
Abstract
Previous studies revealed that oxidative stress and inflammation are the main contributors to secondary injury after traumatic brain injury (TBI). In an earlier study, we reported that lutein/zeaxanthin isomers (L/Zi) exert antioxidative and anti-inflammatory effects by activating the nuclear factor-kappa B (NF-κB) and nuclear factor-erythroid 2-related factor 2 (Nrf2) pathways. However, its precise role and underlying mechanisms were largely unknown after TBI. This study was conducted to investigate the potential mechanism of L/Zi isomers in a TBI model induced by a cold injury model in mice. To investigate the effects of L/Zi, male C57BL/6j mice-induced brain injury using the cold trauma model was allocated into two groups (n = 7): (i) TBI + vehicle group and (ii) TBI + L/Zi group (20 mg/kg BW). Brain samples were collected 24 h later for analyses. L/Zi given immediately after the injury decreased infarct volume and blood–brain barrier (BBB) permeability; L/Zi treatment also significantly reduced proinflammatory cytokines, including interleukin1 beta (IL-1β), interleukin 6 (IL-6), and NF-κB levels and increased growth-associated protein 43 (GAP-43), neural cell adhesion molecule (NCAM), brain-derived neurotrophic factor (BDNF), and Nrf2 levels compared with vehicle control. These data suggest that L/Zi improves mitochondrial function in TBI models, possibly decreasing inflammation and activating the Nrf2 pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carotenoids are molecules found in plants. Lutein and zeaxanthin are xanthophyll carotenoids, which are abundant in green leafy vegetables and brightly colored fruits. They are found in many places in body tissues, but they are most prominent in the central nervous system tissues. For example, they are found in primate retina macaques at concentrations 500 times higher than other body tissues, such as serum (Johnson 2014). These carotenoids filter blue light and function as antioxidants (Snodderly 1995). US-based studies indicate that lutein and zeaxanthin are protective against age-related macular degeneration, one of the leading causes of blindness and blindness (Friedman et al. 2004; Nakajima et al. 2009; Snodderly 1995). Many studies have shown that more carotenoid intake protects against age-related macular degeneration, cancer, and cardiovascular and neurodegenerative diseases (Krinsky and Johnson 2005).
Lutein and zeaxanthin constitute 66–77% of the total carotenoid concentration in human brain tissue (Barker et al. 2011; Craft et al. 2004). The cortical lutein and zeaxanthin are protective and affect interneuronal communication and functions through various mechanisms. Although the molecular basis of these neuroprotective effects of lutein and zeaxanthin is unidentified, multiple mechanisms have been proposed, including reduction of oxidative stress and inflammation and regulation of the functional properties of synaptic membranes (Johnson 2005; Krinsky and Johnson 2005; Kritchevsky et al. 2000; Song et al. 2000). It has been shown that lutein and zeaxanthin may be essential for developing the light-sensing process and the neural circuits in the visual system by strengthening the retina gap junction (Stahl and Sies 2001). Lutein and zeaxanthin as maculopure pigments have also been found to enhance visual processing speed (Hammond and Wooten 2005; Renzi and Hammond 2010a, b). Several studies have also been done to evaluate the relationship between lutein and zeaxanthin concentrations and cognitive function. From these studies, two groups of rhesus monkeys were discerned. One of these groups was treated with lutein alone and a diet containing only zeaxanthin and no other carotenoids, and xanthophyll concentrations were measured by HPLC on extracts after examining retina and brain tissues after the diet. When the values obtained were analyzed, the retinal concentrations of lutein and zeaxanthin showed a significant correlation with the concentrations in the cerebellum. Similarly, concentrations of zeaxanthin in the occipital cortex, frontal cortex, and pons correlate with zeaxanthin levels in the retina (Johnson et al. 2005; Vishwanathan et al. 2013). In light of these results, macular pigment density has been researched as a biomarker of lutein and zeaxanthin in the primate brain. In a study, Cheng et al. (2015) showed that lutein modulates oxidative stress and inflammation and is a protective effect against ischemia/reperfusion injury in the rat skeleton (Cheng et al. 2015). Treatment of lutein administered to rats reduces reactive oxygen species, lipid peroxidation, protein carbonylation, and sulfhydryls, and it has been shown to significantly reduce oxidative stress by increasing nuclear factor erythroid 2 (Nrf-2) levels and antioxidant status. According to the same study results, lutein treatment reduced the expression of nuclear factor kappa B (NF-κB) and cyclooxygenase-2 (COX-2), which increased after ischemia/reperfusion injury (Cheng et al. 2015).
Sun et al. (2014) experimentally treated lutein-treated mice with transient brain ischemia, finding that it is neuroprotective and that this effect may be related to the antioxidant properties of lutein (Sun et al. 2014). Ozawa et al. (2012) showed that oxidative stress in the retina decreases in rats treated with the experimental ocular disease after lutein treatment and the degradation of functional proteins such as rhodopsin and synaptophysin well as brain-derived neurotrophic factor (BDNF) depletion and DNA damage (Ozawa et al. 2012). The results of another study in adult rhesus monkeys also support the hypothesis that lutein may be useful as an antioxidant in the brain (Mohn et al. 2017).
NF-κB p65 and COX-2 decreased the expression of the intracellular adhesion molecule (ICAM)-1 protein after treatment with lutein in severe traumatic brain injury (TBI)-treated rats as well as decreasing Nrf-2 and endothelin-1 protein levels. These findings suggest that lutein has anti-inflammatory and antioxidative effects as well as a protective effect against severe TBI (Tan et al. 2017).
TBI is one of the most common causes of morbidity and mortality (Kelestemur et al. 2016; Keskin et al. 2017). In addition to acute injury in TBI, neuronal damage also occurs due to secondary effects that develop afterward. These include oxidative damage due to inflammation and the formation of free radicals, excessive glutamate release, impaired calcium homeostasis, excitotoxicity, deterioration of the blood–brain barrier, and all-out cell death (Biegon et al. 2004; Kilic et al. 2013; Loane and Faden 2010; Malkesman et al. 2013). In addition to the results of several TBI studies showing that combined treatment modalities for preventing inflammation and oxidative injury are beneficial, changes in the diet can be predicted to contribute to antioxidative and anti-inflammatory treatment strategies (Esenwa and Elkind 2016; Russo and McGavern 2016). Currently, efforts to develop new neuroprotective treatment strategies are underway by evaluating the mechanisms of secondary damage. Many pharmacologic agents cannot have the desired effect on the neuroprotective pathways at the experimental stage, as the possible drug interactions cannot affect the essential physiopathological mechanisms. This has led to the emergence of single-drug modalities in the treatment process (Hawryluk and Bullock 2016). Given the recently described neuroprotective effects in experimental models, carotenoids such as genotoxic, non-mutagenic, and highly therapeutic indole and lutein and zeaxanthin isomers (L/Zi) may contribute to the development of new treatment strategies by conducting further clinical investigations (Ravikrishnan et al. 2011; Thurnham and Howard 2013). Therefore, in the present study, we investigated the effects of L/Zi isomers on the infarct volume, blood–brain barrier (BBB) permeability, proinflammatory cytokines, and brain-derived neurotrophic factor (BDNF) levels in a TBI model induced by a cold injury model in mice.
Methods
Animals
Adult male C57BL/6j mice (n = 7) weighing 23–26 g were obtained from the Laboratory Animal Unit of the Istanbul Medipol University. All experimentations were done following the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and approved by the Animal Research Ethics Committee of Istanbul Medipol University (37/2017). Mice were kept in a constant 12:12-h light: dark regime with ad libitum access to food and water.
Traumatic Brain Injury
Traumatic brain injury was performed as defined earlier in a cold injury-induced trauma model in mice fixed on a stereotaxic device (WPI Instruments, USA) (Johnson 2014). The rectal temperature was kept between 36.5 and 37.0 °C with homoeothermic drapes throughout the trials. The traumatic injury was done by a liquid nitrogen-cooled copper probe (tip diameter 2.5 mm), located on the skull for 60 s, then removed. Immediately after TBI induction, the animals were injected intraperitoneally with L/Zi isomers (20 mg/kg BW) or vehicles (5% EtOH in isotonic saline). The mice were then kept in the feeding room for post-traumatic healing for 24 h. Twenty-four h after trauma, mice were intensely anesthetized with 4% isoflurane (30% O2, remainder N2O), and euthanasia was performed by cervical dislocation. Brain tissues were removed, frozen on dry ice, and sliced with a cryostat into 18-μm sections used in brain volume analysis.
Experimental Design
After 1-week adaptation period, animals were randomly allocated into two groups (n = 7): (i) TBI + vehicle group receiving the vehicle in equal volume (5% EtOH in isotonic saline) or (ii) TBI + L/Zi Isomers group (20 mg/kg BW) at the corresponding time points.
Analysis of Infarct Volume and Brain Edema
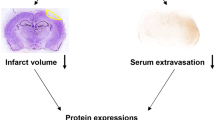
Coronal brain sections from equal brain levels, 1 mm apart, were stained with Cresyl violet. Infarction volume and brain swelling were calculated as described earlier (Johnson 2014). Briefly, brain infarcts were defined by delineating non-lesioned tissue in both hemispheres, where edema corrected infarction areas and infarction volumes are determined using an image analysis system (Image J; NIH, Bethesda, MD, USA). Brain edema was examined by outlining the lesions with and without lesions in both hemispheres, which were deducted from each other and separated by the non-lesioned tissue in the contralateral brain, yielding percentage values of the brain edema.
Western Blot Analysis
As previously described, western blot of brain tissue samples collected from the injured cortex was performed to detect the expression of protein levels due to inflammation and antioxidant mechanism (Friedman et al. 2004; Snodderly 1995). Briefly, samples of the same group were pooled, homogenized, and sonicated in lysis buffer supplemented with 1X protease and phosphatase inhibitor cocktail (#5872, Cell Signaling). According to the manufacturer’s protocol, total protein content was detected with a Qubit 2.0 Fluorometer (Invitrogen, Life Technologies Corporation, Carlsbad, CA, USA). Equal amounts of protein (20 μg) were size-fractionated using any-kD Mini-Protean TGX gel electrophoresis and then transferred to a nitrocellulose membrane using the Trans-Blot Turbo Transfer System (Bio-Rad, Life Sciences Research). After that, membranes were blocked in 5% nonfat milk in Tris-buffered saline (TBS) containing 0.1% Tween (TBS-T; blocking solution) for 1 h at room temperature, washed in TBS-T, and incubated overnight with growth-associated protein-43 (GAP-43), glial fibrillary acidic protein (GFAP), ICAM, neural cell adhesion molecule (NCAM), interleukin (IL1β), IL6, BDNF, NFκB, and Nrf2 antibodies (Abcam, Cambridge, UK). The following day, membranes were washed with TBS-T and then incubated with horseradish peroxidase-conjugated goat anti-rabbit (Abcam, Cambridge, UK) or goat anti-mouse (Abcam, Cambridge, UK) secondary antibody (diluted 1:1000) for 1 h. Individual blots were performed at least three times. Protein loading was controlled by stripping and reprobing the nitrocellulose membrane with an anti-β-actin antibody (Abcam, Cambridge, UK). Protein levels were analyzed densitometrically using an image analysis system (Image J; National Institute of Health, Bethesda, MD, USA), corrected with values determined on β-actin blots, and expressed as relative values compared with the control group.
Statistical Analysis
Differences between groups were analyzed by t-test using SPSS 18 for Windows (SPSS Inc. Chicago, IL, USA). All values are given as mean ± S.D; p values < 0.05 are considered significant.
Results
Evaluation of the Effect of L/Zi Isomers on Reducing Brain Edema Following Traumatic Brain Injury
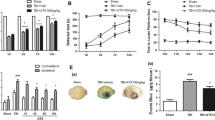
The animals were treated with L/Zi isomers or vehicles immediately after TBI induction. Brain swelling/edema and infarct volume were evaluated by Cresyl violet staining in 18-µm-thick coronal brain sections. In the control group, reproducible brain infarcts were detected 24 h after the onset of TBI. L/Zi isomers significantly reduced brain infarct volume when compared with the control group (P < 0.05) (Fig. 1a). In addition, brain swelling presented some decline with L/Zi isomers administration, but it was not a statistically significant level (Fig. 1b).

Effect of Lutein/Zeaxanthin isomers (L/Zi) on brain infarct volume and edema in traumatic brain injury in mice. L/Zi treatment reduced infarct volume significantly (a) and a moderate decrease in brain edema was observed (b). Each bar represents the mean and standard error of the mean. *p < 0.05
Evaluation of the Effect of L/Zi Isomers on Brain Neurotrophic Factors and Markers Associated with Plasticity and Inflammatory Proteins After Traumatic Brain Injury
We analyzed the levels of GAP-43 (Fig. 2a), a signaling molecule in neuronal growth, and GFAP (Fig. 2b), a main component of the glial scar, by Western blot analysis to evaluate the effect of L/Zi on the glial cell proliferation. We have observed that L/Zi administration significantly increased GAP-43 and reduced GFAP levels compared with the control group (P < 0.001), suggesting that L/Zi can play a role in neuroprotection following TBI by increasing GAP-43 expression and inhibiting the expression of GFAP.

Effect of Lutein/Zeaxanthin isomers (L/Zi) on brain GAP-43 (a), GFAP (b), ICAM (c), NCAM (d), IL-1β (e), IL-6 (f), BDNF (g), NF-κB (h), and Nrf2 (i) in traumatic brain injury in mice. Data are expressed as percent of the control value. Each bar represents the mean and standard error of the mean. Blots were repeated at least 3 times (n = 3). β-Actin was included to ensure equal protein loading. **p < 0.01; ***p < 0.001
Analyzes of the ICAM levels revealed that L/Zi resulted in a significant increase in the protein level of ICAM (P < 0.01; Fig. 2c), which was associated with decreased levels of pro-inflammatory proteins, IL-1β (P < 0.001; Fig. 2e), and IL-6 (P < 0.01; Fig. 2f). In addition, L/Zi administration significantly increased NCAM levels after traumatic brain injury (P < 0.001; Fig. 2d). Both BDNF and Nrf2 (P < 0.001; Fig. 2g, i) levels were significantly increased in L/Zi treated group as compared with the control. In addition, L/Zi administration significantly decreased NF-κB levels after traumatic brain injury (P < 0.001; Fig. 2h).
Discussion
TBI has become a serious health problem due to various accidents or incidents in contemporary life. While some TBI cases may be minor injuries, a considerable proportion of the TBI cases may leave heavy sequelae. In particular, there has been an increase in the frequency of neurodegenerative diseases such as dementia, Alzheimer’s disease, and Parkinson’s disease, especially after TBI (Washington et al. 2016). Secondary damage due to changes in cellular and neurochemical mechanisms that develop immediately after TBI is mainly due to oxidative stress (Abdul-Muneer et al. 2015). Despite all these high morbidity and mortality rates, FDA-approved definitive treatment protocols are unfortunately not yet available (Venegoni et al. 2017). However, the addition of antioxidant-containing foods and molecules to the applied treatment protocols may contribute to alleviating symptoms (de Roos and Duthie 2015). We investigated brain edema, infarct volume, some molecules involved in the inflammatory response, antioxidant mechanism markers, and neuronal plasticity parameters after using L/Zi isomers in TBI treatment in our study.
The experimental TBI model used in this study is a cortical cryogenic injury model and leads to the functional deterioration of the blood–brain barrier in rodents. Immediately after the injury, astrocytic activation and inflammation, which result in the expansion of the area of injury, occur immediately around the first area of injury (Albert-Weissenberger and Sirén 2010). Increased proinflammatory cytokines, including IL-1β, are associated with TBI in secondary TBI-associated secondary damage. IL-1β has been shown to increase in rodent brains following TBI versus exhaling low levels under physiological conditions (Kamm et al. 2006). IL-1β enhances neuronal damage by activating other pro-inflammatory mediators and leading to glutamate-mediated excitotoxicity (Ley et al. 2011). It has also been shown that IL-1β causes edema in the brain following injury (Holmin and Mathiesen 2000). IL-6, another proinflammatory cytokine, cannot be detected in the normal brain but is elevated in response to brain damage (Woodcock and Morganti-Kossmann 2013). According to our results, the decrease in infarct volume and brain edema may be related to the inhibition of L/Zi isomers by IL-1β and IL-6 (Fig. 2e, f).
In both focal and diffuse TBI, glial cells are activated and proliferate due to secondary damage to the membrane (Vos 2011). Woodcock and colleagues reported that IL-6 activates the JAK/STAT pathway in brain injury, thereby increasing GFAP expression; thus, reactive astrogliosis following trauma may be due to IL-6 (Woodcock and Morganti-Kossmann 2013). Another study showed that serum levels of GFAP in TBI patients were increased and could be used to marker the severity of trauma (Nylén et al. 2006). It is thought that inhibition of GFAP in these findings enhances healing and improves the outcomes of TBI patients. According to our study results, the decrease in GFAP levels in the L/Zi isomer-treated group may be due to the inhibition of IL-6 by L/Zi isomers (Fig. 2b).
Many studies are reporting an increase in ICAM expressions after TBI. The increase in ICAM expressions can lead to increased permeability in the blood–brain barrier (Bowes et al. 1993; Knoblach and Faden 2002). This may be a negative situation, but it may also allow the passage of mediators that trigger the healing process. The present study results showed a significant increase in ICAM levels in the group treated with L/Zi isomers (Fig. 2c). This seems to be contradictory to the literature (Tan et al. 2017), which may be due to the coexistence of L/Zi isomers and the differently applied experimental trauma models. In another study, You and colleagues showed an increase in ICAM-1 mRNA levels in response to NF-κB activation in an experimental subarachnoid hemorrhage model (You et al. 2013). NF-κB is one of the primary regulators in the inflammatory process that develops after brain injury, enhancing expression in TBI and other neurodegenerative disorders (Kaltschmidt et al. 2005; Nonaka et al. 1999). The inhibition of NF-κB may positively affect the treatment process when the increase in NF-κB after trauma is considered a precautionary measure. Indeed, a TBI study of the NF-κB inhibitor has been shown to reduce brain edema (Xiao and Wei 2005). In our study, the decrease in NF-κB levels in the group treated with L/Zi isomers promoted the infarct volume and brain edema-reducing effect of L/Zi isomers. Conversely, an increase in ICAM levels while NF-κB expressions are reduced reinforces the notion that TBI mechanisms or factors may still be unknown.
Another parameter we evaluated in our study was Nrf2. Nrf2 is an important regulator of antioxidant and anti-inflammatory processes (Wardyn et al. 2015). Increased Nrf2 mediates the expression of antioxidant genes in TBI, resulting in decreased infarct volume and brain edema by inhibiting IL-1β, IL-6, and NF-κB expression (Wagner et al. 2012). In our study, the results obtained in the group treated with L/Zi isomers are in parallel with this situation. In addition, increases in GAP43 and NCAM expressions were detected in the L/Zi isomers-treated group. GAP43 is an important parameter showing regeneration after TBI (Gao et al. 2008), while NCAM is a molecular agent responsible for post-traumatic reorganization and is used as a neurogenesis marker (Budinich et al. 2012). Brain-derived growth factor (BDNF) is a specific neurotrophin secreted by dendritic structures (Hartmann et al. 2001). BDNF has important neuroprotective properties and is associated with new neuron formation, differentiation, growth, and survival (Yulug et al. 2017). The increase in BDNF levels in the L/Zi isomer-treated group in our study enhanced the hypothesis that L/Zi isomers may have neuroprotective effects following TBI.
Conclusions
In our study, results were obtained that reduce the infarct volume and brain edema of the antioxidant molecule, L/Zi isomers, and reduce IL-1β, IL-6, and GFAP levels through NF-κB downregulation in mice after TBI. In addition, L/Zi isomers have been shown to induce endogenous antioxidant systems via the upregulation of Nrf2, leading to an increase in neuronal plasticity markers GAP43, NCAM, and BDNF, an important neuroprotective molecule. The use of L/Zi isomers as a diet or drug in addition to the current treatment approaches in light of this information may lead to promising developments in the treatment of many neurodegenerative disorders characterized by TBI or oxidative stress.
Availability of Data and Materials
All data are presented in the manuscript. Further results and analysis data are available with the corresponding author on reasonable request.
Abbreviations
- BBB:
-
Blood-brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- COX-2:
-
Cyclooxygenase-2
- ICAM-1:
-
Intracellular adhesion molecule-1
- IL1β:
-
Interleukin 1 beta
- IL6:
-
Interleukin 6
- GAP43:
-
Growth associated protein 43
- L/Zi:
-
Lutein/zeaxanthin
- NCAM:
-
Neural cell adhesion molecule
- NF-κB:
-
Nuclear factor-kappa B
- Nrf2:
-
Nuclear factor-erythroid 2-related factor 2
- TBI:
-
Traumatic brain injury
References
Abdul-Muneer PM, Chandra N, Haorah J (2015) Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol 51:966–979. https://doi.org/10.1007/s12035-014-8752-3
Albert-Weissenberger C, Sirén AL (2010) Experimental traumatic brain injury. Exp Transl Stroke Med 2:16. https://doi.org/10.1186/2040-7378-2-16
Barker FM, Snodderly DM, Johnson EJ, Schalch W, Koepcke W, Gerss J, Neuringer M (2011) Nutritional manipulation of primate retinas, V: effects of lutein, zeaxanthin, and n-3 fatty acids on retinal sensitivity to blue-light-induced damage. Investig Ophthalmol vis Sci 52:3934–3942. https://doi.org/10.1167/iovs.10-5898
Biegon A, Fry PA, Paden CM, Alexandrovich A, Tsenter J, Shohami E (2004) Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: implications for treatment of neurological and cognitive deficits. Proc Natl Acad Sci USA 101:5117–5122. https://doi.org/10.1073/pnas.0305741101
Bowes MP, Zivin JA, Rothlein R (1993) Monoclonal antibody to the ICAM-1 adhesion site reduces neurological damage in a rabbit cerebral embolism stroke model. Exp Neurol 119:215–219. https://doi.org/10.1006/exnr.1993.1023
Budinich CS, Chen H, Lowe D, Rosenberger JG, Bernstock JD, McCabe JT (2012) Mouse brain PSA-NCAM levels are altered by graded-controlled cortical impact injury. Neural Plast 2012:378307. https://doi.org/10.1155/2012/378307
Cheng F, Zhang Q, Yan F-F, Wan J-F, Lin C-S (2015) Lutein protects against ischemia/reperfusion injury in rat skeletal muscle by modulating oxidative stress and inflammation. Immunopharmacol Immunotoxicol 37:329–334. https://doi.org/10.3109/08923973.2015.1049704
Craft NE, Haitema TB, Garnett KM, Fitch KA, Dorey CK (2004) Carotenoid, tocopherol, and retinol concentrations in elderly human brain. J Nutr Heal Aging 8:156–162 (PMID: 15129301)
de Roos B, Duthie GG (2015) Role of dietary pro-oxidants in the maintenance of health and resilience to oxidative stress. Mol Nutr Food Res 59:1229–1248. https://doi.org/10.1002/mnfr.201400568
Esenwa CC, Elkind MS (2016) Inflammatory risk factors, biomarkers and associated therapy in ischaemic stroke. Nat Rev Neurol 12:594–604. https://doi.org/10.1038/nrneurol.2016.125
Friedman DS, O’Colmain BJ, Muñoz B, Tomany SC, McCarty C, DeJong PTVM, Nemesure B, Mitchell P, Kempen J, Congdon N (2004) Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 122:564–572. https://doi.org/10.1001/archopht.122.4.564
Gao B, Kilic E, Baumann CR, Hermann DM, Bassetti CL (2008) Gamma-hydroxybutyrate accelerates functional recovery after focal cerebral ischemia. Cerebrovasc Dis 26:413–419. https://doi.org/10.1159/000151683
Hammond BR, Wooten BR (2005) CFF thresholds: relation to macular pigment optical density. Ophthalmic Physiol Opt 25:315–319. https://doi.org/10.1111/j.1475-1313.2005.00271.x
Hartmann M, Heumann R, Lessmann V (2001) Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20:5887–5897. https://doi.org/10.1093/emboj/20.21.5887
Hawryluk GWJ, Bullock MR (2016) Past, present, and future of traumatic brain ınjury research. Neurosurg Clin N Am 27:375–396. https://doi.org/10.1016/j.nec.2016.05.002
Holmin S, Mathiesen T (2000) Intracerebral administration of interleukin-1 β and induction of inflammation, apoptosis, and vasogenic edema. J Neurosurg 92:108–120. https://doi.org/10.3171/jns.2000.92.1.0108
Johnson EJ (2005) Obesity, lutein metabolism, and age-related macular degeneration: a web of connections. Nutr Rev 63:9–15. https://doi.org/10.1111/j.1753-4887.2005.tb00105.x
Johnson EJ (2014) Role of lutein and zeaxanthin in visual and cognitive function throughout the lifespan. Nutr Rev 72:605–612. https://doi.org/10.1111/nure.12133
Johnson EJ, Neuringer M, Russell RM, Schalch W, Snodderly DM (2005) Nutritional manipulation of primate retinas, III: effects of lutein or zeaxanthin supplementation on adipose tissue and retina of xanthophyll-free monkeys. Investig Ophthalmol vis Sci 46:692–702. https://doi.org/10.1167/iovs.02-1192
Kaltschmidt B, Widera D, Kaltschmidt C (2005) Signaling via NF-κB in the nervous system. Biochim Biophys Acta, Mol Cell Res 1745:287–299. https://doi.org/10.1016/j.bbamcr.2005.05.009
Kamm K, VanderKolk W, Lawrence C, Jonker M, Davis AT (2006) The Effect of traumatic brain injury upon the concentration and expression of interleukin-1?? and interleukin-10 in the rat. J Trauma Inj Infect Crit Care 60:152–157. https://doi.org/10.1097/01.ta.0000196345.81169.a1
Kelestemur T, Yulug B, Caglayan AB, Beker MC, Kilic U, Caglayan B, Yalcin E, Gundogdu RZ, Kilic E (2016) Targeting different pathophysiological events after traumatic brain injury in mice: role of melatonin and memantine. Neurosci Lett 612:92–97. https://doi.org/10.1016/j.neulet.2015.11.043
Keskin I, Gunal MY, Ayturk N, Kilic U, Ozansoy M, Kilic E (2017) Dose-dependent neuroprotective effect of enoxaparin on cold-induced traumatic brain injury. Neural Regen Res 12:761. https://doi.org/10.4103/1673-5374.206646
Kilic U, Yilmaz B, Reiter RJ, Yüksel A, Kilic E (2013) Effects of memantine and melatonin on signal transduction pathways vascular leakage and brain injury after focal cerebral ischemia in mice. Neuroscience 237:268–276. https://doi.org/10.1016/j.neuroscience.2013.01.059
Knoblach SM, Faden AI (2002) Administration of either anti-intercellular adhesion molecule-1 or a nonspecific control antibody improves recovery after traumatic brain injury in the rat. J Neurotrauma 19:1039–1050. https://doi.org/10.1089/089771502760341956
Krinsky NI, Johnson EJ (2005) Carotenoid actions and their relation to health and disease. Mol Aspects Med 26:459–516. https://doi.org/10.1016/j.mam.2005.10.001
Kritchevsky SB, Bush AJ, Pahor M, Gross MD (2000) Serum carotenoids and markers of inflammation in nonsmokers. Am J Epidemiol 152:1065–1071. https://doi.org/10.1093/aje/152.11.1065
Ley EJ, Clond MA, Singer MB, Shouhed D, Salim A (2011) IL6 deficiency affects function after traumatic brain ınjury. J Surg Res 170:253–256. https://doi.org/10.1016/j.jss.2011.03.006
Loane DJ, Faden AI (2010) Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 31:596–604. https://doi.org/10.1016/j.tips.2010.09.005
Malkesman O, Tucker LB, Ozl J, McCabe JT (2013) Traumatic brain injury - modeling neuropsychiatric symptoms in rodents. Front Neurol 4:157. https://doi.org/10.3389/fneur.2013.00157
Mohn ES, Erdman JW, Kuchan MJ, Neuringer M, Johnson EJ (2017) Lutein accumulates in subcellular membranes of brain regions in adult rhesus macaques: relationship to DHA oxidation products. PLoS One 12:e0186767. https://doi.org/10.1371/journal.pone.0186767
Nakajima Y, Shimazawa M, Otsubo K, Ishibashi T, Hara H (2009) Zeaxanthin, a retinal carotenoid, protects retinal cells against oxidative stress. Curr Eye Res 34:311–318. https://doi.org/10.1080/02713680902745408
Nonaka M, Chen XH, Pierce JES, Leoni MJ, McIntosh TK, Wolf JA, Smith DH (1999) Prolonged activation of NF-κB following traumatic brain injury in rats. J Neurotrauma 16:1023–1034. https://doi.org/10.1089/neu.1999.16.1023
Nylén K, Öst M, Csajbok LZ, Nilsson I, Blennow K, Nellgård B, Rosengren L (2006) Increased serum-GFAP in patients with severe traumatic brain injury is related to outcome. J Neurol Sci 240:85–91. https://doi.org/10.1016/j.jns.2005.09.007
Ozawa Y, Sasaki M, Takahashi N, Kamoshita M, Miyake S, Tsubota K (2012) Neuroprotective effects of lutein in the retina. Curr Pharm Des 18:51–56. https://doi.org/10.2174/138161212798919101
Ravikrishnan R, Rusia S, Ilamurugan G, Salunkhe U, Deshpande J, Shankaranarayanan J, Shankaranarayana ML, Soni MG (2011) Safety assessment of lutein and zeaxanthin (LutemaxTM 2020): subchronic toxicity and mutagenicity studies. Food Chem Toxicol 49:2841–2848. https://doi.org/10.1016/j.fct.2011.08.011
Renzi LM, Hammond BR (2010a) The effect of macular pigment on heterochromatic luminance contrast. Exp Eye Res 91:896–900. https://doi.org/10.1016/j.exer.2010.09.015
Renzi LM, Hammond BR (2010b) The relation between the macular carotenoids, lutein and zeaxanthin, and temporal vision. Ophthalmic Physiol Opt 30:351–357. https://doi.org/10.1111/j.1475-1313.2010.00720.x
Russo MV, McGavern DB (2016) Inflammatory neuroprotection following traumatic brain injury. Science 353:783–785. https://doi.org/10.1126/science.aaf6260
Snodderly DM (1995) Evidence for protection against age-related macular degeneration by carotenoids and antioxidant vitamins. Am J Clin Nutr 62:1448S-1461S. https://doi.org/10.1093/ajcn/62.6.1448S
Song JH, Fujimoto K, Miyazawa T (2000) Polyunsaturated (n-3) fatty acids susceptible to peroxidation are increased in plasma and tissue lipids of rats fed docosahexaenoic acid-containing oils. J Nutr 130:3028–3033. https://doi.org/10.1093/jn/130.12.3028
Stahl W, Sies H (2001) Effects of carotenoids and retinoids on gap junctional communication. BioFactors 15:95–98. https://doi.org/10.1002/biof.5520150209
Sun YX, Liu T, Dai XL, Zheng QS, Hui BD, Jiang ZF (2014) Treatment with lutein provides neuroprotection in mice subjected to transient cerebral ischemia. J Asian Nat Prod Res 16:1084–1093. https://doi.org/10.1080/10286020.2014.939584
Tan D, Yu X, Chen M, Chen J, Xu J (2017) Lutein protects against severe traumatic brain injury through anti-inflammation and antioxidative effects via ICAM-1/Nrf-2. Mol Med Rep 16:4235–4240. https://doi.org/10.3892/mmr.2017.7040
Thurnham DI, Howard AN (2013) Studies on meso-zeaxanthin for potential toxicity and mutagenicity. Food Chem Toxicol 59:455–463. https://doi.org/10.1016/j.fct.2013.06.002
Venegoni W, Shen Q, Thimmesch AR, Bell M, Hiebert JB, Pierce JD (2017) The use of antioxidants in the treatment of traumatic brain injury. J Adv Nurs 73:1331–1338. https://doi.org/10.1111/jan.13259
Vishwanathan R, Neuringer M, Max Snodderly D, Schalch W, Johnson EJ (2013) Macular lutein and zeaxanthin are related to brain lutein and zeaxanthin in primates. Nutr Neurosci 16:21–29. https://doi.org/10.1179/1476830512Y.0000000024
Vos PE (2011) Biomarkers of focal and diffuse traumatic brain injury. Crit Care 15:183. https://doi.org/10.1186/cc10290
Wagner AE, Boesch-Saadatmandi C, Dose J, Schultheiss G, Rimbach G (2012) Anti-inflammatory potential of allyl-isothiocyanate—role of Nrf2, NF-(κ) B and microRNA-155. J Cell Mol Med 16:836–843. https://doi.org/10.1111/j.1582-4934.2011.01367.x
Wardyn JD, Ponsford AH, Sanderson CM (2015) Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem Soc Trans 43:621–626. https://doi.org/10.1042/BST20150014
Washington PM, Villapol S, Burns MP (2016) Polypathology and dementia after brain trauma: does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol 275:381–388. https://doi.org/10.1016/j.expneurol.2015.06.015
Woodcock T, Morganti-Kossmann MC (2013) The role of markers of inflammation in traumatic brain injury. Front Neurol 4:18. https://doi.org/10.3389/fneur.2013.00018
Xiao G, Wei J (2005) Effects of beta-Aescin on the expression of nuclear factor-kappaB and tumor necrosis factor-alpha after traumatic brain injury in rats. J Zhejiang Univ Sci B 6:28–32. https://doi.org/10.1631/jzus.2005.B0028
You WC, Wang C, Pan Y, Zhang X, Zhou X, Zhang X, Shi J, Zhou M (2013) Activation of nuclear factor-κB in the brain after experimental subarachnoid hemorrhage and ıts potential role in delayed brain injury. PLoS One 8:e60290. https://doi.org/10.1371/journal.pone.0060290
Yulug B, Hanoglu L, Khanmammadov E, Duz OA, Polat B, Hanoglu T, Gunal MY, Kilic E (2017) Beyond the therapeutic effect of rTMS in Alzheimer’s disease: a possible neuroprotective role of hippocampal BDNF? : a minireview. MiniRev Med Chem 18:1479–1485. https://doi.org/10.2174/1389557517666170927162537
Funding
This research was supported by OmniActive Health Technologies, NJ, USA. This work was also supported in part by the Turkish Academy of Sciences (TUBA) (KS, EK).
Author information
Authors and Affiliations
Contributions
KS and EK conceived the study. MYG, ABC, and AAS done experiments. FE, OEDK, and EK analyzed the data. KS and EK wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
The experimental protocol was approved by the Animal Research Ethics Committee of Istanbul Medipol University (37/2017) and agreed with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Competing Interests
The authors declare no competing interests.
Disclaimer
Both funding bodies provided financial support and were not involved in the study’s design and the collection, analysis, management, and interpretation of data.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gunal, M.Y., Sakul, A.A., Caglayan, A.B. et al. Protective Effect of Lutein/Zeaxanthin Isomers in Traumatic Brain Injury in Mice. Neurotox Res 39, 1543–1550 (2021). https://doi.org/10.1007/s12640-021-00385-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-021-00385-3