in Tables 4, 5). Starting from the freshwater composition (column 1 in Tables 4, 5) saltwater as sampled in the field (column 7 in Tables 4, 5) is added until the Cl− concentration reached those of the selected samples (2 and 3 in Fig. 5) with a ratio of 6.7 and 2.3 %, respectively.
Calcite dissolution
The dissolution of calcite is a process which is often found to alterate the chemical composition of groundwater in coastal aquifers (Custodio and Bruggeman 1987). Especially in freshening aquifers, it is enhanced by the depletion of Ca2+ from ion exchanger reactions (Walraevens et al. 2007; Andersen et al. 2005). Sediment cores from the CAT-Field show varying calcite content, but calcite is present in all layers down to 60 m. Shells of conches can be found in the quaternary layers of marine origin (tidal flat sediments), while glacigene sediments contain disperse calcite (Binot et al. 2002). The increased Ca2+/Cl− ratio compared to the seawater value of 0.019 (Fig. 6) clearly indicates a Ca2+-releasing process. The dissolution of calcite would increase the concentration of dissolved carbon species as well as the Ca2+ concentration (Eq. 1).
This process affects the concentration of both cations and anions. Accordingly, this reaction moves the sample projection in the Piper diamond more or less to the left (Fig. 5). Thus, the tendency of the samples toward a NaHCO3 type cannot be explained by the calcite dissolution alone.

Ion/chloride ratios for different ions of the NaHCO3 − type field samples and samples from the flow chamber experiment compared to typical seawater ratios (line). Fat line in box indicates mean of samples, box edges are first and third quartiles, bars are minimum and maximum values. Ratios above the seawater value indicate an ion releasing process (as for Ca2+; HCO3 − and experimental SO4 2−), ratios below the seawater value a consuming process (SO4 2− field data)
One driving force for the dissolution of calcite is the ionic strength effect. According the Debye-Hückel theory (Atkins 1990) the activity coefficient depends nonlinearly on the ionic strength. Therefore, a mixture of two water types, both initially equilibrated with respect to calcite, may be undersaturated after mixing. Another driving process is the reaction with carbonic acid (H2CO3). Water, equilibrated with respect to calcite under the atmospheric CO2 pressure (10−3.5 atm), enters the aquifer. There it is exposed to a higher CO2 partial pressure due to the respiration of organic matter. According to the new equilibrium conditions, additional protons may associate with the carbonate ion that is released from calcite to form bicarbonate (Appelo and Postma 2005), leading to the overall reaction
Sanford and Konikow (1989) calculated the expected calcite dissolution or precipitation rate for different CO2 pressures, temperatures and freshwater–seawater mixing ratios. Their thermodynamic calculations showed that calcite dissolution is most significant in mixtures containing less than 50% seawater. Supersaturation results for solutions with a composition closer to seawater. But even from supersaturated solutions with a saturation index of up to 0.3, the precipitation of calcite might be inhibited by the presence of magnesium, organic acids or phosphate. The later two affects precipitation even at concentrations below 1 μmol/l (Berner 1975; Berner et al. 1978; Reddy 1977; Tomson 1983; Walter and Hanor 1979). However, for typical samples from the CAT-Field (Table 4) with a low seawater fraction, calcite dissolution is indicated by the negative saturation indices that are resulting from thermodynamic calculations (Table 4).
The effect of calcite dissolution upon the mixtures of fresh and saltwater was calculated in a second step of the numerical simulation (Table 4, column 3). Each sample was assumed to be in equilibrium with the calcite present in the sediment (saturation index was set to zero), and the local CO2 pressure was set as derived from measured concentrations (assuming equilibrium with the carbonate/CO2 system) at the sample location. In case of sample 3, which originates from the organic rich Holocene layer, calculations suggest calcite dissolution as a result of the high CO2 partial pressure of 10−0.3 atm. For the sandy glacigene layer (sample 2), the low CO2 partial pressure of 10−2.3 atm results in a calculated calcite precipitation, since the mixed solution was slightly supersaturated with respect to calcite (SI = 0.3). But with respect to the discussed inhibition of calcite precipitation and the Ca2+/Cl− ratio in the field samples, precipitation of calcite in the CAT-Field aquifer seems to be unlikely. However, for numerical simulation of field and laboratory processes, the equilibration with respect to calcite and CO2 partial pressure is the most important process with respect to the concentrations of Ca2+ and HCO3 −, approaching the concentrations of the corresponding sample.
Sulfate reduction
Besides calcite dissolution, the mineralization of organic carbon (simplified as CH2O) is another source for HCO3 − in the porewater of sediments. The organic matter in the system investigated here is mainly present in the Pleisto–Holocene layers (Table 1). The mineralization product CO2 dissolves in water and converts to carbonic acid according to the present equilibrium conditions. For the oxidation of organic matter an oxidant is required. In the very upper layer, this will be oxygen from the atmosphere or dissolved in the porewater:
Fine sediments in the upper layer hamper the deep penetration of atmospheric gases. The upcoming of groundwater was already expected for the marsh areas (Fulda 2002). This is supported by the field data (Table 4), as no fresh, oxidizing groundwater is recharged in the CAT-Field. The suboxic conditions are reflected in the low EH values and high reduced iron concentrations of the samples that are taken in a depth of 4.5 m. After all oxygen has been consumed by aerobic respiration, anaerobic micro organisms, e.g., a bacteria of the genus Desulfovibrio, may gain energy from dissimilatory sulfate reduction (Jørgensen 1982; Kasten and Jørgensen 2000). The products of sulfate reduction are a broad range of reduced sulfur species, which may be precipitated in a number of metastable metal sulfide minerals, and finally transformed and immobilised to pyrite (Morse et al. 1987). Equation 4 summarizes the system of sulfate reduction reactions:
The oxidation of organic carbon through sulfate reduction or oxidative respiration affects the anion mixing ratios in the Piper plot only. With respect to the Piper diamond of Fig. 3, these reactions would result in a shift of the sample projection parallel to the cation axis (Fig. 5). Source of SO4 2− is seawater, where concentrations are generally two orders of magnitude higher than in freshwater (Bowen 1979). SO4 2− is delivered from sea salt, entrained in the air from the nearby North Sea surface and precipitated onto the uppermost layer in the CAT-Field, or added to deeper layers of the aquifer by seawater intrusion. Accordingly, samples with a high Cl− content, which represent samples close to the shoreline, have the highest SO4 2− concentrations. The SO4 2−/Cl− ratio of NaHCO3 type field samples (Fig. 6) lie well below the typical value for seawater of 0.05, indicating that the decreasing SO4 2− content is not due to conservative mixing only.
Decreasing SO4 2− concentrations are typical for regions with seawater intrusion (Magaritz and Luzier 1985; Mercado 1985; Nadler et al. 1980; Andersen et al. 2005). Since the decrease cannot be explained by conservative mixing only, it suggests reduction processes as a sulfate sink. The cited authors assign the decrease of sulfate to degradation of organic matter through sulfate reducing bacteria. Gomis-Yagües et al. (2000) discuss gypsum precipitation resulting in the decrease of SO4 2− concentration during seawater intrusion. They admit that usually the activities that are reported for SO4 2− and Ca2+ from groundwater samples taken in seawater intrusion studies do not reach the solubility product for gypsum. Thus, the gypsum precipitation can only occur if additional Ca2+ will be available through ion exchange reactions. Accordingly Walraevens et al. (2007) assume gypsum dissolution in a reaction transport modeling of a freshening aquifer with a depletion of Ca 2+ from exchanger reactions. In the CAT-Field the Ca2+/Cl− ratio increased (Fig. 6). However, thermodynamic calculations suggest that gypsum precipitation is not taking place, because the saturation indices are negative for all field samples (Table 4). This confirms the conclusion mentioned before that an progressive intrusion of saltwater can be excluded for the CAT-Field. Consequently, no excessive release of Ca2+ from exchanger sites occurs. On the other hand, in the CAT-Field conditions appear to be favorable for microbiological sulfate reduction:
In the Pleisto–Holocene layers a high amount of organic matter is present (Table 1). According to Eq. 4, 0.6 mmol/l CH2O (which equals to 7.2 mg/l DOC) are necessary to balance the observed sulfate concentration decrease, which averages 0.3 mmol/l SO4 2−. Since samples show a DOC content of 5–50 mg/l sufficient reactive organic matter seems to be available to facilitate the SO4 2− reduction process.
According to the energy budget, sulfate reduction only takes place if no other oxidants delivering more energy are present (Froelich et al. 1979). The EH values of <180 mV (Table 4) in the samples indicate that the lack of suitable oxidants in porewater of the Pleisto–Holocene layers in the CAT-Field support SO4 2− reduction.
The produced H2S should be able to form iron sulfide minerals; consequently, excess Fe2+ should be available and the iron sulfides detectable. Own data (Table 1) as well as investigations from Dellwig et al. (2001) proofed the presence of pyrite and reduced sulfur in sediment cores from the region. Measured iron concentration in the groundwater suggests the supply of Fe2+ through the freshwater flow. The notwithstanding low pyrite content can be explained by reoxidation processes (Lowson 1982; Schulz et al. 1994), resulting in a more complicated model than the straight Froelich scheme (Froelich et al. 1979). According to Giblin and Howarth (1984), an oxidative dissolution of iron sulfides in coastal regions could be induced by saltwater flushing and plant activity. In any case, the characteristic foul smell was noticed easily during the drilling works, giving a qualitative detection of the presence of H2S.
Data of stable isotope ratio would deliver a definite proof for the SO4 2− reduction pathway, since during microbiological SO4 2− reduction 32S is preferentially consumed rather than the heavier 34S (Robertson et al. 1989). Using stable isotopes, (Dellwig et al. (1999, 2001) documented the microbiological reduction of SO4 2− in a number of sediment cores that were collected in a region a few kilometers west of the CAT-Field. Furthermore, they determined seawater as the main source for SO4 2−.
Another option to identify driving processes could be the utilization of radio tracer methods as described by Jørgensen (2000). Neither stable isotopes nor radio tracers were applied in this project. The measured sediment characteristics and the observed sulfate depletion suggest the mineralization of organic carbon by dissimilatory sulfate reduction. As discussed, a series of published studies in the region support the conclusion that the sulfate depletion is a consequence of the sulfate reduction by organic matter decomposing micro organisms. As HCO3 − is a product of this reaction, the water type will be shifted toward a HCO3 − type by the increased HCO3 − content as documented in Fig. 3.
The degradation of organic matter was implemented in the thermodynamic calculations through reduction of the SO4 2− concentration down to that of the corresponding sample (sampled water in column 7; Table 4). The concentrations of HCO3 − and S2− were raised according to Eq. 4. Reflecting the observations in the CAT-Field, the EH decreased in this calculation step and the solution becomes supersaturated with respect to metal sulfides (e.g. pyrite in Table 4). Additionally, the increase of HCO3 − resulted in a precipitation of calcite, which is reflected in the decrease of the Ca2+ concentration.
Exchanger reactions
Cations, adsorbed reversibly to negative charged surfaces, are exchanged with each other due to equilibrium disturbance after the cation mixing ratios in the aqueous phase changed, e.g. through seawater intrusion (Appelo and Greinaert 1991; Beekman and Appelo 1991; Panteleit et al. 2003). The rather weakly bound ions are exchanged almost immediately; thus, a new equilibrium is reached rapidly. Therefore, if exchanger reactions are identified in field measurements in coastal areas, it is generally considered an indication for a recent movement of the saltwater–freshwater interface. Examples for an active salinization or refreshening of coastal aquifers that are showing patterns of exchanger reactions were found all over the world (e.g. Kim et al. 2003; Martinez and Bocanegra 2002; Vandenbohede and Lebbe 2002).
An increasing Na+ content of the porewater samples as observed in the CAT-Field would be the result of the exchange of adsorbed Na+ through Ca2+:
with X representing the negative charged exchanger site on the surface of the aquifer material. Since this exchanger reaction affects the cation ratio in the water only, it would result in a shift of the sample projection in the Piper diamond parallel to the anion axis toward the lower right (Fig. 5). The deviation from the conservative mixing line as observed in the field samples would occur during a refreshening of the aquifer. In this case, Ca2+-rich water displaces a water body with a higher Na+ ratio of the cations and the subsequent exchange reaction responds according to the law of mass action.
Figure 6 displays the ratios of different ions to Cl− as measured in groundwater samples from the CAT-Field in comparison to typical seawater ratios. Assuming the increased Na+ ratio of the cations would result from ion exchange of adsorbed Na+ by Ca2+ during refreshening, then this must be reflected in higher Na+/Cl− ratios as well as in a lower Ca2+/Cl− ratio. However, Fig. 6 shows that the Na+/Cl− ratio matches the seawater value of 0.98, and the Ca2+/Cl− ratio is even higher than the seawater value. Consequently, in the subsoil of the investigation area, the ion exchange reaction did not cause the observed deviation from the conservative mixing between seawater and freshwater.
Even if the position of the transition zone is stable, the processes discussed before modify the composition of the porewater. Consequently, exchanger processes resulting from these alterations were included as a final step in the thermodynamic calculations. For the calculations, the exchanger characteristics derived from samples used in the column experiments (Panteleit et al. 2001a) were employed. The results of the thermodynamic calculation step showed only slight changes in the cation concentrations. The still remaining differences with respect to pH and the cation concentrations can be related to the fact that the material for the column experiment does not originate from the location of the studied water samples, while the complex exchanger characteristics vary significantly, due to natural heterogeneity.
Laboratory simulation
The described chamber experiment was designed and performed to investigate the processes occurring in the transition zone between saltwater and freshwater in high resolution and detail. In this chapter, the presented experimental data will be compared to field data separately for cations and anions.
Since the saltwater has a much higher ion content than the freshwater, the Na+ concentration and content (Fig. 4a) of individual samples reflects the distribution of seawater in the flow chamber. Sediments are free of easily soluble sodium; therefore, seawater is the only source of Na+. The only possible sink for Na+ is its adsorption to exchanger sites, which in turn will cause a release of weakly bound Ca2+. However, these rapid exchange processes occur only during the movement of the saltwater front. Consequently, they were completed during the first phase of the flow chamber experiment (Panteleit et al. 2003).
More complex is the concentration pattern of Ca2+ (Fig. 4b). Two sources can be distinguished: On one hand, due to the higher ion content of the seawater, the Ca2+ concentrations are highest in the saltwater dominated part of the chamber. Additionally, rising concentrations in the upper layer, which is containing Pleisto–Holocene material, identify the silty and clayey material as another source for Ca2+. This experimental result corresponds to the observations from the CAT-Field, where ion strength effects next to enhanced CO2 pressure from the degradation of organic matter were identified as major factors controlling the calcite dissolution.
The spatial distribution of the Ca2+/TEC ratio does not reflect the calcite dissolution and is conditioned by the high Na+ content of the seawater. High shares of Ca2+ were found in the freshwater-filled areas, with decreasing Ca2+ mixing ratios in areas where saltwater turns toward the Na+ type water. This result is found in both the field and laboratory data as reflected in the corresponding allocation of data points in the cation triangle of Fig. 3.
Due to the conservative behavior of Cl−, both the Cl− concentration and mixing ratio distribution depend directly on the amount of saltwater in the porewater. Cl− and Na+ distributions (Fig. 4a) correlate well with seawater, showing a constant Na+/Cl− ratio of 0.98 (Fig. 6).
While processes concerning the chloride and the cation distribution were simulated well through the flow chamber experiment, the measured anion distributions differ significantly between field and laboratory data. These deviations can be explained by different constrains inherent to the laboratory experiment:
The HCO3 − concentration is highest in layer 1 and shows a steep decrease between layers 2 and 3. Layer 1 and 2 contain organic substances in the Pleisto–Holocene material, while layer 3 consists of glacigene sands that are almost free of organic matter. Microbiological degradation of organic matter produces CO2, which dissolves to carbonic acid in the porewater which then partly dissociates according to Eq. 3. Saltwater is a second, though less important, source for HCO3 −, causing the slightly higher concentrations in the saltwater dominated part of layer 3. Due to the high ion content in the saltwater the effective HCO3 − concentrations are slightly higher than in the freshwater.
The HCO3 −/AEC ratios are lowest in the saltwater dominated part of the chamber and increase toward the freshwater area in the flow chamber. Highest ratios are found in the Pleisto–Holocene layers. Field and laboratory observations coincide. However, in the flow experiment high SO4 2− concentrations result in maximum HCO3 − ratios to the anion equivalent concentration (AEC) that stays well below those of the field samples (Table 2; Fig. 4d).
Sulfate correlates negatively with Cl−. Mixing ratios of SO4 2− are lowest in the saltwater, while freshwater has generally higher mixing ratios. The flow chamber experiment shows a steep gradient of SO4 2− with depth. Compared to field data, the freshwater in the chamber holds a very high content of SO4 2− (Table 2). Clearly two different sources for SO4 2− can be distinguished. The first source is the saltwater, intruding into the flow chamber system. The second source is the Pleisto–Holocene deposit, with high SO4 2− concentrations measured in layers 1 and 2. This observation is in contradiction to the very low SO4 2− concentrations measured in the Holocene layers of the CAT-Field. The Pleisto–Holocene sediments hold reduced sulfur species as metal sulfide minerals. During the chamber experiment these reduced species get in contact with oxidizing waters and atmospheric oxygen and reoxidize to SO4 2− (Eq. 6):
This oxygen consuming process may cause the smaller enrichment of HCO3 − in the laboratory experiment compared to field conditions, as reflected in the lower HCO3 −/Cl− ratio in Fig. 6. Accordingly the laboratory data plots more toward the CaCl zone (north in the diamond part of the piper diagram in Fig. 3). The observed high SO4 2− concentrations in the upper two layers of the chamber experiment also result in a higher SO4 2−/Cl− ratio compared to seawater. In contrast, the SO4 2−/Cl− ratio from field samples is well below the seawater value due to sulfate reduction (Fig. 6).
As for the field samples, thermodynamic calculations were performed for three flow-chamber samples, each representing one sediment layer (Table 5). The processes included in the calculations are those discussed above. Thus, the first two steps (mixing and equilibration with calcite and the CO2 pressure) are identical with the field sample calculation. In the third step, SO4 2− is added from sulfide oxidation until the calculation data reach the measured SO4 2− concentration. Finally, the exchanger sites are equilibrated with the new solution composition. In this step, the calculated cation concentrations approach the measured concentrations; however, the exchange capacity seems to be underestimated.
Conclusions
Processes that occur during saltwater intrusion into a freshwater aquifer can be simulated well through the presented flow chamber experiment. Cation distributions correlate well between experimental and field data. Given a stable position of the transition zone between saltwater and freshwater, the cation distribution is controlled by mixing, redox processes, and the dissolution of calcite. The calcite dissolution is a result of two processes: first, the changing ion strength in the mixing zone of two water types, which were previously in equilibrium with the mineral phase; second, the enhanced CO2 concentration of porewater due to the degradation of organic matter contained in the sediments.
Different redox conditions between field and flow chamber experiment affect the anion distribution. In the open chamber experiment, redox sensitive sediments were exposed to oxygen while being under reduced conditions in the field. A closed chamber system could have maintained reducing conditions, simulating field conditions to a certain degree after removing sediments from in situ and packing the chamber.
In both systems, field and flow chamber, the degradation of organic material is a driving process controlling the anion distribution and causing elevated HCO3 − concentrations. Organic material is present in high concentrations in the upper Pleisto–Holocene layer, while it is absent or not easy degradable in the deeper, glacigene sediments. In the CAT-Field oxygen for the reduction of the organic carbon is only available in the uppermost layer. In the deeper part of the Pleisto–Holocene layer sulfate is reduced and fixed in the sediment as sulfide. SO4 2− probably is replenished by seawater intrusion in deeper layers and by seaspray in the surface layer. In the flow chamber experiment, the sulfides are reoxidized in the presence of oxygen, resulting in high sulfate concentrations in the sediment layers containing Pleisto–Holocene material.
References
Andersen MS, Nyvang V, Jakobsen R, Postma D (2005) Geochemical processes and solute transport at the seawater/freshwater interface of a sandy aquifer. Geochim Cosmochim Acta 69:3979–3994. doi:10.1016/j.gca.2005.03.017
Appelo CAJ, Greinaert W (1991) Processes accompanying the intrusion of salt water. In: Breuck WD (ed) Hydrogeology of salt water intrusion—a selection of SWIM papers. Heise, Hannover, pp 291–303
Appelo CAJ, Postma D (2005) Geochemistry, groundwater and pollution, 2nd edn. AA Balkema, Leiden, p 649
Atkins P (1990) Pysikalische Chemie (Physical chemistry). VCH, Weinheim
Beekman HE, Appelo CAJ (1991) Ion chromatography of fresh- and salt-water displacement: laboratory experiments and multicomponent transport modelling. J Contam Hydrol 7:21–37. doi:10.1016/0169-7722(91)90036-Z
Berner RA (1975) The role of magnesium in the crystal growth of calcite and aragonite from sea water. Geochimica Cosmochimica Acta 39:489–504. doi:10.1016/0016-7037(75)90102-7
Berner RA, Westrich JT, Graber R, Smith J, Martens CS (1978) Inhibition of aragonite precipitation from supersaturated seawater: a laboratory and field study. Am J Sci 278:816–837
Binot F, Druivenga G, Eckard H, Fulda C, Große K, Grossmann E, Höltscher F, Kantor W, Kessels W, Neuß M, Panteleit B, Rifai H, Suckow A, and Wonik T (2002) Forschungsbohrung Cuxhaven Lüdingworth 1 und 1A CAT-LUD 1 und CAT-LUD 1A—Ergebnisse (Results from te research drillings Cuxhaven Lüdingworth), Report of the Leibnitz Institute for Applied Geosciences 121520
Bowen HJM (1979) Environmental chemistry of the elements. Academic Press, New York
Bridge ME, Lloyd DR (2005) Is there a significant diffusive “salt-pump mechanism” for the transport of organic contaminants from the marine environment into fresh water aquifers? Environ Geol 49:207–213. doi:10.1007/s00254-005-0048-5
Cook JM, Miles DL (1980) Methods for the chemical analysis of groundwater. Hydrogeology Unit, Institute of Geological Sciences, London
Custodio E, Bruggeman GA (1987) Groundwater problems in coastal areas. UNESCO
Dai Z, Samper J (2006) Inverse modeling of water flow and multicomponent reactive transport in coastal aquifer systems. J Hydrol 327:447–461. doi:10.1016/j.jhydrol.2005.11.052
Dellwig O, Watermann F, Brumsack H-J, Gerdes G (1999) High-resolution reconstruction of a Holocene coastal sequence (NW Germany) using inorganic geochemical data and diatom inventories. Estuar Coast Shelf Sci 48:617–633. doi:10.1006/ecss.1998.0462
Dellwig O, Watermann F, Brumsack H-J, Gerdes G, Krumbein WE (2001) Sulphur and iron geochemistry of Holocene coastal peats (NW Germany): a tool for palaeoenvironmental reconstruction. Palaeogeogr Palaeoclimatol Palaeoecol 167:359–379. doi:10.1016/S0031-0182(00)00247-9
Djabri L, Rouabia A, Hani A, Lamaouroux C, Polidou-Bosch A (2007) Origin of water salinity in a lake and coastal aquifer sytem. Environ Geol. doi:10.1007/s00254-007-0851-2
Domenico PA, Schwartz FW (1998) Physical and Chemical Hydrogeology, 2nd edn. Wiley, New York, 506 pp
Foyle AM, Henry VJ, Alexander CR (2002) Mapping the threat of seawater intrusion in a regional coastal aquifer aquitard system in the southeastern United States. Environ Geol 43:151–159. doi:10.1007/s00254-002-0636-6
Froelich PN, Klinkhammer GP, Bender ML, Luedtke ML, Heath GR, Cullen D, Dauphin P, Hammond D, Hartmann B, Maynard V (1979) Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochim Cosmochim Acta 43:1075–1090. doi:10.1016/0016-7037(79)90095-4
Fulda C (2002) Numerische Studie zur Salz-/Süßwasserverteilung im Rahmen der Cuxhavener Forschungsbohrung (Numeric study of the salt-/freshwater distribution at the research well Cuxhaven). Mitteilungen der Deutschen Geophysikalischen Gesellschaft Sonderband, vol II, pp 10–26
Giblin AE, Howarth RW (1984) Porewater evidence for a dynamic sedimentary iron cycle in salt marshes. Limnol Oceanogr 29:47–63
Gomis-Yagües V, Boluda-Botella N, Ruiz-Beviá F (2000) Gypsum precipitation/dissolution as an explanation of the decrease of sulphate concentration during seawater intrusion. J Hydrol 228:48–55. doi:10.1016/S0022-1694(99)00207-3
Grasshoff K, Ehrhardt M, Kremling K (1999) Methods of seawater analysis. Wiley-VCH, Weinheim
Jørgensen BB (1982) Ecology of the bacteria of the sulphur cycle with special reference to anoxic-oxic interface environments. Phil Trans R Soc Lond B 298:543–561. doi:10.1098/rstb.1982.0096
Jørgensen BB (2000) Bacteria and marine biogeochemistry. In: Schulz HD, Zabel M (eds) Marine geochemistry. Springer, Berlin, pp 173–208
Kasten S, Jørgensen BB (2000) Sulfate reduction in marine sediments. In: Schulz HD, Zabel M (eds) Marine geochemistry. Springer, Berlin, pp 263–282
Kessels W, Dörhöfer G, Fritz J, Fulda C (2000) Das Forschungsprojekt “Bremerhaven-Cuxhavener Rinne” zur Beurteilung von Grundwasservorkommen in Rinnensystemen (The research projekt “Bremerhaven-Cuxhavener Rinne” to evaluate the groundwater sources in buried valleys). Arbeitshefte Wasser 2000(1):189–203
Kim Y, Lee K-S, Koh D-C, Lee D-H, Lee S-G, Park W-B, Koh G-W, Woo M-C (2003) Hydrogeochemical and isotopic evidence of groundwater salinization in a coastal aquifer: a case study in Jeju volcanic island, Korea. J Hydrol 270:282–294. doi:10.1016/S0022-1694(02)00307-4
Kirsch R, Rumpel H-M, Scheer W, Wiederhold H (eds) (2006) Groundwater resources in buried valleys—a challenge for geosciences. LIAG, Hannover, 305 pp
Lowson RT (1982) Aqueous oxidation of pyrite by molecular oxygen. Chem Rev 82:461–497. doi:10.1021/cr00051a001
Magaritz M, Luzier JE (1985) Water-rock interactions and seawater-freshwater mixing effects in the coastal dunes aquifer, Coos Bay, Oregon. Geochim Cosmochim Acta 49:2515–2525. doi:10.1016/0016-7037(85)90119-X
Magaritz M, Goldenberg L, Kafri U, Arad A (1980) Dolomite formation in the seawater-freshwater interface. Nature 287:622–624
Mao X, Prommer H, Barry DA, Langevin CD, Panteleit B, Li L (2006a) Three-dimensional model for multi-component reactive transport with variable density groundwater flow. Environ Model Softw 21:615–628. doi:10.1016/j.envsoft.2004.11.008
Mao X, Enot P, Barry DA, Li L, Binley A, Jeng D-S (2006b) Tidal influence on behaviour of a coastal aquifer adjacent to a low-relief estuary. J Hydrol 327:110–127. doi:10.1016/j.jhydrol.2005.11.030
Martinez ME, Bocanegra EM (2002) Hydrochemistry and cation-exchange processes in the coastal aquifer of Mar Del Plata, Argentinia. Hydrogeol J 10:393–408. doi:10.1007/s10040-002-0195-7
Mercado A (1985) The use of hydrogeochemical patterns in carbonate sand and sandstone aquifers to identify intrusion and flushing of saline water. Ground Water 23:635–645. doi:10.1111/j.1745-6584.1985.tb01512.x
Morse JW, Millero FJ, Cornwell JC, Rickard D (1987) The chemistry of the hydrogen sulfide and iron sulfide systems in natural waters. Earth Sci Rev 24:1–42. doi:10.1016/0012-8252(87)90046-8
Nadler A, Magaritz M, Mazor E (1980) Chemical reactions of seawater with rocks and freshwater: experimental and field observation on brackish waters in Israel. Geochim Cosmochim Acta 44:879–886. doi:10.1016/0016-7037(80)90268-9
Panteleit B, Hammerich T (2005) Hydrochemische Charakteristiken und Prozesse im Küstenbereich bei Cuxhaven (Hydrochemical characteristics and processes at the coastal zone near Cuxhaven/Germany). Z Angew Geol 51:58–64
Panteleit B, Binot F, Kessels W, Schulz HD, Kantor W (2001a) Geological and geochemical characteristics of a salinization-zone in a coastal aquifer. In: Seiler K-P, Wohnlich S (eds) New approaches characterising groundwater flow, vol 2. AA Balkema, Leiden, pp 1237–1241
Panteleit B, Kessels W, Kantor W, Schulz HD (2001b) Geochemical characteristics of salinization-zones in the coastal aquifer test field (CAT-Field) in North Germany. Paper presented at the SWICA-M3 saltwater intrusion and coastal aquifers conference, Essaouira
Panteleit B, Schulz HD, Kessels W (2002) Geochemical processes in the salt-freshwater transition zone—preliminary results of a sand tank experiment. Paper presented at the 17th salt water intrusion meeting, Delft, 6–10 May 2002
Panteleit B, Kessels W, Schulz HD (2003) Geochemical processes in the salt-freshwater transition zone—exchanger reactions in a 2D-sand-tank experiment. In: Hadeler A, Schulz HD (eds) Geochemical Processes in soil and groundwater—measurement–modeling–upscaling. Wiley VCH, Weinheim, pp 596–610
Panteleit B, Binot F, Kessels W (2006) Mud tracer test during soft rock drilling. Water Resour Res 42:W11415. doi:10.1029/2005WR004487
Parkhurst DL, Appelo CAJ (1999) PHREEQC for Windows—a hydrogeochemical transport model. USGS, Denver
Reddy MM (1977) Crystallisation of calcium carbonate in the presence of trace concentrations of phosphorous containing anions. J Cryst Growth 41:287–295. doi:10.1016/0022-0248(77)90057-4
Robertson WD, Cherry JA, Schiff SL (1989) Atmospheric sulfur deposition 1950–1985 inferred from sulfate in groundwater. Water Resour Res 25:1111–1123. doi:10.1029/WR025i006p01111
Sanford WE, Konikow LF (1989) Simulation of calcite dissolution and porosity changes in saltwater mixing zones in coastal aquifers. Water Resour Res 25:655–667. doi:10.1029/WR025i004p00655
Schulz HD, Dahmke A, Schinzel U, Wallmann K, Zabel M (1994) Early diagenetic processes, fluxes, and reaction rates in sediments of the South Atlantic. Geochim Cosmochim Acta 58:2041–2060. doi:10.1016/0016-7037(94)90284-4
Tomson MB (1983) Effect of precipitation inhibitors on calcium carbonate scale formation. J Cryst Growth 62:106–112. doi:10.1016/0022-0248(83)90013-1
Vandenbohede A, Lebbe L (2002) Numerical modelling and hydrochemical characterisation of a fresh-water lens in the Belgian coastal plain. Hydrogeol J 10:576–586. doi:10.1007/s10040-002-0209-5
Walraevens K, Cardenal-Escarcena J, Van Camp M (2007) Reaction transport modelling of a freshening aquifer (Tertiary Ledo-Paniselian Aquifer, Flanders-Belgium). Appl Geochem 22:289–305. doi:10.1016/j.apgeochem.2006.09.006
Walter LM, Hanor JS (1979) Effect of orthophosphate on the dissolution kinetics of biogenic magnesian calcites. Geochim Cosmochim Acta 43:1377–1385. doi:10.1016/0016-7037(79)90128-5
Ward WC, Halley RB (1985) Dolomitization in a mixing zone of near-seawater composition, late Pleistocene, northeastern Yucatan Peninsula. J Sediment Petrol 55(3):407–420
Werner AD, Gallagher MR (2006) Characterisation of seawater-intrusion in the Pioneer Valley, Australia, using hydrochemistry and three-dimensional numerical modelling. Hydrogeol J 14:1452–1469. doi:10.1007/s10040-006-0059-7
Wiederhold H, Binot F, Kessels W (2005) Die Forschungsbohrung Cuxhaven und das Coastal Aquifer Testfield (CAT-Field)—ein Testfeld für angewandte geowissenschaftliche Forschung (The Cuxhaven research borehole and the coastal aquifer test field (CAT-Field)—a test field for applied geoscientific research. Z Angew Geol 51:3–7
Zilberbrand M, Rosenthal E, Shachnai E (2001) Impact of urbanization on hydrochemical evolution of groundwater and on unsaturated-zone gas composition in the coastal city of Tel Aviv, Israel. J Contam Hydrol 50:175–208
Acknowledgments
Antje Weitz is thanked for discussion, debates and helpful reviews which improved the paper and ideas. Toni Wienrich, Yvonne Habenicht, Jens Berger and Markus Helms are thanked for their aid in constructing, sampling and analyzing the flow chamber.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Panteleit, B., Hamer, K., Kringel, R. et al. Geochemical processes in the saltwater–freshwater transition zone: comparing results of a sand tank experiment with field data. Environ Earth Sci 62, 77–91 (2011). https://doi.org/10.1007/s12665-010-0499-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12665-010-0499-1