
Abstract
Lignin, a natural biopolymer and abundant by-product, is a particularly promising feedstock for carbon-based materials and a potentially sustainable alternative to phenolic resins, which are typically derived from crude oil. The source and method used to isolate lignin have a large impact on the thermal properties of the polymer, and can affect resultant materials prepared from lignin. Previous investigations into lignin characterisation often utilise a variety of feedstocks and isolation methods, which can make robust comparisons challenging. We present a systematic investigation into the chemical composition of lignins extracted using an identical Organosolv isolation method but from different biomass feedstocks: hemp hurds, eucalyptus chips, flax straw, rice husk and pine. We show how the aromatic structure of lignin can affect the thermal behaviour of the polymer, which correlates to the structure of resulting carbons. Carbons from lignins with a high syringyl unit content display a pronounced foaming behaviour which, on activation, results in a high-surface area material with hierarchical porosity.

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lignin is a natural biopolymer found in lignocellulosic biomass and is a major by-product of the paper pulping and biofuel production processes. Every year, 70 million metric tons of lignin is produced, [1] and without a large-scale application it is primarily burnt as a fuel [1, 2]. The wide availability, low cost and high aromaticity of lignin have made this an attractive feedstock for a variety of applications. The high carbon content of lignin makes it a particularly promising feedstock for carbon-based materials including carbon fibre and activated carbons [3,4,5,6]. Lignin-derived activated carbons have shown promise in a range of applications from electrode materials in supercapacitors [7,8,9,10] and batteries [11,12,13] to sorbents for the adsorptive storage of gases [14, 15]. In addition, the highly aromatic structure of lignin makes it a good candidate for a sustainable replacement for phenolic resin, a crude oil-derived aromatic polymer used in the synthesis of polymeric carbons and as a thermal binder [16,17,18,19].
Native lignin is found in plant cell walls within a matrix of cellulose and hemicellulose [20,21,22]. The polymer has a highly complex structure that differs depending on its origin. Although due to the complexity of the three-dimensional macromolecule the exact structure of lignin is unknown, [23] it is formed primarily from three aromatic monomers known as monolignols; p-coumaryl, coniferyl, and sinapyl alcohol. These result in the formation of p-hydroxyphenyl (H), guaiacyl (G) and sinapyl (S) residues respectively in the polymer [20, 22].
Studies have previously shown that the thermal properties of lignin, and the structure of lignin-derived carbon materials, can be affected by both the polymer source and isolation method [8, 24,25,26,27,28,29]. Previous investigations into lignin characterisation, however, have frequently utilised a variety of feedstocks and pulping methods, making it challenging to make robust comparisons. Here we present the characterisation of five lignins, four were extracted using an identical organosolv pulping process from different sources; hemp hurds (also called hemp shives or core fibres), eucalyptus chips, flax straw, and rice husk, in addition to an industrial organosolv extracted lignin from pine. The feedstocks were chosen to ensure two types of lignin with differences in aromatic structure were examined, wood lignin (pine, hemp hurds and eucalyptus chips) and grass lignin (flax straw and rice husk). We show how the aromatic lignin structure impacts the thermal behaviour of the polymer, and the high-value application of these materials to produce products such as activated carbons with hierarchical porosity.
Materials and Methods
Materials
The four lignocellulosic feedstocks (hemp hurds, eucalyptus chips, flax straw and rice husk) and an industrial organosolv lignin from pine were provided by MAST Carbon International, Basingstoke. The biomass was used as provided, chopped into pieces up to 3 cm long. All reagents and solvents used were of analytical grade.
Organosolv Lignin Extraction
The organosolv process used to extract lignin was based on the autocatalytic Alcell process [30]. Experimental conditions were chosen according to information from a variety of sources [31,32,33]. Digestion of biomass took place in a Parr pressure vessel. A suspension of biomass-water-ethanol (10 mL solvent per 1 g biomass, solvent was a 60:40 ethanol-deionised water solution by weight) was heated in the sealed vessel at 200 °C for 2 h before cooling to room temperature. The delignified fibre and lignin-rich black liquor were separated by filtration, and fibres rinsed with a 60:40 EtOH-DI solution. The filtrate and black liquor were combined and the ethanol removed under vacuum until the onset of precipitation. Precipitation was achieved by the addition of a 0.1 M sulphuric acid solution to the concentrated liquor (2:1 ratio by volume of acid to liquor) whilst stirring. Lignin was separated by filtration and rinsed with DI water until the filtrate was neutral, then dried in a drying oven at 55 °C. Extractions were performed in duplicate for each feedstock.
Purity Determination of Lignin
Lignin purity is commonly defined by the acid-insoluble (known as Klason) lignin content [34]. A modified procedure described by Effland et al. [35] was followed. The dried lignin sample (200 mg to the nearest 0.1 mg) was dissolved in 72% sulphuric acid (2 mL), held at 30 °C for one hour, and stirred at frequent intervals. The mixture was quantitatively transferred to a conical flask using deionised (DI) water (56 mL). The flask was covered and boiled on a hot plate for 4 h; the volume was kept constant by addition of DI water. The solution was filtered hot through a sintered glass crucible, washed with hot water, and then dried at 105 °C overnight. Samples were cooled in a desiccator before weighing. The Klason content was determined as a percentage of the oven-dried lignin sample. Analyses were run in duplicate for each sample.
Fourier transform infrared (FTIR) spectroscopy was used to confirm the absence of cellulose in lignin samples. A force of 115 N was applied to a small amount of solid sample on a Perkin Elmer Spectrum 100. A spectrum of 32 scans was recorded from 4000 to 600 cm−1 with a resolution of 4 cm−1. The instrument software Spectrum version 10.4.00 was used to remove a background spectrum, which was run at the start of the experiment. A MasterGrind Shield Series Aluminium Herb Grinder was used to grind feedstocks and dried, rinsed pulp samples to a fine powder. Ground samples were then used to record a FTIR spectrum.
Microanalysis of lignin samples were carried out by Des Davies at the University of Bristol Microanalytical Laboratory, UK. Elemental analyses (C, H, N, S) were carried out using combustion, content of O was determined by difference. Ash content was determined by burning the sample in pure oxygen at 900 °C.
Lignin Structure Characterisation
Characterisation of the lignin polymer structure was performed using gel permeation chromatography (GPC) and 2D 1H-13C HSQC NMR spectroscopy. GPC was used to determine the molecular weight and polydispersity of lignin polymers. Acetylation of lignin to enable dissolution in tetrahydrofuran (THF) was not required for the organosolv lignin examined. Samples were dissolved in THF only at a concentration of 2 mg mL−1. Characterisation was performed on an Agilent Technologies 1260 Infinity instrument with a flow rate of 1 mL min−1 at 35 °C, THF was used as the eluent and samples were referenced against a polystyrene standard.
All NMR spectra were acquired on a Bruker Avance III NMR spectrometer operating at 500.13 MHz for 1H and 125.77 MHz for 13C. Lignin samples (approximately 60 mg) were swollen in dimethyl sulfoxide-d6. 2D 1H-13C HSQC spectra were acquired using a spectral width of − 40–180 ppm for 13C, and − 1.28–10.68 ppm for 1H. The standard Bruker pulse sequence hsqcedetgpsp.3 was used with 128 transients for 256 increments and a TD of 1024 in the F2 direction. Data acquisition and processing were performed using Topspin 2.1. A semi-quantitative method was used for analysis of the HSQC cross-peak intensities, as described by del Rio et al. and Wen et al. [36, 37] Inter-unit linkages were estimated from peaks in the aliphatic side-chain region (δC/δH 50–90/2.5–5.5 ppm). Relative abundances of linkages were estimated from Cα-Hα correlations. The S/G ratio was determined from peaks in the aromatic region (δC/δH 95–140/6.0-7.7 ppm). The C2,6-H2,6 peak correlating to S-units and the C2-H2 plus C6-H6 peaks correlating to G-units were used to calculate the S/G ratio. The same area was integrated over all spectra using the MestReNova software. Cross-peaks were assigned using literature data from a variety of sources [27, 32, 36,37,38,39].
Thermal Behaviour Determination
The thermal behaviour of organosolv lignins was examined using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The DSC method was based on that of Huijgen [40] and Buranov [41]. The thermal analysis was performed under a nitrogen flow (50 mL min− 1) at a heating rate of 10 °C min− 1 throughout. The analysis was performed over two cycles, with the first to eliminate any history effects. Samples were heated from 20 to 140 °C, held isothermally for 10 min, quenched from 140 to − 50 °C, and held isothermally for 10 min. Samples were then heated a second time from – 50 to 200 °C. Samples were run in duplicate and the mean values are reported.
TGA was undertaken using a Seteram TGA 92 16.18 analyser at the University of Bath. A small amount of sample (10–15 mg) was weighed into a 100 µL alumina crucible. To obtain a TG trace the samples were run from 20 to 1000 °C at 5 °C min− 1 in nitrogen (99.8% purity, 1.5 bar). To obtain thermal decomposition (Td) values, samples were heated from 20 to 105 °C at 5 °C min− 1 in nitrogen, dried for 30 min, and then heated as before. The dry weight of the sample was taken at 105 °C, and the Td calculated as the temperature at which 95 wt% of the dry sample remained. A scanning electron microscope (JEOL IT300 SEM) was used to obtain micrographs of TGA chars and pyrolysed lignin samples. The microscope was operated in secondary electron mode at an accelerating voltage of 15 kV.
Activated Carbon Preparation and Characterisation
Pyrolysis was performed in a horizontal tube furnace (Lenton, LTF 12/75/750) fitted with a digital PID controller 3216CC. The furnace contained a separate work tube fitted with gas-tight end seals. The industrial organosolv pine lignin (3 g) was placed in an alumina combustion boat (20 ml capacity) and inserted into the centre of the furnace. PureShield argon (99.998% purity) with a flow rate of 500 ml min− 1 was used as the furnace gas. The furnace was purged with inert gas for 30 min, then heated from room temperature to 105 °C and held for a further 30 min. The sample was then heated from 105 °C to 350 or 900 °C, samples were held at temperature for 30 or 360 min, a heating rate of 5 °C min− 1 was used throughout. The char was ground and passed through a 35-mesh sieve prior to activation. The ground char (1 g) was heated to the activation temperature of 1000 °C under argon using the same method as the char pyrolysis. At the activation temperature, the furnace gas was switched to carbon dioxide (99.998% purity) at 100 ml min− 1 for 60 min, then cooled to room temperature in an argon atmosphere (500 ml min− 1).
Carbons were characterised by nitrogen adsorption at 77 K using a 3Flex analyser from Micromeritics. Prior to analysis, chars (1 g) and carbons (100 mg) were degassed at 250 °C and 300 °C respectively for a minimum of 4 h under dynamic 10− 7 mbar vacuum. Isotherm data was collected using filler rods over the range p/po 0.01–0.30. The BET surface area was calculated according to the procedure set out by the International Organisation for Standardisation ISO 9277:2010 [42]. The Rouquerol correction was applied, and the range p/po 0.01–0.03 used to calculate the BET surface area.
Results & Discussion
Extraction & Purity Determination of Organosolv Lignin
For reliable comparison of differences in lignin structure between the four feedstocks, an identical ethanol-based organosolv pulping method was used to isolate lignin from; hemp, flax, rice husk, and eucalyptus. The organosolv process was the chosen isolation method because it results in less modification of the lignin in comparison to other procedures, and allowed facile adaptation of this method to a laboratory scale. The lignin content of biomass depends primarily on the species, growing conditions, and geographic location. The efficiency of the organosolv extraction process for the four starting feedstocks is presented in Table 1, and represented as the yield of lignin produced per weight of starting biomass. The average yield was similar for each feedstock (around 10 wt%), although eucalyptus had a slightly higher yield (nearer 14 wt%). Similar extraction conditions were employed by Wildschut et al. [43] who reported feedstock delignification of 40%. As eucalyptus chips are reported to contain 23 to 35% lignin,[44, 37] flax straw 24 to 31%, [41, 45] hemp hurds between 16 and 28%,[46, 47] and rice husk between 19 and 31%, [48, 49] a delignification rate of 40% means the lignin yields reported here are within the expected range.
High purity lignin samples were required for robust comparisons of lignin structure, and the resulting effects on thermal behaviour. The purity of the lab-isolated and industrial organosolv lignins were assessed using the acid insoluble (known as Klason) lignin content. The lignin undergoes an acid-catalysed hydrolysis, solubilising the carbohydrates and leaving behind an acid-insoluble residue, which is determined gravimetrically [34, 50]. This method is one of the most common measures of lignin content and has been used throughout the literature [27, 43, 45, 51,52,53,54].
The acid insoluble lignin content, ash content and elemental composition of the samples are shown in Table 1. The high purity of all lignin samples was confirmed by their high acid-insoluble lignin content (> 80 wt%), and negligible ash content (< 0.15 wt%). Elemental analysis showed lignin samples were primarily composed of carbon, oxygen, and hydrogen in a ratio of approximately 65:30:6. These lignins also contained trace nitrogen (< 1.20 wt%) and sulphur (< 0.15 wt%). The elemental composition is consistent with values reported for other organosolv lignins in the literature, [22, 27, 55] while the trace sulphur is a result of the acid precipitation step in the extraction procedure.
Furthermore, the successful delignification of the feedstock and extraction of a product free from cellulose was supported by Fourier Transform Infrared (FTIR) spectroscopy. FTIR spectra (not shown) of the four lab-extracted organosolv lignins were free from bands characteristic of the presence of hemicellulose (at 1740 and 1240 cm− 1) and cellulose (at 898 cm− 1) [32, 51, 56].
Characterisation of Lignin Structure
Differences in polymer structure and molecular weight between Organosolv lignin samples was assessed using a combination of 2D NMR spectroscopy and GPC. The aromatic structure and linkages (depicted in Fig. 1) of lignin samples were examined using 2D HSQC NMR 1H-13C spectroscopy, spectra are shown in Fig. 2. Valuable information about the abundance of monolignol monomers can be determined in the aromatic region (δC/δH 95–140/6.0-7.8 ppm). All lignin samples had signals relating to syringyl (S) and guaiacyl (G) units. In addition, rice husk and hemp lignins had a signal characteristic of the p-hydroxyphenyl (H) unit. The S2,6 peak was present in all spectra and is especially broad for the eucalyptus and industrial lignins. The S’2,6 peak of the α-oxidised S unit is present in all spectra except flax lignin. Signals corresponding to G2, G5, and G6 of the G-unit are present in all lignins. The H2,6 signal is only present in the rice husk and hemp lignin spectra, and is far more intense in the former. Cross-peaks relating to cinnamaldehyde end groups Jβ and cinnamyl alcohol end groups, Iα were present in all lignin spectra. Hemp and rice husk lignin also exhibited a cross peak for p-coumaric acid PCA, which is not seen in eucalyptus, flax, and industrial lignins due to the absence of H-units. The samples can be divided into two main types of lignin: high S unit content and high G unit content. Relative values were calculated for the ratio of S and G units (S/G ratio), and are shown in Table 2. Eucalyptus, hemp, and industrial lignins were primarily composed of S units, with S/G ratios of 2.85, 1.18, and 1.37 respectively. Rice husk and flax lignins by contrast have a higher G unit content, with S/G ratios of 0.19 and 0.18 respectively.

The aromatic structures present in lignin; s syringyl unit, s’ oxidised syringyl unit, g guaiacyl unit, h p-hydroxyphenyl unit, fa ferulic acid, i cinnamyl alcohol end group, j cinnamaldehyde end group, and pca p-coumaric acid, and the main linkages present; a β-aryl-ether β-O-4, b resinol β-β, c phenylcoumaran β-5, and d spirodienone

2D-NMR HSQC spectra of the aromatic region (left) (δC/δH 95–140/6.0-7.8 ppm) and aliphatic side-chain region (right) (δC/δH 50–90/2.5-6.0 ppm) of a hemp lignin, b flax lignin, c rice husk lignin, d eucalyptus lignin, and e industrial lignin. Spectra have been recoloured to highlight regions of interest using Inkscape
In the aliphatic side-chain region (δC/δH 50–90/2.5-6.0 ppm) cross-peaks corresponding to methoxy groups are prominent in all lignin samples. Signals corresponding to β-aryl-ether β-O-4 (A), resinol β-β (B), and phenylcoumaran β-5 (C) linkages are evident in all spectra at varying intensities. In particular the β-O-4 linkage appears highly abundant in all spectra (43–54%), which differs from Constant et al. [27] who reported a low β-O-4 content in their organosolv lignins from poplar and spruce. The β-O-4 linkage is usually the most abundant in native lignin but is readily cleaved by depolymerisation/isolation methods. The difference here is likely due to the harsher pulping conditions used by Constant et al. causing extensive cleavage of the β-O-4. The low S/G lignins from rice husk and flax have a high abundance of β-5 (Cα) linkages (~ 40%), which contrasts with the low abundance (10–17%) in high S/G lignins from eucalyptus, hemp, and the industrial pine. Conversely, the high S/G lignins show a higher abundance of β-β (Bα) linkages (29–35%) than the low S/G lignins (13–17%). All lignins except for eucalyptus have signals belonging to p-hydroxycinnamyl alcohol end groups. Spirodienone structures (Dα) appear to be present in eucalyptus lignin only, and rice husk lignin is the only sample to show signals relating to carbohydrates.
Gel permeation chromatography (GPC) was used to determine the molecular weights of lignin samples. The molecular weight of lignin obtained by GPC has previously been demonstrated in the literature to be highly dependent on the experimental set-up [27, 28, 38, 51]. This includes the type of column, eluent chosen, calibration standard, and pre-treatment of the lignin sample. Absolute values of molecular weight should, therefore, be used with caution, but comparisons can be made between different lignins run under the same conditions. The samples were examined using a THF solvent system without acetylation, and compared to a polystyrene standard, the results are illustrated in Fig. 3a. Values range from 800 to 1100 Da for the number average molecular weight Mn, 1500 to 2200 Da for the weight average molecular weight Mw, and 1.94 to 2.28 for the polydispersity index (PDI). Organosolv lignins are known to have an overall lower molecular weight than lignins extracted from alternative methods (such as from the Kraft process), [22, 25, 27, 31] and values of a similar range are reported in this paper. Despite similar values across the lignins examined, there are small differences in the average Mw of samples. Rice husk lignin has the lowest Mw of around 1550 Da, the industrial, flax and hemp lignins have increasing values of 1800, 2000 and 2200 Da respectively, and eucalyptus lignin had the highest Mw value of 2400 Da. A comparison of the size exclusion chromatograms (Fig. 3b) shows the Mw distribution of rice husk and the industrial lignins are shifted towards lower molecular weight values. A high molecular weight shoulder is seen in the other three samples, most prominently in eucalyptus lignin.

a GPC results of lignin samples extracted using the organosolv method. Mn is the number average molecular weight, Mw is the weight average molecular weight, and PDI is the polydispersity index. Each sample was measured three times. Error bars show the greatest difference between the maximum and minimum measurements and the mean. Results show that the lignin extracted from each feedstock has a relatively low molecular weight (Mw 2.4–1.5 kDa). b Size exclusion chromatograms (SEC) of the four lignin samples isolated using the same extraction method from rice husk (Blue solid line), flax (Green dashed line), hemp (Violet dash dotted line), and eucalyptus (Orange dash double dotted line) feedstocks, and an industrial organosolv lignin (Black dotted line). Samples were run in triplicate. The full data set for each lignin type is included in the supplementary information. (Color figure online)
Constant et al. [27] has previously shown that the starting feedstock had little bearing on the average molecular weight of lignin polymers, however it did influence the Mw distribution. Findings by Tejado et al. [25] support the idea that lignin polymers with a higher G-unit content show a higher average Mw. This is reportedly due to the presence of highly stable C-C bonds at the C5 position of the aromatic ring, which are not cleaved during the pulping process. Lignins with S-units are unable to form these bonds due to the C-5 substituted methoxy group. While literature data indicates the extraction method has the most significant influence, our findings show the feedstock also appears to have a small effect on the molecular weight distribution of lignin. In contrast to findings by Tejado et al. however, the lignin with the largest S-unit content exhibited the highest Mw. In addition, hemp and flax which have a high and low S/G ratio respectively both exhibit a similar Mw value and distribution.
Thermal Behaviour of Organosolv Lignins
The behaviour of organosolv lignin samples is important in the context of thermal processing of the polymer. The primary difference between the lignin samples examined in this paper has been shown to be the make-up of the polymer’s aromatic backbone. This should thus be the primary influence on variations in thermal behaviour between the different lignin samples, which was examined using a combination of differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). Temperature- heat flow curves from DSC analysis are illustrated in Fig. 4a. An inflection, related to the glass transition (Tg) temperature, is immediately evident in all lignins examined. The glass transition of lignin represents the segmental mobility of lignin polymeric chains [52] with organosolv lignins typically having a low Tg of 100–120 °C [28, 41, 52]. The Tg values of all samples tested here (values listed in Table 3), fall within the values expected for organosolv lignins.

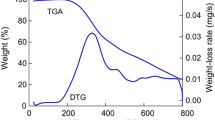
a Differential scanning calorimetry (DSC) curves. The onset temperatures relating to the glass transition are highlighted. Spectra were offset along the heat flow axis for clarity. b Thermogravimetric (TG) trace of rice husk (Blue solid line), flax (Green dashed line), hemp (Violet dash dotted line), eucalyptus (Orange dash double dotted line), and industrial (Black dotted line) organosolv lignins. Samples were run from 20 to 1000 °C in nitrogen at 5 °C min− 1. Inset: Images of post-analysis TG chars, it is evident two types of carbon are formed. Eucalyptus and hemp lignins produced a carbon foam, whereas rice husk and flax lignins produce a carbon char. Samples for both experiments were run in duplicate. (Color figure online)
Lignins from different sources did show variation in Tg. The high S/G (~ 1.3) industrial and hemp lignins have the lowest Tg values (101 and 103 °C respectively), and the low S/G (~ 0.2) flax and rice husk lignins have an increased Tg (111 and 118 °C respectively). Eucalyptus lignin however, with a S/G ratio (2.9) higher than the industrial and hemp lignins, exhibited the highest glass transition (120 °C), indicating Tg is influenced by factors other than the aromatic composition. A variety of factors including the molecular weight distribution, degree of crosslinking, hydrogen bonding, free volume between polymer chains and chemical structure, [24, 57] have previously been shown to impact the Tg. It is therefore difficult to assess which factor has the greatest influence. It is well known within the literature [3, 24, 25, 57] that, as with molecular weight, the lignin extraction procedure has a large influence on the glass transition. Higher Tg ranges are reported for lignins extracted via steam explosion (113–139 °C) and the Kraft process (120–160 °C).
Thermogravimetric traces for thermal decomposition of lignin samples are shown in Fig. 4b, and values are listed Table 3. Lignin decomposition occurs over a wide temperature range, unlike the decomposition of cellulosic components of biomass. The four organosolv lignins all exhibit similar decomposition behaviour, which can be divided into three stages, as noted by Watkins et al. [58] An initial small decline in sample weight is due to water loss by evaporation. A sharp decrease in weight loss is then observed between 200 and 400 °C, when 40–50 wt% of the material is lost. This is due to the degradation of carbohydrate components in the lignin, which are converted to volatile gases including CO, CO2, and CH4. Finally, a more gradual but significant loss of mass continues up to 1000 °C, representing emission of volatile components including phenolics, alcohols and aldehyde acids.
The decomposition temperature of samples (Td), which corresponds to the temperature at which 95 wt% of an initial dry sample remains, [59] is recorded in Table 3. The decomposition temperature of hemp lignin appears to begin at a lower temperature (263 °C) than the other lignin samples (~ 275 °C). This early onset can be observed in the hemp lignin TG trace (Fig. 4b), where the sharp mass loss stage of the sample begins first. Most notably, a swelling of the char was observed during TG analysis of the high S/G hemp, eucalyptus, and industrial lignins, producing a carbon foam, as shown in the inset of Fig. 4b. The low S/G flax and rice husk lignins did not exhibit swelling to the same extent. Consequently the highest char yields were recorded for rice husk and flax lignins (39.8 wt% and 38.6 wt% respectively), followed by the high S/G eucalyptus and industrial lignins (36.2 and 35.6 wt% respectively) and hemp (33.7 wt%) lignin. Similar differences in char yields relating to the aromatic content have been noted by other groups [26, 28]. Lignins from softwoods (with a high G-unit content) had an increased char yield compared to those from hardwoods (containing both G and S units). These results were confirmed by this work, and the increased char yield can be attributed to the higher stability of ether bonds between G-units resulting in higher thermal stability, and the propensity of G-units to undergo condensation and coupling reactions, forming a more cross-linked char and resulting in an increased yield of residue [26, 28].
The peak of the first derivative of the thermogravimetric curves (DTGmax, not shown) corresponds to the thermal decomposition temperature of materials. Values for DTGmax are summarised in Table 3. Vales are similar across the samples analysed, increasing for lignins in the order; eucalyptus and hemp (354 °C), rice husk (359 °C), and flax (368 °C). The values are comparable to those recorded previously for organosolv-extracted wheat straw lignins [52, 55]. In addition to DTGmax, all lignin samples exhibit a low-temperature shoulder between 150 and 250 °C, which was more pronounced in hemp lignin than the other samples. Although not seen on the 2D-NMR, this shoulder is possibly due to residual carbohydrates from hemicellulose in the samples, [55]. The more pronounced shoulder in hemp lignin could explain the earlier onset of decomposition.
Scanning electron microscopy was used to investigate the structure of lignin TG chars further (see Fig. 5). It is clear from the micrographs that each lignin melted to form a smooth carbon. Micrographs of flax and rice husk chars are shown in Fig. 5a, b respectively. The top surface of both chars shows evidence of large bubbles forming and breaking, presumably due to the release of volatiles during carbonisation. These bubbles are also evident in the underside of the rice husk char. The foaming of the hemp and industrial lignin chars can be clearly seen in Fig. 5c, d, with large voids throughout the structure. Despite shattering of the eucalyptus lignin char, the smooth carbon surface and formation of bubbles is also evident in Fig. 5e.

Scanning electron micrographs of chars from the thermogravimetric analysis of a flax, b rice husk, c hemp, d industrial, and e eucalyptus organosolv lignins. Samples were heated from room temperature to 1000 °C at 5°C/min under nitrogen
Despite similar decomposition traces, extensive swelling occurs in some lignin samples but not in others. The swelling behaviour was investigated further by heating small samples (not shown) of the industrial lignin in a conventional oven (Jeio Tech OV-11/12) in air from room temperature to 250 °C. The lignin had formed a melt by 150 °C, and the onset of expansion occurred between 150 and 200 °C, with an increase in sample volume of 210% during this period. From 150 to 250 °C (the highest temperature examined) the sample volume increased by ~ 250%. On removal from the heat source deflation of the char occurred, implying gas escape is the cause of the swelling, and indicates volatiles released during decomposition provide the mechanism for the swelling to occur.
The chemical properties of all lignins investigated were similar; they have a high lignin purity, low ash content, low molecular weight, and similar decomposition trace. The swelling observed in hemp, eucalyptus, and the industrial lignins therefore is thought to be an effect of differences in the aromatic structure. The aforementioned high S/G ratio lignins exhibit swelling of the sample on heating, whereas the low S/G ratio flax and rice husk lignins do not. Foaming of lignocellulosic materials is not common. A plastic phase and swelling behaviour was previously observed by Rodríguez-Mirasol et al. [60] in Kraft eucalyptus lignin. They noted the swelling decreased and disappeared in samples with increasing ash content. Rios et al. [61] prepared a carbon foam by pyrolysis of olive stones under steam, which required a minimum heating rate of 30 °C min− 1 to initiate foam formation. They postulated that for a lignocellulosic precursor to expand the following conditions must be met: (i) softening of the lignocellulosic matrix (lignin in particular) which provides the structural rigidity, (ii) formation of an intermediate ‘melt’ of high viscosity due to depolymerisation of some components, and (iii) evolution of volatiles from the melt before polymerisation of the precursor causes it to solidify.
All lignin samples are seen to form a melt, as evidenced from the plastic-like appearance of the chars and presence of bubbles in the structure in Fig. 5. The foaming in hemp and eucalyptus lignins is likely due to differences in viscosity of the melts between the high and low S/G ratio lignins. Sun et al. [28] noted that low S/G lignins have increased viscosity values compared to high S/G lignins, which was attributed to increased cross-linking in the char, and is supported by the increased char yield reported in this work. Furthermore, 2D-NMR results in this work show that low S/G lignins have an increased quantity (~ 40%) of β-5 linkages compared to high S/G lignins (< 20%). The β-5 linkages limit molecular rotation, [3] which may also lead to increased viscosity of the low S/G lignin melt.
Preparation of Hierarchical Activated Carbons from Lignin
The differences in swelling behaviour between high and low S/G ratio Organosolv lignins has important implications for the application of these materials. The use of high S/G ratio lignins as a binder or for the formation of carbon monoliths for example would not be recommended. Swelling of the lignin during heat treatment would cause deformation and potentially an increasingly brittle product. Therefore the use of a low S/G ratio lignin would be more appropriate. The formation of carbon foams by the high S/G ratio lignins, however, leads to exciting possibilities as a new feedstock for carbon materials with a hierarchical pore structure. Applications including water filtration and supercapacitance require a pore structure with micropores, mesopores, and macropores, to allow rapid diffusion and transport of the adsorptive species.
To investigate the viability of these materials, industrial organosolv lignin was used to prepare activated carbons. Lignin was carbonised using both mild (350 °C for 30 min) and relatively harsh conditions (900 °C for 6 h) to prepare the 350-C and 900-C carbon foams respectively. Activation of foams in carbon dioxide at 1000 °C produced the 350-1000-AC and 900-1000-AC activated carbon samples. The burn-off values and adsorptive characteristics of the lignin chars and activated carbons are listed in Table 4. Nitrogen sorption was used to examine the surface area of the char samples. The BET surface area of char 350-C was too low for accurate measurement (the measurement limit of the instrument is 30 m2 g− 1), and the N2 uptake of the sample (at p/po = 0.30) was well below 1 cm3 g− 1 (STP). The 900 °C char by contrast had a BET surface area of 150 m2 g− 1 and N2 uptake (at p/po = 0.30) above 30 cm3 g− 1 (STP). These results indicate the foaming effect produces carbon with large macropores, but is essentially non-porous at 350 °C.
Below 350 °C low molecular weight volatiles (CO, CO2, H2O, MeOH, HCHO) are released in the initial stages of pyrolysis, [26, 62,63,64,65] in quantities that, as shown previously, contribute to the swelling of the char to form a carbon foam. Research by Liu et al. [63] shows that increasing the carbonisation temperature greatly increases the amount of low molecular weight volatiles released. This is due to devolatilization of higher molecular weight compounds (including phenols and hydrocarbons) and secondary pyrolysis of these products to release CO, CO2 and CH4. Increasing the carbonisation temperature to 900 °C thus increases the quantity of volatiles emitted, enhancing the rudimentary pore structure of the char and leading to an increase in BET surface area.
Full nitrogen isotherms (not shown) of the 350-1000-AC and 900-1000-AC activated carbons were used to extract information on the surface area, gas uptake and pore volume of materials. Both samples exhibited primarily type 1 isotherms, indicative of highly microporous materials. The isotherm of 350-1000-AC also showed a small uptake of nitrogen between p/po = 0.2–0.9, indicating the presence of mesopores in the sample. Both materials had a large BET surface area; 1400 m2 g− 1 for 350-1000-AC and 1060 m2 g − 1 for 900-1000-AC. The total pore volume (Vtot) of 350-1000-AC (0.70 cm3 g − 1) was higher than 900-1000-AC (0.46 cm3 g − 1). The micropore volumes (Vmicro) of 350-1000-AC (0.31 cm3 g − 1) and 900-1000-AC (0.38 cm3 g − 1) were similar. However, Vmicro as a percentage of Vtot (44% and 83% respectively) was highest for 900-1000-AC.
The two different carbonisation conditions of the industrial lignin have produced high surface area activated carbons. Further optimisation of the activation conditions could be used to further enhance the quantity of mesopores in samples, dependent on the desired application. Activation of the mildly treated carbon foam produced an activated carbon with a higher surface area and pore volume than the more severely treated carbon foam. In addition 350-1000-AC has a hierarchical structure, with the isotherm indicating the presence of both micro-and mesopores, which is not seen in 900-1000-AC. The lower proportion of micropores to the total pore volume in comparison to 900-1000-AC can be attributed to the presence of this mesoporosity in 350-1000-AC.
The lower surface area and total pore volume exhibited by 900-1000-AC, despite a higher proportion of micropores, is likely due to the severity of the carbonisation conditions utilised. In general, increasing the carbonisation severity (temperature and time) leads to an improvement in surface area and pore volume of subsequent activated carbons. This is due to an increased release of low molecular weight volatiles, enhancing the development of the char’s rudimentary porous structure. This can be seen with the increased BET surface area of the severely treated char in comparison to the mild char.
Activation of the char should further enhance this porosity, improving the adsorptive characteristics. If the conditions are too severe however, redeposition of volatiles and constriction of the porous structure can occur, which restricts the access of the activating gas into the chars porous structure and results in reduced adsorptive characteristics [66, 67]. The restriction of the activating gas into the pore structure of 900-1000-AC could also be attributed to the absence of mesoprosity, despite the high burn-off level. Meso- and macroporsoity is usually enhanced with increasing burn-off levels and decrease in micropore volume. This is due to the micropore widening and collapse of pore walls becoming the dominant process over the formation of new micropores [68,69,70].
Conclusion
The ratio of aromatic monomers (S/G ratio) in lignin was linked to the polymer’s thermal behaviour. High S/G ratio samples produced a lower char yield than low S/G lignins, and swelled during heating producing a carbon foam. This is an important parameter to consider when choosing lignin for an application. High S/G ratio lignins showed excellent potential to produce high surface area (1400 m2 g− 1) activated carbons with hierarchical porosity; macropores resulted from the swelling, and activation with CO2 introduced meso- and microporosity.
References
Dutta, S., Wu, K.C.-W., Saha, B.: Emerging strategies for breaking the 3D amorphous network of lignin. Catal. Sci. Technol. 4, 3785–3799 (2014)
Singh, J., Suhag, M., Dhaka, A.: Augmented digestion of lignocellulose by steam explosion, acid and alkaline pretreatment methods: a review. Carbohydr. Polym. 117, 624–631 (2015)
Souto, F., Calado, V., Pereira, N.: Lignin-based carbon fiber: a current overview. Mater. Res. Express 5, 072001 (2018)
Chatterjee, S., Clingenpeel, A., McKenna, A., Rios, O., Johs, A.: Synthesis and characterization of lignin-based carbon materials with tunable microstructure. RSC Adv. 4, 4743–4753 (2014)
Rosas, J.M., Berenguer, R., Valero-Romero, M.J., Rodríguez-Mirasol, J., Cordero, T.: Preparation of different carbon materials by thermochemical conversion of lignin. Front. Mater. 1(29), 1–17 (2014)
Rodríguez, J.J., Cordero, T., Rodríguez-Mirasol, J.: Prod. biofuels chem. from lignin, Fang, Z., Smith, R.L. (eds.) (Springer, Singapore, 2016), pp. 217–262
Zhang, W., Zhao, M., Liu, R., Wang, X., Lin, H.: Hierarchical porous carbon derived from lignin for high performance supercapacitor. Colloids Surf. A 484, 518–527 (2015)
Jeon, J.-W., Zhang, L., Lutkenhaus, J.L., Laskar, D.D., Lemmon, J.P., Choi, D., Nandasiri, M.I., Hashmi, A., Xu, J., Motkuri, R.K., Fernandez, C.A., Liu, J., Tucker, M.P., McGrail, P.B., Yang, B., Nune, S.K.: Controlling porosity in lignin-derived nanoporous carbon for supercapacitor applications. ChemSusChem 8, 428–432 (2014)
Hu, S., Lo Hsieh, Y.: Lignin derived activated carbon particulates as an electric supercapacitor: carbonization and activation on porous structures and microstructures. RSC Adv. 7, 30459–30468 (2017)
Guo, N., Li, M., Sun, X., Wang, F., Yang, R.: Enzymatic hydrolysis lignin derived hierarchical porous carbon for supercapacitors in ionic liquids with high power and energy densities. Green Chem. 19, 2595–2602 (2017)
Zhang, H., Jia, D., Yang, Z., Yu, F., Su, Y., Wang, D., Shen, Q.: Alkaline lignin derived porous carbon as an efficient scaffold for lithium-selenium battery cathode. Carbon 122, 547–555 (2017)
Yu, F., Li, Y., Jia, M., Nan, T., Zhang, H., Zhao, S., Shen, Q.: Elaborate construction and electrochemical properties of lignin-derived macro-/micro-porous carbon-sulfur composites for rechargeable lithium-sulfur batteries: the effect of sulfur-loading time. J. Alloys Compd. 709, 677–685 (2017)
Xi, Y., Yang, D., Qiu, X., Wang, H., Huang, J., Li, Q.: Renewable lignin-based carbon with a remarkable electrochemical performance from potassium compound activation. Ind. Crops Prod. 124, 747–754 (2018)
Sangchoom, W., Mokaya, R.: Valorization of lignin waste: carbons from hydrothermal carbonization of renewable lignin as superior sorbents for CO2 and hydrogen storage. ACS Sustain. Chem. Eng. 3, 1658–1667 (2015)
Hao, W., Björnerbäck, F., Trushkina, Y., Oregui Bengoechea, M., Salazar-Alvarez, G., Barth, T., Hedin, N.: High-performance magnetic activated carbon from solid waste from lignin conversion processes. 1. Their use as adsorbents for CO2. ACS Sustain. Chem. Eng. 5, 3087–3095 (2017)
Chatterjee, S., Saito, T.: Lignin-derived advanced carbon materials. ChemSusChem 8, 3941–3958 (2015)
Effendi, A., Gerhauser, H., Bridgwater, A.V.: Production of renewable phenolic resins by thermochemical conversion of biomass: a review. Renew. Sustain. Energy Rev. 12, 2092–2116 (2008)
Simitzis, J., Sfyrakis, J.: Pyrolysis of lignin biomass-novolac resin for the production of polymeric carbon adsorbents. J. Anal. Appl. Pyrol. 26, 37–52 (1993)
Simitzis, J., Sfyrakis, J.: Activated carbon from lignocellulosic biomass-phenolic resin. J. Appl. Polym. Sci. 54, 2091–2099 (1994)
Calvo-Flores, F.G., Dobado, J.A.: Lignin as renewable raw material. ChemSusChem 3, 1227–1235 (2010)
Carrott, P.J.M., Suhas, M.L., Carrott, M.R., Guerrero, C.I., Delgado, L.A.: Reactivity and porosity development during pyrolysis and physical activation in CO2 or steam of kraft and hydrolytic lignins. J. Anal. Appl. Pyrol. 82, 264–271 (2008)
Monteil-Rivera, F., Phuong, M., Ye, M., Halasz, A., Hawari, J.: Isolation and characterization of herbaceous lignins for applications in biomaterials. Ind. Crops Prod. 41, 356–364 (2013)
Laskar, D.D., Yang, B., Wang, H., Lee, J.: Pathways for biomass-derived lignin to hydrocarbon fuels. Biofuels, Bioprod. Biorefin. 7, 602–626 (2013)
Glasser, W. G.: Classification of lignin according to chemical and molecular structure. ACS Symp. Ser. 742, 216–238 (2000)
Tejado, A., Peña, C., Labidi, J., Echeverria, J.M., Mondragon, I.: Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis. Bioresour. Technol. 98, 1655–1663 (2007)
Wang, S., Wang, K., Liu, Q., Gu, Y., Luo, Z., Cen, K., Fransson, T.: Comparison of the pyrolysis behavior of lignins from different tree species. Biotechnol. Adv. 27, 562–567 (2009)
Constant, S., Wienk, H.L.J., Frissen, A.E., de Peinder, P., Boelens, R., van Es, D.S., Grisel, R.J.H., Weckhuysen, B.M., Huijgen, W.J.J., Gosselink, R.J.A., Bruijnincx, P.C.A.: New insights into the structure and composition of technical lignins: a comparative characterisation study. Green Chem. 18, 4888–4907 (2016)
Sun, Q., Khunsupat, R., Akato, K., Tao, J., Labbé, N., Gallego, N.C., Bozell, J.J., Rials, T.G., Tuskan, G.A., Tschaplinski, T.J., Naskar, A.K., Pu, Y., Ragauskas, A.J.: A study of poplar organosolv lignin after melt rheology treatment as carbon fiber precursors. Green Chem. 18, 5015–5024 (2016)
Wang, S., Ru, B., Lin, H., Sun, W., Luo, Z.: Pyrolysis behaviors of four lignin polymers isolated from the same pine wood. Bioresour. Technol. 182, 120–127 (2015)
Pye, E.K., Lora, J.H.: The Alcell process: a proven alternative to kraft pulping. Tappi J. 74, 113–118 (1991)
Botello, J.I., Gilarranz, M.A., Rodriguez, F., Oliet, M.: Recovery of solvent and by-products from organosolv black liquor. Sep. Sci. Technol. 34, 2431–2445 (1999)
Gandolfi, S., Ottolina, G., Consonni, R., Riva, S., Patel, I.: Fractionation of hemp hurds by organosolv pretreatment and its effect on production of lignin and sugars. ChemSusChem 7, 1991–1999 (2014)
Lora, J.H., Katzen, R., Cronlund, M., Wu, C.F.: Recovery of lignin. US4764596 (16 August 1988)
T 222 om-11. Test method for acid-insoluble lignin in wood and pulp. TAPPI Test Methods: (2011)
Effland, M.J.: Modified procedure to determine acid-insoluble lignin in wood and pulp. Tappi 60, 143–144 (1977)
del Río, J.C., Rencoret, J., Marques, G., Gutiérrez, A., Ibarra, D., Santos, J.I., Jiménez-Barbero, J., Zhang, L., Martínez, A.T.: Highly acylated (acetylated and/or p-coumaroylated) native lignins from diverse herbaceous plants. J. Agric. Food Chem. 56, 9525–9534 (2008)
Wen, J.L., Sun, S.L., Yuan, T.Q., Xu, F., Sun, R.C.: Structural elucidation of lignin polymers of eucalyptus chips during organosolv pretreatment and extended delignification. J. Agric. Food Chem. 61, 11067–11075 (2013)
Salanti, A., Zoia, L., Orlandi, M., Zanini, F., Elegir, G.: Structural characterization and antioxidant activity evaluation of lignins from rice husk. J. Agric. Food Chem. 58, 10049–10055 (2010)
Yuan, T.-Q., Sun, S.-N., Xu, F., Sun, R.-C.: Characterization of lignin structures and lignin-carbohydrate complex (LCC) linkages by quantitative 13C and 2D HSQC NMR spectroscopy. J. Agric. Food Chem. 59, 10604–10614 (2011)
Huijgen, W.J.J., Reith, J.H., den Uil, H.: Pretreatment and fractionation of wheat straw by an acetone-Based organosolv process. Ind. Eng. Chem. Res. 49, 10132–10140 (2010)
Buranov, A.U., Ross, K.A., Mazza, G.: Isolation and characterization of lignins extracted from flax shives using pressurized aqueous ethanol. Bioresour. Technol. 101, 7446–7455 (2010)
ISO: Determination of the specific surface area of solids by gas adsorption. BET Method. ISO 9277, 1–24 (2010)
Wildschut, J., Smit, A.T., Reith, J.H., Huijgen, W.J.J.: Ethanol-based organosolv fractionation of wheat straw for the production of lignin and enzymatically digestible cellulose. Bioresour. Technol. 135, 58–66 (2013)
Rodrigues, J., Faix, O., Pereira, H.: Determination of lignin content of eucalyptus globulus wood using FTIR spectroscopy. Holzforschung 52, 46–50 (1998)
Ross, K., Mazza, G.: Characteristics of lignin from flax shives as affected by extraction conditions. Int. J. Mol. Sci. 11, 4035–4050 (2010)
Gümüşkaya, E., Usta, M., Balaban, M.: Carbohydrate components and crystalline structure of organosolv hemp (Cannabis sativa L.) bast fibers pulp. Bioresour. Technol. 98, 491–497 (2007)
Thakur, V.K.: Green Composites from Natural Resources. CRC Press, Boca Raton (2013)
Banerjee, S., Sen, R., Pandey, R.A., Chakrabarti, T., Satpute, D., Giri, B.S., Mudliar, S.: Evaluation of wet air oxidation as a pretreatment strategy for bioethanol production from rice husk and process optimization. Biomass Bioenergy 33, 1680–1686 (2009)
Mansaray, K.G., Ghaly, A.E.: Thermal degradation of rice husks in nitrogen atmosphere. Bioresour. Technol. 65, 13–20 (1998)
Dence, C.W.: Methods Lignin Chem., Lin, S.Y., Dence, C.W. (eds.) (Springer, Berlin, 1992), pp. 33–58
Gosselink, R.J.A., Abächerli, A., Semke, H., Malherbe, R., Käuper, P., Nadif, A., van Dam, J.E.G.: Analytical protocols for characterisation of sulphur-free ligni. Ind. Crops Prod. 19, 271–281 (2004)
Huijgen, W.J.J., Telysheva, G., Arshanitsa, A., Gosselink, R.J.A., de Wild, P.J.: Characteristics of wheat straw lignins from ethanol-based organosolv treatment. Ind. Crops Prod. 59, 85–95 (2014)
Ratnaweera, D.R., Saha, D., Pingali, S.V., Labbé, N., Naskar, A.K., Dadmun, M.: The impact of lignin source on its self-assembly in solution. RSC Adv. 5, 67258–67266 (2015)
Wörmeyer, K., Ingram, T., Saake, B., Brunner, G., Smirnova, I.: Comparison of different pretreatment methods for lignocellulosic materials. Part II: influence of pretreatment on the properties of rye straw lignin. Bioresour. Technol. 102, 4157–4164 (2011)
de Wild, P.J., Huijgen, W.J.J., Heeres, H.J.: Pyrolysis of wheat straw-derived organosolv lignin. J. Anal. Appl. Pyrol. 93, 95–103 (2012)
Rana, R., Langenfeld-Heyser, R., Finkeldey, R., Polle, A.: FTIR spectroscopy, chemical and histochemical characterisation of wood and lignin of five tropical timber wood species of the family of Dipterocarpaceae. Wood Sci. Technol. 44, 225–242 (2009)
Li, H., McDonald, A.G.: Fractionation and characterization of industrial lignins. Ind. Crops Prod. 62, 67–76 (2014)
Watkins, D., Nuruddin, M., Hosur, M., Tcherbi-Narteh, A., Jeelani, S.: Extraction and characterization of lignin from different biomass resources. J. Mater. Res. Technol. 4, 26–32 (2015)
Aldaeus, F., Sjöholm., E.: Cost action FP0901 round robins of lignin samples Part 1: lignin content. Innventia Rep. No. IR 108, 1–23 (2011)
Rodríguez-Mirasol, J., Cordero, T., Rodríguez, J.J.: Preparation and characterization of activated carbons from eucalyptus kraft lignin. Carbon 31, 87–95 (1993)
Rios, R.V.R.A., Martínez-Escandell, M., Molina-Sabio, M., Rodríguez-Reinoso, F.: Carbon foam prepared by pyrolysis of olive stones under steam. Carbon 44, 1448–1454 (2006)
Faix, O., Jakab, E., Till, F., Székely, T.: Study on low mass thermal degradation products of milled wood lignins by thermogravimetry-mass-spectrometry. Wood Sci. Technol. 22, 323–334 (1988)
Liu, Q., Wang, S., Zheng, Y., Luo, Z., Cen, K.: Mechanism study of wood lignin pyrolysis by using TG-FTIR analysis. J. Anal. Appl. Pyrol. 82, 170–177 (2008)
Brebu, M., Tamminen, T., Spiridon, I.: Thermal degradation of various lignins by TG-MS/FTIR and Py-GC-MS. J. Anal. Appl. Pyrol. 104, 531–539 (2013)
Chen, L., Wang, X., Yang, H., Lu, Q., Li, D., Yang, Q., Chen, H.: Study on pyrolysis behaviors of non-woody lignins with TG-FTIR and Py-GC/MS. J. Anal. Appl. Pyrol. 113, 499–507 (2015)
Jia, Q., Lua, A.C.: Effects of pyrolysis conditions on the physical characteristics of oil-palm-shell activated carbons used in aqueous phase phenol adsorption. J. Anal. Appl. Pyrol. 83, 175–179 (2008)
Yang, T., Lua, A.C.: Characteristics of activated carbons prepared from pistachio-nut shells by physical activation. J. Colloid Interface Sci. 267, 408–417 (2003)
Chang, C., Chang, C., Tsai, W.: Effects of burn-off and activation temperature on preparation of activated carbon from corn cob agrowaste by CO2 and steam. J. Colloid Interface Sci. 232, 45–49 (2000)
Daud, W.M.A.W., Ali, W.S.W., Sulaiman, M. Z.: Effects of carbonization temperature on pore development in palm-shell-based activated carbon. Carbon 38, 1925–1932 (2000)
Bang, J.H., Lee, H.-M., An, K.-H., Kim, B.-J.: A study on optimal pore development of modified commercial activated carbons for electrode materials of supercapacitors. Appl. Surf. Sci. 415, 61–66 (2017)
Acknowledgements
The Authors thank Des Davies at the University of Bristol (UoB) for performing the elemental analysis, Sean Davis and the ACCIS technical team at UoB for their support with the SEM and DSC respectively, Mi Tian at the University of Bath for performing thermogravimetric analysis on the industrial organosolv lignin, Helena Quilter at the University of Bath for her early assistance with the GPC-SEC and Noemi Holzleg and Elizabeth Randell on the University of Bath MEng programme for their help during the initial stages of this project. VPT thanks the Engineering and Physical Sciences Research Council (EPSRC) for support via EP/R01650X/1. The early stages of this work were supported by the EPSRC Centre for Doctoral Training in Sustainable Chemical Technologies (EP/G03768X/1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rowlandson, J.L., Woodman, T.J., Tennison, S.R. et al. Influence of Aromatic Structure on the Thermal Behaviour of Lignin. Waste Biomass Valor 11, 2863–2876 (2020). https://doi.org/10.1007/s12649-018-0537-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12649-018-0537-x