
Abstract
Tissue accumulation of L-2-hydroxyglutaric acid (L-2-HG) is the biochemical hallmark of L-2-hydroxyglutaric aciduria (L-2-HGA), a rare neurometabolic inherited disease characterized by neurological symptoms and brain white matter abnormalities whose pathogenesis is not yet well established. L-2-HG was intracerebrally administered to rat pups at postnatal day 1 (P1) to induce a rise of L-2-HG levels in the central nervous system (CNS). Thereafter, we investigated whether L-2-HG in vivo administration could disturb redox homeostasis and induce brain histopathological alterations in the cerebral cortex and striatum of neonatal rats. L-2-HG markedly induced the generation of reactive oxygen species (increase of 2′,7′-dichloroflurescein-DCFH-oxidation), lipid peroxidation (increase of malondialdehyde concentrations), and protein oxidation (increase of carbonyl formation and decrease of sulfhydryl content), besides decreasing the antioxidant defenses (reduced glutathione-GSH) and sulfhydryl content in the cerebral cortex. Alterations of the activities of various antioxidant enzymes were also observed in the cerebral cortex and striatum following L-2-HG administration. Furthermore, L-2-HG-induced lipid peroxidation and GSH decrease in the cerebral cortex were prevented by the antioxidant melatonin and by the classical antagonist of NMDA glutamate receptor MK-801, suggesting the involvement of reactive species and of overstimulation of NMDA receptor in these effects. Finally, L-2-HG provoked significant vacuolation and edema particularly in the cerebral cortex with less intense alterations in the striatum that were possibly associated with the unbalanced redox homeostasis caused by this metabolite. Taken together, it is presumed that these pathomechanisms may underlie the neurological symptoms and brain abnormalities observed in the affected patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
L-2-Hydroxyglutaric aciduria (L-2-HGA; MIM#236792) is a rare autosomal recessive inborn error of metabolism caused by mutations in the L-2-hydroxyglutarate dehydrogenase gene (L2HGDH), resulting in highly elevated levels of L-2-hydroxyglutaric acid (L-2-HG) in tissues and biological fluids (urine, plasma, and cerebrospinal fluid). The L2HGDH gene is expressed in multiple tissues, with the strongest expression in the brain (Topcu et al. 2004). To date, there are over 300 documented cases of this disease in the medical literature (Balaji et al. 2014; Barbot et al. 1997; Barth et al. 1993; Steenweg et al. 2010; Steenweg et al. 2009; Topcu et al. 2005). The main neurological manifestations include progressive mental retardation, variable motor impairment, and cerebellar ataxia. Most patients also present macrocephaly, pyramidal and extrapyramidal signs, dystonia, and a typical MRI profile with striking subcortical leucoencephalopathy and basal ganglia abnormalities (Steenweg et al. 2009; Wajner and Goodman 2011). Characteristic brain MRI findings in L-2-HGA comprise alterations in the subcortical cerebral white matter, dentate nucleus, globus pallidus, putamen, and caudate nucleus (Steenweg et al. 2009). As the disease progresses, alterations of white matter and basal ganglia signal intensities become more diffuse, followed by cerebral white matter atrophy (Barbot et al. 1997). A strong correlation between the clinical disability and the extent of pathologic changes on MRI has been reported, but this is still under debate (Steenweg et al. 2009).
The mechanisms of the neurological symptoms presented by L-2-HGA patients are still poorly understood. However, increased concentrations of L-2-HG were associated with the appearance of neurological symptoms, indicating that this organic acid may be potentially neurotoxic in this disorder (Jovic et al. 2014; Marcel et al. 2012; Patay et al. 2012; Yilmaz 2009). In this context, it was verified that L-2-HG inhibits creatine kinase activity in rat cerebellum homogenates (da Silva et al. 2003), induces oxidative stress (Latini et al. 2003), and increases glutamate uptake in synaptosomes and synaptic vesicles (Junqueira et al. 2003). Furthermore, it was demonstrated that intracerebral administration of L-2-HG to rats induces oxidative stress by decreasing the antioxidant defenses and raising reactive oxygen species in the striatum and cerebellum of rats, besides causing marked brain histopathological alterations (da Rosa et al. 2015).
Considering that the brain is highly susceptible to free radical attack due to its low antioxidant defenses and high oxygen consumption potentially leading to a proportionally high mitochondrial production of reactive species (Halliwell 2015), in the present study, we investigated the effects of intracerebral administration of L-2-HG on a large spectrum of redox homeostasis parameters in the cerebral cortex and striatum of neonatal rats. Lipid oxidative damage (malondialdehyde levels), protein oxidation (carbonyl formation and sulfhydryl content), reactive oxygen species (DCFH oxidation), and the non-enzymatic (GSH levels) and enzymatic antioxidant defenses (glutathione peroxidase, glutathione reductase, glutathione S-transferase, glucose-6-phosphate dehydrogenase, catalase, and superoxide dismutase) were evaluated. We also investigated whether L-2-HG could provoke histopathological changes in the brain of these animals.
Material and Methods
Animals
Neonatal Wistar rats obtained from the Central Animal House of the Department of Biochemistry, ICBS, Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brazil, were used. Six whole litters of 12 animals each were used for each experimental in vivo condition. The animals had free access to water and 20% (w/w) protein commercial chow (SUPRA, Porto Alegre, RS, Brazil) and were maintained on a 12:12-h light/dark cycle in an air-conditioned constant temperature (22 ± 1 °C) colony room. The experimental protocol was approved by the Ethics Committee for animal research of the Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil, and followed the “Guide for the Care and Use of Laboratory Animals” (NIH publication 85–23, revised 2011). All efforts were made to minimize the number of animals used and their suffering.
Reagents
All chemicals were purchased from Sigma Chemical Co., St. Louis, MO, USA. L-2-HG was dissolved in phosphate-buffered saline (PBS) on the day of the experiments, and the pH was adjusted to 7.4.
L-2-HG Administration and Drug Treatment
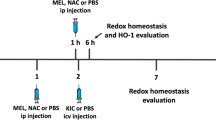
Seventy-two neonatal pups were used in the experiments. Each littermate was injected intracerebrally into the cisterna magna, as previously described (Olivera-Bravo et al. 2011, 2014), between 12 and 48 h after birth (P1) with L-2-HG (0.75 μmol/g, pH 7.4) or vehicle (phosphate-buffered saline (PBS), 0.01 M, pH 7.4). After injection, animals were allowed to recover during 15–30 min and returned to their mother. In some experiments, the antioxidant melatonin (MEL; 20 mg/kg body weight) (Olivier et al. 2009) or the classical NMDA receptor antagonist MK-801 (0.25 mg/kg body weight) (da Rosa et al. 2015) was administered intraperitoneally 60 min before L-2-HG administration.
Sample Preparation
Six hours after L-2-HG injection, animals were euthanized by decapitation, and the brain was immediately removed and kept on an ice Petri plate. The olfactory bulb, pons, and medulla were discarded and the cerebral cortex and striatum were dissected, weighed, and kept chilled until homogenization. These structures were homogenized (1:10 w/v) in 20 mM sodium phosphate, pH 7.4, containing 140 mM chloride potassium (1:10 w/v), centrifuged at 750g for 10 min and the supernatants used for the assays. For hematoxylin and eosin and immunohistochemical analyses, rats were sacrificed 24 h after injections. The whole brain was removed and postfixed in 10% formaldehyde-buffered solution, pH 7.0, for 48 h at room temperature and processed for paraffin-embedded sectioning. Three-micrometer-thick brain slices were obtained with a microtome (MICROM HM 360). The cortical and striatal sections utilized were obtained from four PBS- and L-2-HG-injected animals. Three sections were prepared per animal and five fields per section were analyzed (a total of 15 fields were observed). Medians obtained from five randomly selected fields per animal were used for the statistical calculations. Quantification of histological data was analyzed with × 400 magnification.
Malondialdehyde Levels
MDA concentrations were measured according to the method described by Yagi (1998) with some modifications. Briefly, 100 μL of cerebral cortex or striatum supernatants containing 0.3 mg of protein was treated with 200 μL of 10% trichloroacetic acid and 300 μL of 0.67% thiobarbituric acid in 7.1% sodium sulfate and incubated for 1 h in a boiling water bath. The tubes containing the mixture were allowed to cool on running tap water for 5 min. The resulting pink-stained complex was extracted with 400 μL of butanol. Fluorescence of the organic phase was read at 515 and 553 nm as excitation and emission wavelengths, respectively. A calibration curve was performed using 1,1,3,3-tetramethoxypropane and subjected to the same treatment as supernatants. MDA levels were calculated as nanomole MDA per milligram protein.
Sulfhydryl Group Content
This assay was performed according to Aksenov and Markesbery (2001). It is based on the reduction of 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) by thiol groups, generating a yellow-stained compound TNB whose absorption is measured spectrophotometrically at 412 nm. Thirty microliters of 0.1 mM DTNB was added to 120 μL of cerebral cortex or striatum supernatants containing 0.36 mg of protein. This was followed by 30-min incubation at room temperature in a dark room. Absorption was measured at 412 nm. Results were calculated as nanomole TNB per milligram protein.
Protein Carbonyl Formation
Protein carbonyl formation was measured according to the method of Reznick and Packer (1994). The assay is based on the reaction of the carbonyl groups with 2,4-dinitrophenylhydrazine (DNPH) generating a corresponding dinitrophenyl hydrazone, whose absorbance is read in a spectrophotometer at 370 nm. Briefly, 200 μL of cerebral cortex or striatum supernatants was treated with 400 μL of 10 mM DNPH dissolved in 2.5 M HCl or with 2.5 M HCl (blank) and left in the dark for 1 h. Samples were then precipitated with 600 μL of 20% TCA and centrifuged for 5 min at 9000×g. The pellet was washed with 1 mL of ethanol:ethyl acetate (1:1, v/v) and suspended in 550 μL of 6 M guanidine prepared in 2.5 N HCl. The difference between the absorbance of the samples treated with DNPH and with HCl (blank) was used to calculate the carbonyl content. The results were calculated as nanomole carbonyl groups per milligram of protein, using the extinction coefficient of 22,000 × 106 nmol/mL for aliphatic hydrazones.
Reduced Glutathione Concentrations
This parameter was measured according to Browne and Armstrong (1998) with some modifications. One hundred and 85 μL of 100 mM sodium phosphate buffer, pH 8.0, containing 5 mM EDTA, and 15 μL of o-phthaldialdehyde (1 mg/mL) were added to 30 μL of sample (0.3–0.5 mg of protein) previously deproteinized with metaphosphoric acid. This mixture was incubated at room temperature in a dark room for 15 min. Fluorescence was measured using excitation and emission wavelengths of 350 and 420 nm, respectively. Calibration curve was prepared with standard GSH (0.001–1 mM), and the concentrations were calculated as nanomole GSH per milligram protein.
2′,7′-Dichlorofluorescein Oxidation
DCFH oxidation was determined according to the method of LeBel et al. (1992) with slight modifications. Homogenates (0.04–0.07 mg protein) were incubated with 2′,7′-dichlorofluorescein diacetate (DCF-DA) (5 μM) during 1 h at 37 °C. DCF-DA is permeable to the cell membrane and is deacetylated by esterases to DCFH in the intracellular medium. DCFH is then converted by reactive species into the highly fluorescent product dichlorofluorescein (DCF). DCF fluorescence was measured using wavelengths of 480 nm (excitation) and 535 nm (emission). The calibration curve was performed with standard DCF (0.01–0.75 μM). The production of reactive species was calculated as micromole DCF per milligram protein.
Nitrate and Nitrite Content
Reactive nitrogen species (RNS) formation was evaluated by measuring nitrate and nitrite content according to Navarro-Gonzalvez and collaborators (1998), with some modifications. One hundred and 50 μL of supernatants (containing approximately 1.2 mg of protein) was deproteinized by adding 125 mL of 75 mM ZnSO4 solution, followed by centrifugation at 9000×g for 2 min at 25 °C. The supernatant obtained was neutralized with 55 mM NaOH solution and diluted in 5 vol of glycine buffer solution, pH 9.7. Copper-coated cadmium granules (600–1000 mg) were added to the supernatants to convert all nitrates into nitrite in the biological samples. Aliquots of 200 μL were then treated with 200 μL of Griess reagent (2% sulfanilamide in 5% HCl and 0.1% N-1-(naphtyl)ethylenediamine in H2O) and incubated at room temperature for 10 min. The absorbance was read at 505 nm. A calibration curve was prepared with NaNO2 at concentrations ranging from 1 to 125 mM. The final results were expressed in micromole nitrate and nitrite per milligram protein.
Glutathione Peroxidase Activity
Glutathione peroxidase (GPx) activity was measured according to Wendel (1981) using tert-butylhydroperoxide as substrate. The enzyme activity was determined by monitoring the NADPH disappearance at 340 nm in a medium containing 600 μL of buffer (100 mM potassium phosphate containing 1 mM EDTA, pH 7.0), 10 μL of 40 mM sodium azide, 15 μL of 100 mM GSH, 15 μL of 10 U/mL glutathione reductase, 10 μL of 10 mM NADPH, and 10 μL of sample (3 μg of protein). One GPx unit (U) is defined as 1 μmol of NADPH consumed per minute. The specific activity was calculated as U per milligram protein.
Glutathione Reductase Activity
Glutathione reductase (GR) activity was measured as previously described by Carlberg and Mannervik (1985). The enzyme activity was determined by monitoring NADPH consumption at 340 nm in a medium with 200 mM sodium phosphate buffer, pH 7.5, containing 6.3 mM ethylenediaminetetraacetic acid, 1 mM GSSG, 0.1 mM NADPH, and approximately 3 μg of protein. One GR U is defined as 1 μmol of GSSG reduced per minute. The specific activity was calculated as U per milligram protein.
Glutathione S-Transferase Activity
Glutathione S-transferase (GST) activity was measured according to Mannervik and Guthenberg (1981), with slight modifications, by monitoring the rate of formation of dinitrophenyl-S-glutathione at 340 nm in a medium containing 50 mM potassium phosphate, pH 6.5, 1 mM GSH, 1 mM 1-chloro-2,4-dinitrobenzene (CDNB) as substrate, and tissue supernatants (approximately 0.045 mg of protein). The results were calculated as U per milligram protein.
Glucose-6-Phosphate Dehydrogenase Activity
Glucose-6-phosphate dehydrogenase (G6PDH) activity was measured according to Leong and Clark (1984), following NADPH formation at 25 °C at 340 nm. The reaction medium contained tissue supernatants (approximately 45 μg of protein), 100 mM Tris-HCl buffer, pH 7.5, 1.0 mM MgCl2, 0.05 mM NADP+, and 0.1 mM glucose-6-phosphate. The results were calculated as U per milligram protein.
Catalase Activity
Catalase (CAT) activity was assayed according to Aebi (1984) by measuring the absorbance decrease at 240 nm in a reaction medium containing 20 mM H2O2, 0.1% Triton X-100, 10 mM potassium phosphate buffer, pH 7.0, and approximately 1 μg of protein. One U of the enzyme is defined as 1 μmol of H2O2 consumed per minute. The specific activity was calculated as U per milligram protein.
Superoxide Dismutase Activity
Superoxide dismutase (SOD) activity was assayed according to Marklund (1985) and is based on the capacity of pyrogallol to autoxidize, a process highly dependent on superoxide anion, which is the substrate for SOD. The inhibition of the autoxidation of pyrogallol occurs in the presence of SOD and, therefore, is proportional to the activity of the SOD present in homogenates. The reaction medium contained 50 mM Tris buffer/1 mM ethylenediaminetetraacetic acid, pH 8.2, 80 U/mL CAT, 0.38 mM pyrogallol, and approximately 1 μg of protein, and the absorbance was read at 420 nm. A calibration curve was performed with purified SOD as standard, in order to calculate the activity of SOD present in the samples. The specific activity was calculated as U per milligram protein.
Histological Analysis
We performed histological analysis in the brain with hematoxylin and eosin staining and immunohistochemical for NeuN. Four rats of each group (PBS- or L-2-HG-injected animals) were euthanized 24 h after PBS or L-2-HG (0.75 μmol/g) administration. Brain sections were stained in hematoxylin solution for 5 min and then washed in running tap water. The sections were then placed in eosin solution for 3 min, dehydrated with 100% ethanol, and cleared with xylene for 2 min. Image analysis (magnification of × 100 and × 400) was performed using the Q Capture Pro Software (Olympus). Image J software was utilized to count the numbers of vacuoles (magnification of × 400).
For immunohistochemistry analysis, samples were deparaffinized and immersed in a mixture of 3% hydrogen peroxide and 10% methanol for 15 min to inhibit the endogenous peroxidase activity. After being washed with PBS, they were incubated with common horse serum for 1 h and then overnight with primary antibody at 4 1C in a wet chamber. The following primary antibodies were used: anti-GFAP (1:400 dilution, Cellmarque, clone EP672Y) and anti-NeuN (1:400 dilution, Zetacorporation, clone A60). After overnight incubation with the primary antibodies, the slides were washed three times with PBS, incubated with DAKO secondary polymer for 40 min, followed by tertiary joint horseradish peroxidase for 30 min, and finally with DAB. All slides were counterstained with hematoxylin for 10 s. Positive controls were always used. Quantification of NeuN was performed using the Image J software by counting the numbers of immunoreactive cells (magnification of × 400). Four rats were used per group.
Protein Determination
Protein content was measured by the method of Lowry et al. (1951), using bovine serum albumin as a standard.
Statistical Analysis
Results are presented as mean ± standard deviation. Assays were performed in triplicate, and the mean was used for statistical calculations. Data were analyzed using Student’s t test for unpaired samples or one-way analysis of variance (ANOVA) followed by the post-hoc Duncan multiple range test when F was significant. Only significant values are shown in the text. Differences between groups were considered significant at P < 0.05. All analyses were carried out using the GraphPad Prism 5 software.
Results
In Vivo L-2-HG administration provokes lipid and protein oxidative damage in the neonatal rat cerebral cortex
We initially observed that L-2-HG markedly enhanced (75%) MDA levels in the cerebral cortex of neonatal rats 6 h after its administration [t(7) = 4.321; P < 0.01] (Fig. 1A). Since MDA is an end product of membrane polyunsaturated fatty acid peroxidation, these data indicate that L-2-HG causes lipid oxidative damage. L-2-HG also increased carbonyl formation (11%) [t(5) = 3.317; P < 0.05] (Fig. 1B) and decreased sulfhydryl content (34%) [t(5) = 5.020; P < 0.01] (Fig. 1C). Since carbonyl group generation is currently used as a marker of free radical-mediated protein oxidation and two thirds of total cellular sulfhydryl groups are protein bound (Levine et al. 1994; Requejo et al. 2010), our results indicate that L-2-HG induces protein oxidative damage in cerebral cortex.

Effect of intracerebral administration of L-2-hydroxyglutaric acid (L-2-HG; 0.750 μmol/g) on malondialdehyde (MDA) levels (a), carbonyl formation (b), and sulfhydryl content (c) in the cerebral cortex of neonatal rats. Data are represented as mean ± SD for four to six independent experiments (animals) performed in triplicate and are expressed as nmol/mg protein. *P < 0.5, **P < 0.01, compared to control (Student’s t test for unpaired samples)
L-2-HG Decreases the Antioxidant Defenses in the Rat Cerebral Cortex
Next, we observed that L-2-HG decreased the concentrations of GSH (25%) [t(8) = 5.655; P < 0.001] (Fig. 2), implying an impairment of brain antioxidant defenses by this organic acid.

Effect of intracerebral administration of L-2-hydroxyglutaric acid (L-2-HG; 0.750 μmol/g) on concentrations of GSH in the cerebral cortex of neonatal rats. Data are represented as mean ± SD for four to six independent experiments (animals) performed in triplicate and are expressed as nmol/mg protein. ***P < 0.001, compared to control (Student’s t test for unpaired samples)
L-2-HG Induces Reactive Oxygen Species Generation in the Rat Cerebral Cortex
We also investigated whether L-2-HG could induce reactive species generation that could possibly lead to protein and lipid oxidative damage as well as to reduction of antioxidant defenses. Thus, we tested the effects of L-2-HG on reactive oxygen species (ROS) production, determined by DCFH oxidation in the cerebral cortex, and on RNS generation measured by nitrate and nitrite concentrations. L-2-HG significantly increased DCFH oxidation (up to 25%) [t(5) = 2.914; P < 0.05] (Fig. 3A) but, in contrast, did not change the levels of nitrates and nitrites (Fig. 3B).

Effect of intracerebral administration of L-2-hydroxyglutaric acid (L-2-HG; 0.750 μmol/g) on DCFH oxidation (a) in the cerebral cortex and nitrate and nitrite concentrations (b) in the forebrain of neonatal rats. Data are represented as mean ± SD for four to six independent experiments (animals) performed in triplicate and are expressed as μmol/mg protein. *P < 0.5, compared to control (Student’s t test for unpaired samples)
L-2-HG-Induced Lipid Peroxidation and Decreased Antioxidant Defenses Are Prevented by Melatonin and MK-801 in the Neonatal Rat Cerebral Cortex
Since L-2-HG induced ROS formation, we tested whether the reactive species scavenger melatonin (MEL) could prevent lipid oxidation and the decrease of the antioxidant defenses caused by this metabolite. We observed that MEL totally prevented MDA increase [t(4) = 2.788; P < 0.05] (Fig. 4A) and GSH decrease [t(6) = 7.476; P < 0.001] (Fig. 4B) caused by L-2-HG. These data support the involvement of ROS in these effects and may offer a new perspective of treatment with antioxidants. Furthermore, the effects on MDA levels [t(5) = 3.995; P < 0.05] and GSH concentrations [t(6) = 4.208; P < 0.01] were similarly abolished by the NMDA receptor antagonist MK-801 (Fig. 4A, B), indicating overactivation of NMDA receptors by L-2-HG.

Effect of melatonin (MEL; 20 mg/Kg) and MK-801 (0,25 mg/Kg) on L-2-hydroxyglutaric acid (L-2-HG 0.750 μmol/g)-induced increase of MDA (a) and decrease of GSH concentrations (b) in the cerebral cortex of neonatal rats. Data are represented as mean ± SD for four to six independent experiments (animals) performed in triplicate and are expressed as nmol/mg protein. *P < 0.05, compared to control; #P < 0.05, ##P < 0.01, ###P < 0.001, compared to L-2-HG (Student’s t test for unpaired samples)
L-2-HG Modulates the Activities of Antioxidant Enzymes in the Neonatal Rat Cerebral Cortex
The effects of L-2-HG intracerebroventricular administration on the enzymatic antioxidant defenses were further studied. We verified that L-2-HG reduced G6PDH activity (14%) [t(6) = 2.775; P < 0.05] (Fig. 5A) and increased GPx (18%) [t(5) = 4.326; P < 0.05] (Fig. 5B), GR (20%) [t(6) = 4.326; P < 0.01] (Fig. 5C), and GST (39%) [t(6) = 3.914; P < 0.01] (Fig. 5D) activities. In contrast, CAT (Fig. 5E) and SOD (Fig. 5F) activities were not altered by the metabolite.

Effect of intracerebral administration of L-2-hydroxyglutaric acid (L-2-HG; 0.750 μmol/g) on the activities of glucose-6-phosphate dehydrogenase (G6PDH) (a), glutathione peroxidase (GPx) (b), glutathione reductase (GR) (c), glutathione S-transferase (GST) (d), catalase (CAT) (e), and superoxide dismutase (SOD) in the cerebral cortex of neonatal rats. Data are represented as mean ± SD for four to six independent experiments (animals) performed in triplicate and are expressed as U/mg protein. *P < 0.05, **P < 0.01 compared to control (Student’s t test for unpaired samples)
L-2-HG Mildly Disrupts Redox Homeostasis in the Neonatal Rat Cerebral Striatum
We also observed that L-2-HG administration decreased sulfhydryl content (50%) [t(4) = 10.13; P < 0.001] (Fig. 6A) but did not alter MDA levels, GSH concentrations, and ROS formation (DCFH oxidation) in the rat striatum. It was also verified that L-2-HG modulated the enzymatic antioxidant defenses by increasing GPx (35%) [t(5) = 3.255; P < 0.05] (Fig. 6B), G6PDH (20%) [t(5) = 3.354; P < 0.05] (Fig. 6C), and SOD (22%) [t(7) = 4.633; P < 0.01] (Fig. 6D) activities, with no changes in the activities of GR and GST.

Effect of intracerebral administration of L-2-hydroxyglutaric acid (L-2-HG; 0.750 μmol/g) on sulfhydryl content (a) and the activities of glutathione peroxidase (GPx) (b), glucose-6-phosphate dehydrogenase (G6PDH) (c), and superoxide dismutase (SOD) (d) in striatum of neonatal rats. *P < 0.05, **P < 0.01, ***P < 0.001, compared to control (Student’s t test for unpaired samples)
L-2-HG Provokes Extensive Vacuolation in the Cerebral Cortex and Moderate Vacuolation in the Striatum of Neonatal Rat
The effect of L-2-HG intracerebroventricular administration on the brain histological integrity was then studied by HE staining. We observed that L-2-HG administration provoked intense edema and vacuolation in the cerebral cortex (fourfold) [t(6) = 10.80, P < 0.001] (Fig. 7) with less significant effects in the striatum (twofold) [t(6) = 7.80, p < 0.001] (Fig. 8). In contrast, no significant differences in NeuN labeling were found in any experimental conditions (PBS and L-2-HG), neither in the striatum nor in the cerebral cortex, thus discarding significant neuronal death 24 h after injections.

Light microscopy images showing hematoxylin and eosin (HE) staining of the cerebral cortex from control (PBS) (a, c) and L-2-hydroxylglutaric acid (L-2-HG)-injected neonatal rats (b, d). Animals were sacrificed 24 h after L-2-HG intracerebroventricular administration. Representative images were obtained from four rats per group. HE staining with magnification of × 100 and × 400. Arrows in panel d indicate vacuolation. e Quantification of the number of vacuoles was performed with × 400 magnification by using the median of five randomly selected fields from three cerebral cortex sections per brain. Values are mean ± standard deviation for four rats per group. ***P < 0.001 compared to PBS-injected rats (Student’s t test for unpaired samples)

Light microscopy images showing hematoxylin and eosin (HE) staining of striatum from control (PBS) (a, c) and L-2-hydroxylglutaric acid (L-2-HG)-injected neonatal rats (b, d). Animals were sacrificed 24 h after intracerebroventricular administration. Representative images were obtained from four rats per group. HE staining with magnification of × 100 and × 400. Arrows in panel d indicate vacuolation. e Quantification of the number of vacuoles was performed with × 400 magnification by using the median of five randomly selected fields from three striatum sections per brain. Values are mean ± standard deviation for four rats per group. ***P < 0.001 compared to PBS-injected rats (Student’s t test for unpaired samples)
Discussion
L-2-HGA is clinically characterized by predominant neurological findings, such as psychomotor delay/mental deficiency, extrapyramidal and pyramidal symptoms, macrocephaly, seizure demyelination of white matter, cerebral and cerebellar atrophy, seizures, leukoencephalopathy, and other neurologic manifestations (Steenweg et al. 2010; Steenweg et al. 2009; Topcu et al. 2005). In some patients, this disorder leads to the formation of brain tumors (Aghili et al. 2009; Haliloglu et al. 2008; Moroni et al. 2004), whose etiology is still poorly known. Accumulation of L-2-HG in tissues, predominantly in the brain, is the biochemical hallmark of this disease, and this may be because the mutated gene is highly expressed in the brain (Topcu et al. 2004). Although the disorder is predominantly neurologic, its pathogenesis is not yet established. However, it was suggested that the pathologic phenotypes of affected patients result from the toxic effects of L-2-HG on central nervous system (Rzem et al. 2006). In this context, the structural resemblance of L-2-HG to L-glutamate and α-ketoglutarate, two major metabolites of the glutamatergic system, makes it likely that the pathophysiology of the disease may involve alterations of this system or pathways of α-ketoglutarate metabolism. Furthermore, previous in vitro and in vivo studies have shown disruption of redox homeostasis provoked by L-2-HG, as well as alterations of the glutamatergic system in developing rats and affected patients with L-2-HGA (da Rosa et al. 2015; Junqueira et al. 2003; Latini et al. 2003; Rodrigues et al. 2017).
It is of note that nothing has been reported on the brain toxicity of L-2-HG in neonatal brain. Therefore, in the present study, we investigated whether L-2-HG in vivo administration could disturb redox status and induce histopathological alterations in the cerebral cortex and striatum of neonatal rats. It is emphasized that these cerebral structures are mainly affected in L-2-HGA, as observed in cerebral imaging by leukoencephalopathy/spongiosis of white matter, subcortical abnormalities, and caudate and putamen alterations (Anghileri et al. 2016; Fourati et al. 2016).
We verified that L-2-HG administration markedly increased MDA levels in the cerebral cortex, corroborating the in vitro and in vivo findings previously reported and showing that L-2-HG increases the levels of thiobarbituric acid-reactive substances in the brain of 30-day-old rats (da Rosa et al. 2015; Latini et al. 2003). Since MDA is an end product of membrane fatty acid peroxidation, these data indicate that L-2-HG induces lipid oxidative damage. L-2-HG also markedly increased carbonyl groups and decreased sulfhydryl content. Since carbonyl groups are formed by oxidation of amino acid side chain protein residues (Pro, Arg, Lys, and Thr) and two thirds of sulfhydryl groups in cells are protein bound (Levine et al. 1994; Requejo et al. 2010), it is assumed that L-2-HG induces protein oxidative damage in the brain.
L-2HG also caused a significant decrease of GSH concentrations in the cerebral cortex that could be tentatively attributed to its oxidation by increased reactive species generation. Since GSH represents the major naturally occurring tissue antioxidant and plays a key role in maintaining the intracellular redox state protecting crucial cellular sulfhydryl groups (Halliwell 2015), our present findings suggest that L-2-HG impairs the tissue non-enzymatic antioxidant defenses.
DCFH oxidation was also increased by L-2-HG, but, in contrast, this organic acid did not change nitrate and nitrite content, reflecting induction of ROS generation. It could be also hypothesized that the increase of ROS formation could lead secondarily to increased lipid and protein oxidation and decreased GSH concentrations. On the other hand, since GSH is considered an important defense against lipid oxidative damage by scavenging reactive species responsible for the initiation of this process, it is feasible that the decrease of this antioxidant could make lipids and proteins more vulnerable to the oxidative damage caused by L-2-HG through ROS formation.
Moreover, the free radical scavenger melatonin prevented L-2-HG-induced reduction of GSH levels and increase of MDA concentrations, strongly indicating that disturbance of cellular redox status was secondary to reactive species production provoked by L-2-HG. It is likely that hydroxyl and peroxyl radicals were involved in these effects, since these reactive species are scavenged by melatonin (Anisimov et al. 2006; Halliwell 2015; Reiter et al. 2013). On the other hand, it is feasible that NMDA receptor overstimulation was involved in the L-2-HG-induced impairment of cell redox since the classical antagonist of these receptors MK-801 prevented lipid oxidative damage and antioxidant defense decrease.
We also observed that L-2-HG increased GPx, GR, and GST activities and inhibited G6PDH activity in the cerebral cortex. We presume that the increased activities of GPx and GST might have occurred due to the overexpression of these enzymes at gene level in order to compensate the increased formation of lipid peroxides, which is in line with the fact that L-2-HG increases MDA levels, a lipid peroxidation end product. In this particular, it is known that GST also detoxifies 4-hydroxynonenal and isoprostanes that are lipid peroxidation products (Halliwell 2015). Furthermore, increased GR activity also suggests overexpression of this enzyme protein aiming to recycle GSH once the concentrations of this antioxidant are decreased. As regards to the L-2-HG-induced decrease of G6PDH activity, we speculate that it occurred due to reactive species attack causing modifications in the enzyme protein structure as previously shown for this and other antioxidant enzymes (Grings et al. 2017; Seminotti et al. 2016).
Since oxidative stress results from an imbalance between the total antioxidant defenses and the reactive species generated in a tissue, our present data strongly indicate that L-2-HG induces oxidative stress in vivo in the rat cerebral cortex. It must be emphasized that ROS are capable to oxidize various molecules, including lipids, proteins, sugars, and DNA, representing a key event in the pathogenic cascade leading to necrotic and apoptotic cell death (Circu and Aw 2010; Niizuma et al. 2009). Furthermore, oxidative stress is a very deleterious condition especially to the brain due to its high rate of oxidative metabolism coupled to ROS production, lower activity of antioxidant enzymes, and higher peroxidation potential (high content of polyunsaturated fatty acids) (Halliwell 2015).
It was also demonstrated in this study that L-2-HG provoked extensive edema and vacuolation in the cerebral cortex of the animals, with less intense effects in the striatum. Although not proven, we assume that these brain abnormalities could be possibly related to the disruption of redox homeostasis elicited by this organic acid (Cubells et al. 1994; da Rosa et al. 2015; Saxena et al. 2010; Wilt et al. 2000). However, L-2-HG did not cause apparent change in striatal and cortical neuronal density (neuronal death) 24 h after injection, as denoted by the pan-neuronal marker NeuN. This is in accordance with previous reports showing low neuronal vulnerability in another cerebral organic acidemia, despite the intense vacuolation (Koeller et al. 2002, 2004).
It is difficult at present to determine the pathophysiological relevance of our data since brain concentrations of L-2-HG in L-2-HGA are still unknown. We cannot rule out the possibility that intracerebral L-2-HG concentrations in the present study were supraphysiological, so that our findings must be taken carefully. However, the concentrations of organic acids accumulating within neural cells in organic acidemias were reported to be usually much higher than those of plasma and cerebrospinal fluid (trapping theory) of patients with various organic acidemias (Hoffmann et al. 1993). Furthermore, L-2-HGA is considered a cerebral organic acidemia because L-2-HG is trapped in the CNS (Sauer et al. 2006, 2010), where at high concentrations, it potentially can exert its neurotoxic effects disturbing cellular redox homeostasis.
As regards to the association between our present results and the neurodegeneration encountered in patients with L-2-HGA, we observed that edema and vacuolation were more pronounced in the cerebral cortex relatively to the striatum, which follows the neuropathological pattern of the affected patients, including subcortical leukoencephalopathy and signal changes in the striatum (Barbot et al. 1997). Therefore, it is presumed that our present data may possibly contribute to clarify the neuropathology in this disorder.
In conclusion, this is the first report showing that L-2-HG administration to neonatal rats induces in vivo lipid and protein oxidative damage and disrupts the antioxidant defenses mainly in the cerebral cortex. These effects were prevented by the potent antioxidant melatonin and the NMDA antagonist MK-801, implying that reactive species generation and overstimulation of these glutamate receptors may underlie the altered cellular redox status. These in vivo data allied to previous in vitro (Latini et al. 2003) and in vivo (da Rosa et al. 2015) experimental animal and humans studies (Jellouli et al. 2014; Rodrigues et al. 2017) strongly indicate that imbalance of redox homeostasis may potentially contribute at least in part to the neurologic alterations found in L-2-HGA. Moreover, it is conceivable that sustained elevated levels of L-2-HG found in the patients may lead to chronic oxidative stress. Another novel and important finding of the present study is that this organic acid provoked histopathological alterations more evident in the cerebral cortex probably due to the induced disruption of redox homeostasis. It may be presumed that our present findings acting synergistically with other intrinsic (severity of mutations) and extrinsic factors may contribute to explain the pathophysiology of the neurologic symptoms and brain abnormalities observed in the affected patients. In case our present in vivo results can be confirmed in tissues (blood and cultivated fibroblasts) from individuals affected by L-2HGA, it is therefore tempting to speculate that antioxidants could be supplemented to the affected patients as a potential adjuvant therapy to prevent or attenuate oxidative damage.
References
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126. https://doi.org/10.1016/S0076-6879(84)05016-3
Aghili M, Zahedi F, Rafiee E (2009) Hydroxyglutaric aciduria and malignant brain tumor: a case report and literature review. J Neuro-Oncol 91(2):233–236. https://doi.org/10.1007/s11060-008-9706-2
Aksenov MY, Markesbery WR (2001) Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci Lett 302(2-3):141–145. https://doi.org/10.1016/S0304-3940(01)01636-6
Anghileri E, Bertolino N, Salsano E, Antelmi L, Carpinelli P, Castellotti B, Zucca I, Gellera C, Bisogno R, Caccia C, Cuccarini V (2016) In-vivo brain H1-MR-spectroscopy identification and quantification of 2-hydroxyglutarate in L-2-Hydroxyglutaric aciduria. Brain Res 1648(Pt A):506–511. https://doi.org/10.1016/j.brainres.2016.08.013
Anisimov VN, Popovich IG, Zabezhinski MA, Anisimov SV, Vesnushkin GM, Vinogradova IA (2006) Melatonin as antioxidant, geroprotector and anticarcinogen. Biochim Biophys Acta 1757(5-6):573–589. https://doi.org/10.1016/j.bbabio.2006.03.012
Balaji P, Viswanathan V, Chellathurai A, Panigrahi D (2014) An interesting case of metabolic dystonia: L-2 hydroxyglutaric aciduria. Ann Indian Acad Neurol 17(1):97–99. https://doi.org/10.4103/0972-2327.128565
Barbot C, Fineza I, Diogo L, Maria M, Melo J, Guimarães A, Melo Pires M, Luis Cardoso M, Vila rinho L (1997) L-2-Hydroxyglutaric aciduria: clinical, biochemical and magnetic resonance imaging in six Portuguese pediatric patients. Brain Dev 19(4):268–273. https://doi.org/10.1016/S0387-7604(97)00574-3
Barth PG, Hoffmann GF, Jaeken J, RJA W, Duran M, Jansen GA, Jakobs C, Lehnert W, Hanefeld F, Valk J, RBH S, Trefz FK, Hartung HP, Chamoles NA, Sfaello Z, Caruso U (1993) L-2-hydroxyglutaric acidaemia: clinical and biochemical findings in 12 patients and preliminary report on L-2-hydroxyacid dehydrogenase. J Inherit Metab Dis 16(4):753–761. https://doi.org/10.1007/BF00711907
Browne RW, Armstrong D (1998) Reduced glutathione and glutathione disulfide. Methods Mol Biol 108:347–352. https://doi.org/10.1385/0-89603-472-0:347
Carlberg I, Mannervik B (1985) Glutathione reductase. Methods Enzymol 113:484–490. https://doi.org/10.1016/S0076-6879(85)13062-4
Circu ML, Aw TY (2010) Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 48(6):749–762. https://doi.org/10.1016/j.freeradbiomed.2009.12.022
Cubells JF, Rayport S, Rajendran G, Sulzer D (1994) Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci 14(4):2260–2271
da Rosa MS, João Ribeiro CA, Seminotti B, Teixeira Ribeiro R, Umpierrez Amaral A, de Moura Coelho D, de Oliveira FH, Leipnitz G, Wajner M (2015) In vivo intracerebral administration of L-2-hydroxyglutaric acid provokes oxidative stress and histopathological alterations in striatum and cerebellum of adolescent rats. Free Radic Biol Med 83:201–213. https://doi.org/10.1016/j.freeradbiomed.2015.02.008
da Silva CG, ARF B, Schuck PF, Leipnitz G, CAJ R, CMD W, ATS W, Wajner M (2003) L-2-hydroxyglutaric acid inhibits mitochondrial creatine kinase activity from cerebellum of developing rats. Int J Dev Neurosci 21(4):217–224. https://doi.org/10.1016/S0736-5748(03)00035-2
Fourati H, Ellouze E, Ahmadi M, Chaari D, Kamoun F, Hsairi I, Triki C, Mnif Z (2016) MRI features in 17 patients with l2 hydroxyglutaric aciduria. Eur J Radiol Open 3:245–250. https://doi.org/10.1016/j.ejro.2016.09.001
Grings M, Moura AP, Parmeggiani B, Pletsch JT, Cardoso GMF, August PM, Matté C, Wyse ATS, Wajner M, Leipnitz G (2017) Bezafibrate prevents mitochondrial dysfunction, antioxidant system disturbance, glial reactivity and neuronal damage induced by sulfite administration in striatum of rats: implications for a possible therapeutic strategy for sulfite oxidase deficiency. Biochim Biophys Acta 1863(9):2135–2148. https://doi.org/10.1016/j.bbadis.2017.05.019
Haliloglu G, Jobard F, Oguz K, Anlar B, Akalan N, Coskun T, Sass J, Fischer J, Topcu M (2008) L-2-hydroxyglutaric aciduria and brain tumors in children with mutations in the L2HGDH gene: neuroimaging findings. Neuropediatrics 39(02):119–122. https://doi.org/10.1055/s-2008-1081217
Halliwell BGJ (2015) Cellular responses to oxidative stress: adaptation, damage, repair, senescence and death. In: Free radicals in biology and medicine. Oxford University Press Inc, Oxford, pp 199–283. https://doi.org/10.1093/acprof:oso/9780198717478.003.0005
Hoffmann GF, Meier-Augenstein W, Stockler S, Surtees R, Rating D, Nyhan WL (1993) Physiology and pathophysiology of organic acids in cerebrospinal fluid. J Inherit Metab Dis 16(4):648–669. https://doi.org/10.1007/BF00711898
Jellouli NK et al (2014) Founder effect confirmation of c.241A>G mutation in the L2HGDH gene and characterization of oxidative stress parameters in six Tunisian families with L-2-hydroxyglutaric aciduria. J Hum Genet 59(4):216–222. https://doi.org/10.1038/jhg.2014.4
Jovic NJ, Kosac A, Koprivsek K (2014) L-2-Hydroxyglutaric aciduria: a case report. Srp Arh Celok Lek 142:337–341
Junqueira D, Brusque AM, Porciúncula LO, Rotta LN, CAJ R, MES F, CSD F, CMD W, ATS W, Souza DO, Wajner M (2003) Effects of L-2-hydroxyglutaric acid on various parameters of the glutamatergic system in cerebral cortex of rats. Metab Brain Dis 18(3):233–243. https://doi.org/10.1023/A:1025559200816
Koeller DM, Woontner M, Crnic LS, Kleinschmidt-DeMasters B, Stephens J, Hunt EL, Goodman SI (2002) Biochemical, pathologic and behavioral analysis of a mouse model of glutaric acidemia type I. Hum Mol Genet 11(4):347–357. https://doi.org/10.1093/hmg/11.4.347
Koeller DM, Sauer S, Wajner M, de Mello CF, Goodman SI, Woontner M, Mühlhausen C, Okun JG, Kölker S (2004) Animal models for glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27(6):813–818. https://doi.org/10.1023/B:BOLI.0000045763.52907.5e
Latini A, Scussiato K, Borba Rosa R, Leipnitz G, Llesuy S, Belló-Klein A, Dutra-Filho CS, Wajner M (2003) Induction of oxidative stress by L-2-hydroxyglutaric acid in rat brain. J Neurosci Res 74(1):103–110. https://doi.org/10.1002/jnr.10735
LeBel CP, Ischiropoulos H, Bondy SC (1992) Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5(2):227–231. https://doi.org/10.1021/tx00026a012
Leong SF, Clark JB (1984) Regional development of glutamate dehydrogenase in the rat brain. J Neurochem 43(1):106–111. https://doi.org/10.1111/j.1471-4159.1984.tb06684.x
Levine RL, Williams JA, Stadtman ER, Shacter E (1994) Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol 233:346–357
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Mannervik B, Guthenberg C (1981) Glutathione transferase (human placenta). Methods Enzymol 77:231–235. https://doi.org/10.1016/S0076-6879(81)77030-7
Marcel C, Mallaret M, Lagha-Boukbiza O, Kremer S, Echaniz-Laguna A, Tranchant C (2012) L-2-hydroxyglutaric aciduria diagnosed in a young adult with progressive cerebellar ataxia and facial dyskinesia. Rev Neurol 168(2):187–191. https://doi.org/10.1016/j.neurol.2011.06.002
Marklund SL (1985) Product of extracellular-superoxide dismutase catalysis. FEBS Lett 184(2):237–239. https://doi.org/10.1016/0014-5793(85)80613-X
Moroni I, Bugiani M, D’Incerti L, Maccagnano C, Rimoldi M, Bissola L, Pollo B, Finocchiaro G, Uziel G (2004) L-2-hydroxyglutaric aciduria and brain malignant tumors: a predisposing condition? Neurology 62(10):1882–1884. https://doi.org/10.1212/01.WNL.0000125335.21381.87
Navarro-Gonzalvez JA, Garcia-Benayas C, Arenas J (1998) Semiautomated measurement of nitrate in biological fluids. Clin Chem 44:679–681
Niizuma K, Endo H, Chan PH (2009) Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem 109(Suppl 1):133–138. https://doi.org/10.1111/j.1471-4159.2009.05897.x
Olivera-Bravo S, Fernandez A, Sarlabos MN, Rosillo JC, Casanova G, Jimenez M, Barbeito L (2011) Neonatal astrocyte damage is sufficient to trigger progressive striatal degeneration in a rat model of glutaric acidemia-I. PLoS One 6(6):e20831. https://doi.org/10.1371/journal.pone.0020831
Olivera-Bravo S, Isasi E, Fernández A, Rosillo JC, Jiménez M, Casanova G, Sarlabós MN, Barbeito L (2014) White matter injury induced by perinatal exposure to glutaric acid. Neurotox Res 25(4):381–391. https://doi.org/10.1007/s12640-013-9445-9
Olivier P, Fontaine RH, Loron G, van Steenwinckel J, Biran V, Massonneau V, Kaindl A, Dalous J, Charriaut-Marlangue C, Aigrot MS, Pansiot J, Verney C, Gressens P, Baud O (2009) Melatonin promotes oligodendroglial maturation of injured white matter in neonatal rats. PLoS One 4(9):e7128. https://doi.org/10.1371/journal.pone.0007128
Patay Z, Mills JC, Lobel U, Lambert A, Sablauer A, Ellison DW (2012) Cerebral neoplasms in L-2 hydroxyglutaric aciduria: 3 new cases and meta-analysis of literature data. AJNR Am J Neuroradiol 33:940–943. https://doi.org/10.3174/ajnr.A2869
Reiter RJ, Tan DX, Rosales-Corral S, Manchester LC (2013) The universal nature, unequal distribution and antioxidant functions of melatonin and its derivatives. Min Rev Med Chem 13(3):373–384
Requejo R, Chouchani ET, Hurd TR, Menger KE, Hampton MB, Murphy MP (2010) Measuring mitochondrial protein thiol redox state. Methods Enzymol 474:123–147. https://doi.org/10.1016/S0076-6879(10)74008-8
Reznick AZ, Packer L (1994) Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol 233:357–363
Rodrigues DGB, de Moura Coelho D, Sitta Â, CED J, Hauschild T, Manfredini V, Bakkali A, Struys EA, Jakobs C, Wajner M, Vargas CR (2017) Experimental evidence of oxidative stress in patients with l-2-hydroxyglutaric aciduria and that l-carnitine attenuates in vitro DNA damage caused by d-2-hydroxyglutaric and l-2-hydroxyglutaric acids. Toxicol in Vitro 42:47–53. https://doi.org/10.1016/j.tiv.2017.04.006
Rzem R, Van Schaftingen E, Veiga-da-Cunha M (2006) The gene mutated in l-2-hydroxyglutaric aciduria encodes l-2-hydroxyglutarate dehydrogenase. Biochimie 88(1):113–116. https://doi.org/10.1016/j.biochi.2005.06.005
Sauer SW, Okun JG, Fricker G, Mahringer A, Müller I, Crnic LR, Mühlhausen C, Hoffmann GF, Hörster F, Goodman SI, Harding CO, Koeller DM, Kölker S (2006) Intracerebral accumulation of glutaric and 3-hydroxyglutaric acids secondary to limited flux across the blood-brain barrier constitute a biochemical risk factor for neurodegeneration in glutaryl-CoA dehydrogenase deficiency. J Neurochem 97(3):899–910. https://doi.org/10.1111/j.1471-4159.2006.03813.x
Sauer SW, Opp S, Mahringer A, Kamiński MM, Thiel C, Okun JG, Fricker G, Morath MA, Kölker S (2010) Glutaric aciduria type I and methylmalonic aciduria: simulation of cerebral import and export of accumulating neurotoxic dicarboxylic acids in in vitro models of the blood-brain barrier and the choroid plexus. Biochim Biophys Acta 1802(6):552–560. https://doi.org/10.1016/j.bbadis.2010.03.003
Saxena G, Bharti S, Kamat PK, Sharma S, Nath C (2010) Melatonin alleviates memory deficits and neuronal degeneration induced by intracerebroventricular administration of streptozotocin in rats. Pharmacol Biochem Behav 94(3):397–403. https://doi.org/10.1016/j.pbb.2009.09.022
Seminotti B, Amaral AU, Ribeiro RT, MDN R, Colín-González AL, Leipnitz G, Santamaría A, Wajner M (2016) Oxidative stress, disrupted energy metabolism, and altered signaling pathways in glutaryl-CoA dehydrogenase knockout mice: potential implications of quinolinic acid toxicity in the neuropathology of glutaric acidemia type I. Mol Neurobiol 53(9):6459–6475. https://doi.org/10.1007/s12035-015-9548-9
Steenweg ME, Salomons GS, Yapici Z, Uziel G, Scalais E, Zafeiriou DI, Ruiz-Falco ML, Mejaški-Bošnjak V, Augoustides-Savvopoulou P, Wajner M, Walter J, Verhoeven-Duif NM, Struys EA, Jakobs C, van der Knaap MS (2009) L-2-Hydroxyglutaric aciduria: pattern of MR imaging abnormalities in 56 patients. Radiology 251(3):856–865. https://doi.org/10.1148/radiol.2513080647
Steenweg ME, Jakobs C, Errami A, van Dooren SJM, Adeva Bartolomé MT, Aerssens P, Augoustides-Savvapoulou P, Baric I, Baumann M, Bonafé L, Chabrol B, JTR C, Clayton P, Coker M, Cooper S, Falik-Zaccai T, Gorman M, Hahn A, Hasanoglu A, King MD, de Klerk HBC, Korman SH, Lee C, Meldgaard Lund A, Mejaški-Bošnjak V, Pascual-Castroviejo I, Raadhyaksha A, Rootwelt T, Roubertie A, Ruiz-Falco ML, Scalais E, Schimmel U, Seijo-Martinez M, Suri M, Sykut-Cegielska J, Trefz FK, Uziel G, Valayannopoulos V, Vianey-Saban C, Vlaho S, Vodopiutz J, Wajner M, Walter J, Walter-Derbort C, Yapici Z, Zafeiriou DI, Spreeuwenberg MD, Celli J, den Dunnen JT, van der Knaap MS, Salomons GS (2010) An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: a genotype-phenotype study. Hum Mutat 31(4):380–390. https://doi.org/10.1002/humu.21197
Topcu M et al (2004) L-2-Hydroxyglutaric aciduria: identification of a mutant gene C14orf160, localized on chromosome 14q22.1. Hum Mol Genet 13(22):2803–2811. https://doi.org/10.1093/hmg/ddh300
Topcu M et al (2005) L-2-hydroxyglutaric aciduria: a report of 29 patients. Turk J Pediatr 47(1):1–7
Wajner M, Goodman SI (2011) Disruption of mitochondrial homeostasis in organic acidurias: insights from human and animal studies. J Bioenerg Biomembr 43(1):31–38. https://doi.org/10.1007/s10863-011-9324-0
Wendel A (1981) Glutathione peroxidase. Methods Enzymol 77:325–333. https://doi.org/10.1016/S0076-6879(81)77046-0
Wilt SG, Dugger NV, Hitt ND, Hoffman PM (2000) Evidence for oxidative damage in a murine leukemia virus-induced neurodegeneration. J Neurosci Res 62(3):440–450. https://doi.org/10.1002/1097-4547(20001101)62:3<440::AID-JNR14>3.0.CO;2-M
Yagi K (1998) Simple procedure for specific assay of lipid hydroperoxides in serum or plasma. Methods Mol Biol 108:107–110. https://doi.org/10.1385/0-89603-472-0:107
Yilmaz K (2009) Riboflavin treatment in a case with l-2-hydroxyglutaric aciduria. Eur J Paediatr Neurol 13(1):57–60. https://doi.org/10.1016/j.ejpn.2008.01.003
ᅟ
Funding
This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico #404883/2013-3, Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul #2266-2551/14-2, Pró-Reitoria de Pesquisa/Universidade Federal do Rio Grande do Sul #PIBIC 27613, FIPE/HCPA, and Financiadora de Estudos e Projetos/Rede Instituto Brasileiro de Neurociência #01.06.0842-00.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Approval
The experimental protocol was approved by the Ethics Committee for Animal Research of the UFRGS, Porto Alegre, Brazil, and followed the “National Institutes of Health Guide for the Care and Use of Laboratory Animals” (NIH Publications No. 85–23, revised 2011).
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ribeiro, R.T., Zanatta, Â., Amaral, A.U. et al. Experimental Evidence that In Vivo Intracerebral Administration of L-2-Hydroxyglutaric Acid to Neonatal Rats Provokes Disruption of Redox Status and Histopathological Abnormalities in the Brain. Neurotox Res 33, 681–692 (2018). https://doi.org/10.1007/s12640-018-9874-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-018-9874-6