
Abstract
Sequential endoproteolytic cleavages operated by the γ-secretase and the β-secretase (BACE1) on the β-amyloid precursor protein result in the production of the β-amyloid (Aβ) species, with two C-terminal variants, at residue 40 or at residue 42. Accumulation in brain tissue of aggregates of Aβ42 is the major pathogenetic event in Alzheimer’s disease (AD). The causes of Aβ accumulation in the common sporadic form of AD are not completely understood, but they are likely to include oxidative stress (OS). Data reviewed here shed light on how Aβ generation, oxidative stress, and secretase functions are intimately related in sporadic AD. According to our hypothesis, in sporadic AD, OS resulted from several cellular insults such as aging, hypoxia, hyperglycemia, and hypercholesterolemia—that are well-known risk factors for AD development—can determine a primary induction of γ-secretase and BACE1. The loop proceeds with the generation of Aβ42 and its signaling to BACE1 transcription.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common age-related disease (Bachman et al. 1993); therefore, the risk of AD increases dramatically in individuals above the age of 70 and it is predicted that the incidence of the disease could increase a further 3-fold over the next 50 years. AD can be classified in two forms: sporadic AD, which accounts for the majority of cases, and a rare familial early onset form (FAD), in which gene mutations have been observed.
The β-amyloid peptide (Aβ) is generated following the sequential cleavage of its precursor, the β-amyloid precursor protein (AβPP). AβPP is an integral membrane protein with a single membrane spanning domain, a large, extracellular, N-terminus and a shorter, cytoplasmic, C-terminus. The amyloidogenic processing of AβPP involves two sequential cleavages operated by the β- and the γ-secretases at the N-and C-termini of Aβ sequence, respectively. The β-secretase (BACE1) cleaves AβPP at the beginning of the Aβ sequence, generating an extracellular soluble fragment, called sβAPP, and an intracellular C-terminal named C99. The last fragment is further cleaved by the γ-secretase that produces Aβ peptides of different lengths, these being predominantly Aβ40 and Aβ42.
The central role of Aβ in the pathogenesis of AD is supported by several lines of evidence. Aggregates of Aβ are neurotoxic and initiate a series of events, including the hyperphosphorylation of tau, which results in neuronal cell dysfunction and death (Yankner 1996); moreover, Aβ soluble oligomers have been found to alter memory function in different murine AD models (Dodart et al. 2002; Lesné et al. 2006). All genes bearing mutations that cause FAD, APP, and presenilins 1 and 2, which represent the catalytic core of the γ-secretase, mediate the accumulation of Aβ42, increasing its production and accumulation (Citron et al. 1992; Citron et al. 1997; Lemere et al.1996).
One of the most well-known and studied effects of Aβ is its capacity to induce, and be induced by, oxidative stress (OS); thus, Aβ induces OS in vivo and in vitro (Hensley et al. 1994; Mark et al. 1997; Murakami et al. 2005; Tabner et al. 2005) and OS is itself able to induce the increased production of Aβ (Paola et al. 2000; Tamagno et al. 2002; Tong et al. 2005).
The cause of modified AβPP processing and Aβ42 accumulation in sporadic AD is unclear but is likely to include OS.
OS increase is believed to be an early event in AD (Nunomura et al. 2001; Cutler et al. 2004) as also confirmed by the extensive oxidative damage observed in mild cognitive impairment (MCI), considered a transition stage between normal aging and dementia (Lovell and Markesbery 2001).
Moreover, the identification of oxidatively membrane damage, cytoskeleton derangement and, consequently, cell death (Perry et al. 2000) suggest that OS plays an important role in AD pathogenesis.
Our studies strengthen the hypothesis that OS may also be a basic common pathway of Aβ accumulation, common to different AD risk factors, such as aging, hypoxia, hyperglycemia, and hypercholesterolemia.
Oxidative Stress in AD
Of note, the major non-genetic risk factor involved in the pathogenesis of sporadic late onset AD is aging, which is strictly related to the damage resulting from reactive oxygen species (ROS) indicative of OS (Markesbery and Carney 1999).
ROS are a by-products of cellular metabolism and are generated during mitochondrial oxidative phosphorylation as molecules with unpaired electrons such as superoxide (O2 −).
The brain is particularly vulnerable to OS because of its high consumption of oxygen, high levels of polyunsaturated fatty acids, and relatively low levels of antioxidants (Floyd and Hensley 2002). Accumulation of oxidative damage in the brain is particularly dangerous since it is a post-mitotic tissue with neurons exhibiting only a weak self-renewal potential due to their low proliferative capacity.
An increased oxidative burden has been observed in the brain of non-demented elderly and of sporadic AD patients (Behl and Moosmann 2002; Moosmann and Behl 2002).
The free radical hypothesis of aging implies the accumulation of ROS resulting in the damage of the major cell components: nucleus, mitochondrial DNA, membranes, and cytoplasmic proteins (Harman 1992).
Membrane lipids are commonly attacked by ROS, and lipid peroxidation is the most frequently oxidative marker that appears to increase during aging (Zhu et al. 2006). The oxidative modification of membrane fatty acids leads to a structural damage and to the generation of aldehydic end products, such as 4-hydroxynonenal (HNE), which have a high oxidative potential themselves and can impair cellular functions (Keller and Mattson 1998).
Post-mortem analysis of the brains of AD subjects found increased levels of lipid peroxidation in brain regions that are affected by an early neurodegeneration (Mielke and Lyketsos 2006).
Several studies show that also protein oxidation increases with the aging of the brain (Abd El Mohsen et al. 2005) as well as the levels of oxidized proteins in AD patients and this increase strongly correlate with cognitive performance (Keller et al. 2005).
Another well-known age-dependent modification mediated by OS is the DNA oxidative damage. It is well known that mutations in mitochondrial DNA cause a respiratory chain dysfunction that leads to an increase in ROS production. These mitochondrial mutations accumulate during brain aging and neurodegenerative disorders (Corral-Debrinski et al. 1992; Wang et al. 2005).
OS increases with age not only through variations in ROS generation but also in their elimination (Barja 2004). Indirect evidence of cellular OS is the increased expression of molecules involved in oxidant defense, such as heme oxygenase, superoxide dismutases, glutathione transferases, and catalase (Markesbery and Carney 1999); then these neurons do not necessarily succumb to OS, but dynamically regulate their defense mechanisms in response to oxidants.
In the case of neurodegenerative disorders where OS is postulated to play a role, the normal balance between production of and defense against OS has been modified and OS impairs the cellular defences causing a vicious cycle.
These challenges become clear during the AD process, and include a strong presence of increased sulfhydryls, induction of heme oxygenase-1 and increased expression of Cu/Zn superoxide dismutase (Nunomura et al. 1999; Smith et al. 1995; Pappolla et al. 1992), which indicates loss of homeostasis. Moreover, the protein oxidation that increases during aging is strictly associated with the decreased capacity of antioxidant defence machinery (Rodrigues Siqueira et al. 2005).
OS may be an early event in the progression from normal aging to AD pathology. Thus, extensive oxidative damage it has been shown in MCI, a transition stage between normal aging and dementia.
It has been shown that in MCI subjects, plasma levels of non-enzymatic antioxidants and activity of antioxidant enzymes appeared to be decreased if compared to those of normal aging subjects (Guidi et al. 2006; Sultana et al. 2009). Moreover, levels of oxidative markers appeared to be increased.
Proteomic analysis demonstrated a large number of protein-bound HNE in MCI brain (Song et al. 2009). F2-isoprostanes (F2-IsoPs) levels and neuroprostanes were also significantly increased in MCI patients and in late-stage AD (Markesbery et al. 2005). Moreover, in brains from patients with MCI, acrolein has been found to be elevated in hippocampus and temporal cortex where OS is high. Due to its high reactivity, acrolein is not only a marker of lipid peroxidation but also an initiator of OS by adducting cellular nucleophilic groups found in proteins, lipids, and nucleic acids (Singh et al. 2010). Thus, extensive oxidative damage observed in MCI brain regions suggests that OS may be an early event in progression from normal aging to AD.
Based on these notions, it seems likely that increased production of ROS may act as important mediators of synaptic loss and eventually promote neurofibrillary tangles and senile plaques formation (Kern and Behl 2009).
In summary, the oxidative burden observed in healthy brain aging and in early stages of dementia confirms that the accumulation of oxidatively modified biomolecules is a general hallmark of brain aging and could be an early event in the progression of AD.
Oxidative Stress and Major AD Risk Factors
Our studies, as described below, strengthen the hypothesis that OS may be considered a basic common pathway of AD progression, common to most important AD risk factors.
Hypoxia
It is well known that patients with stroke and cerebral infarction are at risk of AD (Rocchi et al. 2009).
Recent studies have shown that a history of stroke can increase AD prevalence by approximately 2-fold in elderly patients (Schneider et al. 2003; Vermeer et al. 2003). The risk is higher when stroke is concomitant with atherosclerotic vascular risk factor (Jellinger 2002).
Recently it has been proposed that hypoxia can alter APP processing, increasing the activity of the β- and the γ-secretases, resulting in the acceleration of Aβ production and plaques formation in vivo and in vitro (Sun et al. 2006; Zhang et al. 2007).
We recently showed, both in vivo and in vitro, that hypoxia up-regulates BACE1 expression in a biphasic manner, through two distinct mechanisms: (a) an early release of ROS from mitochondria and (b) a late activation of HIF-1α, a molecule that regulates oxygen homeostasis (Guglielmotto et al. 2009). The data suggest that the early post-hypoxic up-regulation of BACE1 depends on the generation of ROS mediated by sudden interruption of the mitochondrial electron transport chain. Although it is generally accepted that intracellular ROS levels change during hypoxia, the direction in which this change occurs is still debated. Many Authors reported that the level of intracellular ROS increases under hypoxia and suggested that mitochondria are the source of ROS involved in this cellular response (Chandel et al. 1998; Turrens 2003; Klimova and Chandel 2008). It is now accepted that hypoxia increases ROS via the mitochondrial transport chain and specifically by the function of complex III (Bell et al. 2007).
Hyperglycemia
Diabetes mellitus, a metabolic disorder characterized by hyperglycemia, is an important risk factor for AD, and multiple mechanisms connecting the two diseases have been proposed.
Hyperglycemia enhances the formation of advanced glycation end-products (AGEs) senescent protein derivatives that result from the auto-oxidation of glucose and fructose (Bucala and Cerami 1992; Brownlee et al. 1988; Takeuchi and Makita 2001).
The involvement of AGEs in brain aging and AD was reported many years ago in studies showing that the microtubule associated protein tau and Aβ were substrates of glycation (Ledesma et al. 1994; Smith et al. 1994; Vitek et al. 1994; Yan et al. 1994; Yan et al. 1995).
More recently it has been reported that AGEs have other pathological effects at cellular and molecular levels. Among these are the production of ROS, especially superoxide and hydrogen peroxide (Carubelli et al. 1995; Muscat et al. 2007). Moreover, glycated proteins increase the rate of free radical production compared to native proteins (Neeper et al. 1992).
Another mechanism through which AGEs mediate the production of OS is the interaction with RAGE, which is a multiligand receptor of the immunoglobulin superfamily of cell surface molecules (Qin et al. 2008). The role of RAGE in the pathogenesis of AD has been extensively studied because it also binds Aβ (Yan et al. 1996).
We recently demonstrated a novel pathogenic mechanism of AGEs, which contributes to Aβ accumulation (Guglielmotto et al. 2010). In streptozotocin rats, as well as in SK-N-BE differentiated neuroblastoma cells, two different AGEs, pentosidine and glyceraldehyde derived pyridinium (GLAP), were able to up-regulate BACE1 expression through their binding with RAGE which is followed by a strong production of ROS and by an activation of the NF-κB pathway, which is a representative transcription factor activated by RAGE (Granic et al. 2009). Moreover, NF-κB has been recently identified as a molecular intermediate involved in the Aβ-mediated control of BACE1. Thus, the inhibitor of IκB kinase that blocks NF-κB transcriptional activity fully reverses the Aβ42-induced increase of BACE1 promoter transactivation (Buggia-Prevot et al. 2008). These data agree with a previous report showing that BACE1 promoter transactivation could be regulated by NF-κB in neuronal cells (Bourne et al. 2007).
The role of RAGE on BACE1 up-regulation was reported before in RAGE-injected brains of AD animal model and in cultured cells (Cho et al. 2009). Our results confirmed and extended this finding by showing that natural ligands of RAGE, AGEs, mediate BACE1 up-regulation in a diabetic animal model as well as in culture model. Thus, activation of AGEs/RAGE axis, as a result of hyperglycemia, driving the up-regulation of the key enzyme for Aβ production, provides a mechanistic link between diabetes mellitus and AD.
Hypercholesterolemia
Recent studies indicate that alterations in cholesterol metabolism influence some molecular mechanisms involved in AD. Three important lines of evidence have implicated cholesterol in AD; (a) hypercholesterolemia is recognized to be a risk factor for sporadic AD (Puglielli et al. 2003; Panza et al. 2007); (b) epidemiological studies showed that APO-E4 allele is strictly associated with an increased risk of AD (Corder et al. 1993; Evans et al. 2004); (c) feeding cholesterol to rabbits produces some of the pathological hallmarks of AD, including amyloid plaques (Kandiah and Feldman 2009).
Moreover, early epidemiological studies indicated that cholesterol-lowering drugs, belonging to the family of statins, reduce the prevalence of AD (Jick et al. 2000), a conclusion not fully accepted because of other contradictory clinical studies (Kandiah and Feldman 2009; Ishii et al. 2003; Serrano-Pozo et al. 2010).
Thus, if altered cholesterol metabolism in the brain has repeatedly been suggested to be implicated in the pathogenesis of AD, the molecular mechanisms underlying its involvement are still largely unknown.
Cholesterol is essential for normal brain functions; most of it is present in the free form and derives from de novo synthesis by astrocytes, as plasma lipoproteins cannot cross the blood–brain barrier (Puglielli et al. 2003), but cholesterol can only be transformed in oxysterols, cholesterol oxidation products that are important to balance the local synthesis of sterols (Lütjohann et al. 1996). We suggested that oxysterols might represent a link between altered cholesterol metabolism in the brain and AD (Gamba et al. 2011). Thus, we recently demonstrated an enhancement of Aβ binding to neuronal cells exerted by oxysterols relevant in brain physiology such as 27-OH cholesterol, 24-OH cholesterol, and 7β-OH cholesterol. The increase has been related to the strongly oxysterols-mediated up-regulation of CD36 and β1 integrin, a well characterized multireceptor complex, able to stimulate the Aβ binding to neuronal cells.
We have also recently demonstrated an up-regulation of BACE1 mediated by the sterol regulatory element binding protein 2 (SREBP2) transcription factor, that tightly regulates the brain cholesterol metabolism, in rats fed with a high fat diet mimicking cholesterol-rich western diet as well as in differentiated SK-N-BE neuroblastoma cells exposed to high cholesterol levels (Mastrocola et al. 2011).
Since it is well known that BACE1 activity located in cholesterol-rich rafts may be positively modulated by cholesterol levels (Cordy et al. 2003; Wolozin 2004), our data on the direct regulation of BACE1 by SREBP2 transcriptional activity adds new insights on the complex molecular mechanisms underlying the implication of cholesterol in the pathogenesis of AD.
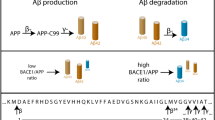
The data here reported strongly supported the hypothesis that OS could be a basic common pathway of Aβ accumulation, as determined by different age-related risk factors (Fig. 1).

Diagram sketching of the hypothesis that OS could be a basic common pathway of Aβ accumulation, as determined by different age-related risk factors
Aβ Production is a Major Link Between Oxidative Stress and Pathogenesis of AD
According to the amyloid cascade hypothesis of AD, Aβ is considered to be the primary motor of neuronal degeneration, although the pathway leading to neuronal death is extremely complicated (Hardy and Allsop 1991).
It is still controversial whether the most deleterious form of Aβ peptides in the early stage of AD is represented by the fibrillar or the soluble oligomeric peptide form (Drouet et al. 2000).
Although neuronal degeneration occurs in proximity of the amyloid plaques, some studies have suggested that intermediate Aβ aggregates such as protofibrils or simple oligomers are also involved in AD pathogenesis and even appear to be the most dangerous species (Gong et al. 2003; Kayed et al. 2003; Resende et al. 2008; Tamagno et al. 2006). These data may help to explain, for example, why neurodegeneration and specific spatial learning deficits may occur in AD animal models before the appearance of amyloid plaques (Chui et al. 1999; Koistinaho et al. 2001).
More attention has thus been focused on the early stages of amyloid production and on its maturation from small soluble molecules to oligomers and fibrils with high molecular weight.
One of the most known and studied effects of Aβ is its ability to induce, and be induced by, OS. Several by-products of protein, lipid, and glucose oxidation seem to be elevated in the brain of patients with AD and to a lesser extent in the brains of healthy aged controls, as the burden of free radicals builds up proportionally to the duration of the disease (Borghi et al. 2007; Butterfield et al. 2001; Markesbery and Lovell 1998; Sayre et al. 1997). Both amyloid deposits and soluble Aβ seem to drive the accumulation of ROS (Behl 2005; Praticò 2008).
Transition metals, Cu(II), Zn(II), and mainly Fe(III) favor the neurotoxicity of Aβ, through their reduction, that yields hydrogen peroxide (H2O2) (Huang et al. 1999). It has been shown that Aβ residue Tyr-10 is a pivotal residue in driving the catalytic production of H2O2 in the presence of Cu(II). The phenoxy radical of this residue produced by the reaction with ROS causes neurotoxicity and acceleration in Aβ peptides aggregation (Barnham et al. 2004).
Another crucial residue of Aβ is Met-35, thus the substitution of Met with cysteine resulted in no protein oxidation in C. Elegans model (Yatin et al. 1999). An interesting downstream effect of Aβ-induced OS on lipids is the interference with membrane stability and the generation of calcium flow into the cells, leading to enhanced toxicity (Kirkitadze and Kowalska 2005). Moreover, lipid peroxidation induced by Aβ peptides impairs the function of ion-motive ATPase, glucose, and glutamate transporters and of GTP- binding protein, as the results of their covalent modification by aldehydic end products such as HNE.
Aβ is also able to induce oxidative modifications of proteins involved in cellular defence mechanisms against noxious stimuli as well as in energy pathways. In a murine experimental knock-in model of AD, harboring APP and PS1 mutations, a direct correlation between the excessive production of Aβ peptides and impairment of antioxidant enzymes, with consequent mitochondrial dysfunction, was reported (Anantharaman et al. 2006). These are further demonstrations of how increased OS induced by Aβ can lead to increased oxidative modifications of proteins and lipids leading to impaired cellular functions and cell death, and consequently cognitive impairment and AD pathology (Sultana et al. 2009).
Furthermore, Aβ can strike the production of pro-inflammatory mediators, such as TNF-α or IL-1β, leading to microglia activation, OS induction, and enhanced processing of APP to generate more Aβ (Akiyama et al. 2000; Atwood et al. 2003). In this connection, of particular interest is the report that APP promoter responds positively to inflammatory mediators; the inflammatory response leads to enhanced APP and Aβ productions and the involvement of OS is suggested by the finding that the iron chelation is able to reverse the phenomenon (Rogers et al. 2002).
OS may also be the cause of Aβ accumulation. Oxidant agents and oxidative products increase APP expression (Cheng and Trombetta 2004; Patil et al. 2006) and intracellular and secreted Aβ levels in many neuronal and non-neuronal cellular types (Frederikse et al. 1996; Misonou et al. 2000; Atwood et al. 2003; Murray et al. 2007).
We have extensively analyzed the transcriptional activation of BACE1, the key enzyme for Aβ production, first in response to OS, both in vivo and in vitro.
BACE1 gene promoter has a complex structure, carrying several transcription factor binding sites, such as for SP1, AP1, AP2, CREB, glucocorticoid receptor, NF-κB, and others (Sambamurti et al. 2004).
Different signaling pathways, such as SP1 (Christensen et al. 2004), JNK/AP1 (Tamagno et al. 2008), NF-κB (Buggia-Prevot et al. 2008), and p25/cdk5/STAT3 (Wen et al. 2008) have been suggested to control BACE1 transcription.
We and others have shown that the expression and activity of BACE1 is increased by oxidant agents (Tamagno et al. 2002; Tamagno et al. 2003; Tamagno et al. 2005; Kao et al. 2004; Tong et al. 2005). Several reports show that levels of BACE1 protein and activity are increased in brain of sporadic AD patients, compared to normal age controls (Fukumoto et al. 2002; Holsinger et al. 2002; Yang et al. 2003; Zhao et al. 2007); moreover, there is a significant correlation of BACE1 activity and oxidative markers in sporadic AD tissues (Borghi et al. 2007).
We have proposed a sequence of events that link OS, BACE1 induction, and apoptotic cell death mediated by an overproduction of Aβ. First, we have shown that oxidant agents and HNE significantly increase the expression, protein levels, and activity of BACE1 in NT2 neurons (Tamagno et al. 2002; Tamagno et al. 2003). These events are followed by both an overproduction of Aβ peptides and morphological signs of apoptotic cell death (Tamagno et al. 2005).
Then we have found that OS increases the γ-secretase activity in cultured cells and in vivo, and that the increased expression of BACE1 induced by OS is regulated by the γ-secretase activity (Tamagno et al. 2008). These results have important implications for the pathogenesis of sporadic AD. First, they suggest that OS, secondary to different AD risk factors, can increase the expression of both secretases, thereby enhancing Aβ production. Second, our data revealed the existence of a positive feedback loop in which γ-secretase activity results in up-regulation of BACE1 expression. Previously Minopoli et al. (2007) had shown a correlation between the induction of OS and the increase of γ-secretase cleavage on APP and then Jo et al. (2010) confirmed the finding that γ-secretase up-regulates BACE1 expression.
An increasing body of evidence implicates brain inflammation related to OS in the up-regulation of BACE1. Thus, the BACE 1 promoter has also a binding site for the transcriptional regulator proliferator-activated receptor γ (PPAR γ) (Sastre et al. 2006). Activation of PPARγ by non-steroidal anti-inflammatory drugs (NSAIDs) or PPARγ agonists cause repression of BACE1 gene promoter activity, while pro-inflammatory cytokines that reduces PPARγ levels lead to increased BACE1 mRNA (Sastre et al. 2006).
Several different mechanisms appear to be involved in the OS-mediated BACE1 up-regulation, and probably they overlap to promote the activation observed.
Our data strongly suggest that OS contributes to the pathogenesis of the common, sporadic, late-onset form of AD, and that, being able to up-regulate γ-secretase and BACE1, can be the molecular link between the two secretases.
These findings led us to think that either Aβ peptides or the APP intracellular domain (AICD) could be the APP derivatives from the γ-secretase cleavage responsible for BACE1 induction.
We investigated which derivative was responsible for BACE1 up-regulation. We first analyzed the effect of the AICD fragments 57 ad 59, the APP derivatives resulting from the γ-secretase cleavage of APP, as well as AICD 50 and 51 that are the ε cleavage derivatives (Passer et al. 2000; Tagami et al. 2008; Fukumori et al. 2006).
Transfection of different cell lines with the corresponding AICD constructs determined no change in BACE1 expression, thus our experiments ruled out the role of AICD in the over-expression of BACE1 and pointed to Aβ peptides (Giliberto et al. 2009). Thus, it is well known that mutant PS1 determines a drastic increase of Aβ42 species (Duff et al. 1996) and familial AD cases with PS1 mutations have mostly an increased ratio of Aβ42/40. We found that over-expression of PS1 mutants could alone determine an increase of BACE1 expression (Giliberto et al. 2009).
Instead, treating neuronal and neuroblastoma cells with 1 μM soluble Aβ42 increased BACE1 transcription, which was reverted if anti Aβ42 antibody was added to culture medium (Giliberto et al. 2009). Of note, Aβ40 has much less effect on BACE1 expression (Guglielmotto et al. 2011).
These findings reveal a novel function of Aβ peptides showing that Aβ42 is the player of a positive feedback loop from the γ-secretase cleavage on the BACE1 cleavage of APP (Fig. 2).

Pathogenetic hypothesis in which OS fosters the expression and activity of BACE1 through the release of Aβ42 peptides and the activation of JNK/AP-1 pathway
It is not clear how Aβ42 could reach BACE1 transcription apparatus. There are many signaling pathways that seem to be regulated or at least affected by Aβ peptides. Among these Buggia-Prevot et al. (2008) found that Aβ42 was able to modulate BACE1 promoter transactivation and activity through an NF-κB-dependent pathway. Thus, an IkappaB kinase inhibitor prevents Aβ42-induced BACE1 promoter transactivation suggesting that NFkappaB could mediate this Aβ42-associated phenotype (Buggia-Prevot et al. 2008).
We recently found that also JNK/cjun pathway activation may be involved in the BACE1 up-regulation mediated by Aβ42 (Guglielmotto et al. 2011).
This pathway links all the pathological hallmarks of AD. JNK activation has been reported to regulate the phosphorylation of APP, leading to modulation of Aβ levels (Colombo et al. 2007; Colombo et al. 2009), as well as to mediate the phosphorylation of tau protein in vitro (Yoshida et al. 2004). Furthermore, the JNK pathway is shown activated in preclinical models of AD, including Tg2576 and Tg575/PS1P264L transgenic mice (Flood et al. 2002; Puig et al. 2004), as well as in brains of AD patients (Zhu et al. 2003a; Zhu et al. 2003b; Lagalwar et al. 2006).
Indeed, we had previously found a significant activation of JNK/AP1 pathway by OS both in vivo and in vitro models, and the up-regulation of BACE1 was abolished when the pathway had been genetically or pharmacologically inhibited (Tamagno et al. 2008).
It remains to be determined how Aβ42 activates JNK pathway. Aβ is known to alter intracellular calcium hosmeostasis (Demuro et al. 2010), and JNK could be activated by the calcium/calmodulin dependent protein kinase II (CAMKII) (Wu et al. 2009) or by a PI3K inducing signal, mediated by calcium release (Assefa et al. 1999). Furthermore, it has been observed that Aβ-induced increase in intracellular calcium concentration stimulates BACE1 expression, resulting in accelerated Aβ generation, and that this process is mediated by the calcineurin-NFAT1 signaling pathway. NFAT1 is normally dephosphorylated by calcium-dependent manner by calcineurin, while it is phosphorylated and inactivated by JNK (Cho et al. 2008). Results of Ortega-Perez et al. (2005), however, demonstrated that, unlike other NFAT members, the transcriptional activity of NFATc2 appears up-regulated by JNK. Based on these notions, JNK-mediated phosphorylation of NFATs could play a differential physiological role among NFAT family members.
Cell surface receptors may be involved as well. Numerous proteins have been described to interact with Aβ directly or indirectly, such as AβPP itself, TrkA, p75NTR, some G proteins, NMDA, and AMPA receptors and many more. Thereby, the interaction of Aβ42 with multiple receptors is likely to produce the activation of the JNK/c-jun pathway. AβPP has been shown to function as a cell surface receptor that mediates neuronal cell death through the activation of JNK pathway (Hashimoto et al. 2003). Moreover, JNK pathway is shown involved in Aβ-driven signaling, as downstream of Aβ-RAGE interaction (Yan et al. 1996), TrKA/p75NTR (Yaar et al. 2007; Costantini et al. 2005), NMDA, and AMPA receptors (Di et al. 2010; Vieira et al. 2010). In general, the JNK signaling seems activated in AD (Lagalwar et al. 2006) and strongly correlates with the OS in AD models (Tamagno et al. 2005).
Also, besides being a means of scavenging Aβ from tissues and having a role in modulating APP endocytic trafficking, LRP family receptors and apolipoprotein E, of which the ε4 allele has a strong linkage with AD, could represent a way for Aβ to enter into neuronal cells (Bu et al. 2006; Jaeger and Pietrzik 2008) and interact with other still unidentified molecules to strike the signaling pathway that leads to BACE1 regulation.
Thus, the effects of Aβ on the cell are extremely complex. Being able to understand the specific signaling and to understand how soluble or insoluble Aβ induces its own production by up-regulating BACE1 expression would lead to new tools to interrupt the vicious circle, with potential therapeutics consequences.
References
Abd El Mohsen MM, Iravani MM, Spencer JP, Rose S, Fahim AT, Motawi TM, Ismail NA, Jenner P (2005) Age-associated changes in protein oxidation and proteasome activities in rat brain: modulation by antioxidants. Biochem Biophys Res Commun 336:386–391
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421
Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK (2006) Beta-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am J Pathol 168:1608–1618
Assefa Z, Valius M, Vántus T, Agostinis P, Merlevede W, Vandenheede JR (1999) JNK/SAPK activation by platelet-derived growth factor in A431 cells requires both the phospholipase C-gamma and the phosphatidylinositol 3-kinase signaling pathways of the receptor. Biochem Biophys Res Commun 261:641–645
Atwood CS, Perry G, Smith MA (2003) Cerebral hemorrhage and amyloid-beta. Science 299:1014
Bachman DL, Wolf PA, Linn RT, Knoefel JE, Cobb JL, Belanger AJ, White LR, D’Agostino RB (1993) Incidence of dementia and probable Alzheimer’s disease in a general population: the Framingham Study. Neurology 43:515–519
Barja G (2004) Free radicals and aging. Trends Neurosci 27:595–600
Barnham KJ, Haeffner F, Ciccotosto GD, Curtain CC, Tew D, Mavros C, Beyreuther K, Carrington D, Masters CL, Cherny RA, Cappai R, Bush AI (2004) Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease beta-amyloid. FASEB J 18:1427–1429
Behl C (2005) Oxidative stress in Alzheimer’s disease: implications for prevention and therapy. Subcell Biochem 38:65–78
Behl C, Moosmann B (2002) Antioxidant neuroprotection in Alzheimer’s disease as preventive and therapeutic approach. Free Radic Biol Med 33:182–191
Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS (2007) The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177:1029–1036
Borghi R, Patriarca S, Traverso N, Piccini A, Storace D, Garuti A, Cirmena G, Odetti P, Tabaton M (2007) The increased activity of BACE1 correlates with oxidative stress in Alzheimer’s disease. Neurobiol Aging 28:1009–1014
Bourne KZ, Ferrari DC, Lange-Dohna C, Rossner S, Wood TG, Perez-Polo JR (2007) Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J Neurosci Res 85:1194–1204
Brownlee M, Cerami A, Vlassara H (1988) Advanced products of nonenzymatic glycosylation and the pathogenesis of diabetic vascular disease. Diabetes Metab Rev 4:437–451
Bu G, Cam J, Zerbinatti C (2006) LRP in amyloid-beta production and metabolism. Ann N Y Acad Sci 1086:35–53
Bucala R, Cerami A (1992) Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv Pharmacol 23:1–34
Buggia-Prevot V, Sevalle J, Rossner S, Checler F (2008) NFkappaB-dependent control of BACE1 promoter transactivation by Abeta42. J Biol Chem 283:10037–10047
Butterfield DA, Drake J, Pocernich C, Castegna A (2001) Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med 7:548–554
Carubelli R, Schneider JE Jr, Pye QN, Floyd RA (1995) Cytotoxic effects of autooxidative glycation. Free Radic Biol Med 18:265–269
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 95:11715–11720
Cheng SY, Trombetta LD (2004) The induction of amyloid precursor protein and alpha-synuclein in rat hippocampal astrocytes by diethyldithiocarbamate and copper with or without glutathione. Toxicol Lett 146:139–149
Cho HJ, Jin SM, Youn HD, Huh K, Mook-Jung I (2008) Disrupted intracellular calcium regulates BACE1 gene expression via nuclear factor of activated T cells 1 (NFAT 1) signaling. Aging Cell 7:137–147
Cho HJ, Son SM, Jin SM, Hong HS, Shin DH, Kim SJ, Huh K, Mook-Jung I (2009) RAGE regulates BACE1 and Abeta generation via NFAT1 activation in Alzheimer’s disease animal model. FASEB J 23:2639–2649
Christensen MA, Zhou W, Qing H, Lehman A, Philipsen S, Song W (2004) Transcriptional regulation of BACE1, the beta-amyloid precursor protein beta-secretase, by Sp1. Mol Cell Biol 24:865–874
Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T (1999) Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med 5:560–564
Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ (1992) Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360:672–674
Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ (1997) Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med 3:67–72
Colombo A, Repici M, Pesaresi M, Santambrogio S, Forloni G, Borsello T (2007) The TAT-JNK inhibitor peptide interferes with beta amyloid protein stability. Cell Death Differ 14:1845–1848
Colombo A, Bastone A, Ploia C, Sclip A, Salmona M, Forloni G, Borsello T (2009) JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol Dis 33:518–525
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923
Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ (2003) Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci USA 100:11735–11740
Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC (1992) Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet 2:324–329
Costantini C, Della-Bianca V, Formaggio E, Chiamulera C, Montresor A, Rossi F (2005) The expression of p75 neurotrophin receptor protects against the neurotoxicity of soluble oligomers of beta-amyloid. Exp Cell Res 311:126–134
Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA 101:2070–2075
Demuro A, Parker I, Stutzmann GE (2010) Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem 285:12463–12468
Di X, Yan J, Zhao Y, Zhang J, Shi Z, Chang Y, Zhao B (2010) L-theanine protects the APP (Swedish mutation) transgenic SH-SY5Y cell against glutamate-induced excitotoxicity via inhibition of the NMDA receptor pathway. Neuroscience 168:778–786
Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci 5:452–457
Drouet B, Pinçon-Raymond M, Chambaz J, Pillot T (2000) Molecular basis of Alzheimer’s disease. Cell Mol Life Sci 57:705–715
Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S (1996) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383:710–713
Evans RM, Hui S, Perkins A, Lahiri DK, Poirier J, Farlow MR (2004) Cholesterol and APOE genotype interact to influence Alzheimer disease progression. Neurology 62:1869–1871
Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW (2002) FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. Neurobiol Aging 23:335–348
Floyd RA, Hensley K (2002) Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging 23:795–807
Frederikse PH, Garland D, Zigler JS Jr, Piatigorsky J (1996) Oxidative stress increases production of beta-amyloid precursor protein and beta-amyloid (Abeta) in mammalian lenses, and Abeta has toxic effects on lens epithelial cells. J Biol Chem 271:10169–10174
Fukumori A, Okochi M, Tagami S, Jiang J, Itoh N, Nakayama T, Yanagida K, Ishizuka-Katsura Y, Morihara T, Kamino K, Tanaka T, Kudo T, Tanii H, Ikuta A, Haass C, Takeda M (2006) Presenilin-dependent gamma-secretase on plasma membrane and endosome is functionally distinct. Biochemistry 45:4907–4914
Fukumoto H, Cheung BS, Hyman BT, Irizarry MC (2002) Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol 59:1381–1389
Gamba P, Leonarduzzi G, Tamagno E, Guglielmotto M, Testa G, Sottero B, Gargiulo S, Biasi F, Mauro A, Viña J, Poli G (2011) Interaction between 24-hydroxycholesterol, oxidative stress, and amyloid-β in amplifying neuronal damage in Alzheimer’s disease: three partners in crime. Aging Cell 10:403–417
Giliberto L, Borghi R, Piccini A, Mangerini R, Sorbi S, Cirmena G, Garuti A, Ghetti B, Tagliavini F, Mughal MR, Mattson MP, Zhu X, Wang X, Guglielmotto M, Tamagno E, Tabaton M (2009) Mutant presenilin 1 increases the expression and activity of BACE1. J Biol Chem 284:9027–9038
Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL (2003) Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA 100:10417–10422
Granic I, Dolga AM, Nijholt IM, van Dijk G, Eisel UL (2009) Inflammation and NF-kappaB in Alzheimer’s disease and diabetes. J Alzheimers Dis 16:809–821
Guglielmotto M, Aragno M, Autelli R, Giliberto L, Novo E, Colombatto S, Danni O, Parola M, Smith MA, Perry G, Tamagno E, Tabaton M (2009) The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J Neurochem 108:1045–1056
Guglielmotto M, Aragno M, Tamagno E, Vercellinatto I, Visentin S, Medana C, Catalano MG, Smith MA, Perry G, Danni O, Boccuzzi G, Tabaton M (2010) AGEs/RAGE complex upregulates BACE1 via NF-kappaB pathway activation. Neurobiol Aging [Epub ahead of print]
Guglielmotto M, Monteleone D, Giliberto L, Fornaro M, Borghi R, Tamagno E,Tabaton M (2011) Amyloid-β42 Activates the Expression of BACE1 Through the JNK Pathway. J Alzheimers Dis [Epub ahead of print]
Guidi I, Galimberti D, Lonati S, Novembrino C, Bamonti F, Tiriticco M, Fenoglio C, Venturelli E, Baron P, Bresolin N, Scarpini E (2006) Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 27:262–269
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12:383–388
Harman D (1992) Free radical theory of aging. Mutat Res 275:257–266
Hashimoto Y, Tsuji O, Niikura T, Yamagishi Y, Ishizaka M, Kawasumi M, Chiba T, Kanekura K, Yamada M, Tsukamoto E, Kouyama K, Terashita K, Aiso S, Lin A, Nishimoto I (2003) Involvement of c-Jun N-terminal kinase in amyloid precursor protein-mediated neuronal cell death. J Neurochem 84:864–877
Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA (1994) A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA 91:3270–3274
Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G (2002) Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann Neurol 51:783–786
Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI (1999) Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem 274:37111–37116
Ishii K, Tokuda T, Matsushima T, Miya F, Shoji S, Ikeda S, Tamaoka A (2003) Pravastatin at 10 mg/day does not decrease plasma levels of either amyloid-beta (Abeta) 40 or Abeta 42 in humans. Neurosci Lett 350:161–164
Jaeger S, Pietrzik CU (2008) Functional role of lipoprotein receptors in Alzheimer’s disease. Curr Alzheimer Res 5:15–25
Jellinger KA (2002) The pathology of ischemic-vascular dementia: an update. J Neurol Sci 203–204:153–157
Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA (2000) Statins and the risk of dementia. Lancet 356:1627–1631
Jo DG, Arumugam TV, Woo HN, Park JS, Tang SC, Mughal M, Hyun DH, Park JH, Choi YH, Gwon AR, Camandola S, Cheng A, Cai H, Song W, Markesbery WR, Mattson MP (2010) Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol Aging 31:917–925
Kandiah N, Feldman HH (2009) Therapeutic potential of statins in Alzheimer’s disease. J Neurol Sci 283:230–234
Kao SC, Krichevsky AM, Kosik KS, Tsai LH (2004) BACE1 suppression by RNA interference in primary cortical neurons. J Biol Chem 279:1942–1949
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489
Keller JN, Mattson MP (1998) Roles of lipid peroxidation in modulation of cellular signaling pathways, cell dysfunction, and death in the nervous system. Rev Neurosci 9:105–116
Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR (2005) Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 64:1152–1156
Kern A, Behl C (2009) The unsolved relationship of brain aging and late-onset Alzheimer disease. Biochim Biophys Acta 1790:1124–1132
Kirkitadze MD, Kowalska A (2005) Molecular mechanisms initiating amyloid beta-fibril formation in Alzheimer’s disease. Acta Biochim Pol 52:417–423
Klimova T, Chandel NS (2008) Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ 15:660–666
Koistinaho M, Ort M, Cimadevilla JM, Vondrous R, Cordell B, Koistinaho J, Bures J, Higgins LS (2001) Specific spatial learning deficits become severe with age in beta–amyloid precursor protein transgenic mice that harbor diffuse beta -amyloid deposits but do not form plaques. Proc Natl Acad Sci USA 98:14675–14680
Lagalwar S, Guillozet-Bongaarts AL, Berry RW, Binder LI (2006) Formation of phospho-SAPK/JNK granules in the hippocampus is an early event in Alzheimer disease. J Neuropathol Exp Neurol 65:455–464
Ledesma MD, Bonay P, Colaço C, Avila J (1994) Analysis of microtubule-associated protein tau glycation in paired helical filaments. J Biol Chem 269:21614–21619
Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, Yamaguchi H, Ruiz A, Martinez A, Madrigal L, Hincapie L, Arango JC, Anthony DC, Koo EH, Goate AM, Selkoe DJ, Arango JC (1996) The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med 2:1146–1150
Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440:352–357
Lovell MA, Markesbery WR (2001) Ratio of 8-hydroxyguanine in intact DNA to free 8-hydroxyguanine is increased in Alzheimer disease ventricular cerebrospinal fluid. Arch Neurol 58:392–396
Lütjohann D, Breuer O, Ahlborg G, Nennesmo I, Sidén A, Diczfalusy U, Björkhem I (1996) Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci USA 93:9799–9804
Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP (1997) A role for 4 hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem 68:255–264
Markesbery WR, Carney JM (1999) Oxidative alterations in Alzheimer’s disease. Brain Pathol 9:133–146
Markesbery WR, Lovell MA (1998) Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging 19:33–36
Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD (2005) Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol 58:730–735
Mastrocola R, Guglielmotto M, Medana C, Catalano MG, Cutrupi S, Borghi R, Tamagno E, Boccuzzi G, Aragno M (2011) Dysregulation of SREBP2 induces BACE1 expression. Neurobiol Dis 44:116–124
Mielke MM, Lyketsos CG (2006) Lipids and the pathogenesis of Alzheimer’s disease: is there a link? Int Rev Psychiatry 18:173–186
Minopoli G, Stante M, Napolitano F, Telese F, Aloia L, De Felice M, Di Lauro R, Pacelli R, Brunetti A, Zambrano N, Russo T (2007) Essential roles for Fe65, Alzheimer amyloid precursor-binding protein, in the cellular response to DNA damage. J Biol Chem 282:831–835
Misonou H, Morishima-Kawashima M, Ihara Y (2000) Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 39:6951–6959
Moosmann B, Behl C (2002) Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs 11:1407–1435
Murakami K, Irie K, Ohigashi H, Hara H, Nagao M, Shimizu T, Shirasawa T (2005) Formation stabilization model of the 42-mer Abeta radical: implications for the long-lasting oxidative stress in Alzheimer’s disease. J Am Chem Soc 127:15168–15174
Murray IV, Liu L, Komatsu H, Uryu K, Xiao G, Lawson JA, Axelsen PH (2007) Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid beta proteins. J Biol Chem 282:9335–9345
Muscat S, Pelka J, Hegele J, Weigle B, Münch G, Pischetsrieder M (2007) Coffee and Maillard products activate NF-kappaB in macrophages via H2O2 production. Mol Nutr Food Res 51:525–535
Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A (1992) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 267:14998–15004
Nunomura A, Perry G, Hirai K, Aliev G, Takeda A, Chiba S, Smith MA (1999) Neuronal RNA oxidation in Alzheimer’s disease and Down’s syndrome. Ann N Y Acad Sci 893:362–364
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60:759–767
Ortega-Pérez I, Cano E, Were F, Villar M, Vázquez J, Redondo JM (2005) c-Jun N-terminal kinase (JNK) positively regulates NFATc2 transactivation through phosphorylation within the N-terminal regulatory domain. J Biol Chem 280:20867–20878
Panza F, Capurso C, D’Introno A, Colacicco AM, De Candia D, Capurso A, Solfrizzi V (2007) Total cholesterol levels and the risk of mild cognitive impairment and Alzheimer’s disease. J Am Geriatr Soc 55:133–135
Paola D, Domenicotti C, Nitti M, Vitali A, Borghi R, Cottalasso D, Zaccheo D, Odetti P, Strocchi P, Marinari UM, Tabaton M, Pronzato MA (2000) Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem Biophys Res Commun 268:642–646
Pappolla MA, Omar RA, Kim KS, Robakis NK (1992) Immunohistochemical evidence of oxidative stress in Alzheimer‘s disease. Am J Pathol 14:621–628
Passer B, Pellegrini L, Russo C, Siegel RM, Lenardo MJ, Schettini G, Bachmann M, Tabaton M, D’Adamio L (2000) Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer’s amyloid beta protein precursor. J Alzheimers Dis 2:289–301
Patil S, Sheng L, Masserang A, Chan C (2006) Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons. Neurosci Lett 406:55–59
Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, Smith MA (2000) Oxidative damage in Alzheimer’s disease: the metabolic dimension. Int J Dev Neurosci 18:417–421
Praticò D (2008) Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci 29:609–615
Puglielli L, Tanzi RE, Kovacs DM (2003) Alzheimer’s disease: the cholesterol connection. Nat Neurosci 6:345–351
Puig B, Gómez-Isla T, Ribé E, Cuadrado M, Torrejón-Escribano B, Dalfó E, Ferrer I (2004) Expression of stress-activated kinases c-Jun N-terminal kinase (SAPK/JNK-P) and p38 kinase (p38-P), and tau hyperphosphorylation in neurites surrounding betaA plaques in APP Tg2576 mice. Neuropathol Appl Neurobiol 30:491–502
Qin J, Goswami R, Dawson S, Dawson G (2008) Expression of the receptor for advanced glycation end products in oligodendrocytes in response to oxidative stress. J Neurosci Res 86:2414–2422
Resende R, Ferreiro E, Pereira C, Resende de Oliveira C (2008) Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1–42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 155:725–737
Rocchi A, Orsucci D, Tognoni G, Ceravolo R, Siciliano G (2009) The role of vascular factors in late-onset sporadic Alzheimer’s disease. Genetic and molecular aspects. Curr Alzheimer Res 6:224–237
Rodrigues Siqueira I, Fochesatto C, da Silva Torres IL, Dalmaz C, Alexandre Netto C (2005) Aging affects oxidative state in hippocampus, hypothalamus and adrenal glands of Wistar rats. Life Sci 78:271–278
Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR (2002) An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem 277:24558–45518
Sambamurti K, Kinsey R, Maloney B, Ge YW, Lahiri DK (2004) Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J 18:1034–1036
Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT (2006) Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci USA 103:443–448
Sayre LM, Zagorski MG, Surewicz WK, Krafft GA, Perry G (1997) Mechanisms of neurotoxicity associated with amyloid beta deposition and the role of free radicals in the pathogenesis of Alzheimer’s disease: a critical appraisal. Chem Res Toxicol 10:518–526
Schneider JA, Wilson RS, Cochran EJ, Bienias JL, Arnold SE, Evans DA, Bennett DA (2003) Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology 60:1082–1088
Serrano-Pozo A, Vega GL, Lütjohann D, Locascio JJ, Tennis MK, Deng A, Atri A, Hyman BT, Irizarry MC, Growdon JH (2010) Effects of simvastatin on cholesterol metabolism and Alzheimer disease biomarkers. Alzheimer Dis Assoc Disord 24:220–226
Singh M, Nam DT, Arseneault M, Ramassamy C (2010) Role of by-products of lipid oxidation in Alzheimer’s disease brain: a focus on acrolein. J Alzheimers Dis 21:741–756
Smith MA, Taneda S, Richey PL, Miyata S, Yan SD, Stern D, Sayre LM, Monnier VM, Perry G (1994) Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci USA 91:5710–5714
Smith MA, Rudnicka-Nawrot M, Richey PL, Praprotnik D, Mulvihill P, Miller CA, Sayre LM, Perry G (1995) Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer’s disease. J Neurochem 64:2660–2666
Song F, Poljak A, Smythe GA, Sachdev P (2009) Plasma biomarkers for mild cognitive impairment and Alzheimer’s disease. Brain Res Rev 61:69–80
Sultana R, Perluigi M, Butterfield DA (2009) Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Acta Neuropathol 118:131–150
Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, Song W (2006) Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci USA 103:18727–18732
Tabner BJ, El-Agnaf OM, Turnbull S, German MJ, Paleologou KE, Hayashi Y, Cooper LJ, Fullwood NJ, Allsop D (2005) Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem 280:35789–35792
Tagami S, Okochi M, Fukumori A, Jiang J, Yanagida K, Nakayama T, Morihara T, Tanaka T, Kudo T, Takeda M (2008) Processes of beta-amyloid and intracellular cytoplasmic domain generation by presenilin/gamma-secretase. Neurodegener Dis 5:160–162
Takeuchi M, Makita Z (2001) Alternative routes for the formation of immunochemically distinct advanced glycation end-products in vivo. Curr Mol Med 1:305–315
Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M (2002) Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis 10:279–288
Tamagno E, Guglielmotto M, Bardini P, Santoro G, Davit A, Di Simone D, Danni O, Tabaton M (2003) Dehydroepiandrosterone reduces expression and activity of BACE in NT2 neurons exposed to oxidative stress. Neurobiol Dis 14:291–301
Tamagno E, Parola M, Bardini P, Piccini A, Borghi R, Guglielmotto M, Santoro G, Davit A, Danni O, Smith MA, Perry G, Tabaton M (2005) Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem 92:628–636
Tamagno E, Bardini P, Guglielmotto M, Danni O, Tabaton M (2006) The various aggregation states of beta-amyloid 1–42 mediate different effects on oxidative stress, neurodegeneration, and BACE-1 expression. Free Radic Biol Med 41:202–212
Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, Tabaton M (2008) Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem 104:683–695
Tong Y, Zhou W, Fung V, Christensen MA, Qing H, Sun X, Song W (2005) Oxidative stress potentiates BACE1 gene expression and Abeta generation. J Neural Transm 112:455–469
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552:335–344
Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM (2003) Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med 348:1215–1222
Vieira M, Fernandes J, Burgeiro A, Thomas GM, Huganir RL, Duarte CB, Carvalho AL, Santos AE (2010) Excitotoxicity through Ca2+-permeable AMPA receptors requires Ca2+-dependent JNK activation. Neurobiol Dis 40:645–655
Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A (1994) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA 91:4766–47670
Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA (2005) Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem 93:953–962
Wen Y, Yu WH, Maloney B, Bailey J, Ma J, Marié I, Maurin T, Wang L, Figueroa H, Herman M, Krishnamurthy P, Liu L, Planel E, Lau LF, Lahiri DK, Duff K (2008) Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron 57:680–690
Wolozin B (2004) Cholesterol and the biology of Alzheimer’s disease. Neuron 41:7–10
Wu CY, Hsieh HL, Sun CC, Yang CM (2009) IL-1beta induces MMP-9 expression via a Ca2+-dependent CaMKII/JNK/c-JUN cascade in rat brain astrocytes. Glia 57:1775–1789
Yaar M, Zhai S, Panova I, Fine RE, Eisenhauer PB, Blusztajn JK, Lopez-Coviella I, Gilchrest BA (2007) A cyclic peptide that binds p75(NTR) protects neurones from beta amyloid(1–40)-induced cell death. Neuropathol Appl Neurobiol 33:533–543
Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS, Scott CW, Caputo C, Frappier T, Smith MA (1994) Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci USA 91:7787–7791
Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P (1995) Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med 1:693–699
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382:685–691
Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y (2003) Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 9:3–4
Yankner BA (1996) Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron 16:921–932
Yatin SM, Varadarajan S, Link CD, Butterfield DA (1999) In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid beta-peptide (1-42). Neurobiol Aging 20:325–330
Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M (2004) Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J Neurochem 90:352–358
Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, Xu H, Zhang YW (2007) Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem 282:10873–10880
Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O’Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R (2007) Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci 27:3639–3649
Zhu X, Ogawa O, Wang Y, Perry G, Smith MA (2003a) JKK1, an upstream activator of JNK/SAPK, is activated in Alzheimer’s disease. J Neurochem 85:87–93
Zhu X, Raina AK, Lee HG, Chao M, Nunomura A, Tabaton M, Petersen RB, Perry G, Smith MA (2003b) Oxidative stress and neuronal adaptation in Alzheimer disease: the role of SAPK pathways. Antioxid Redox Signal 5:571–576
Zhu Y, Carvey PM, Ling Z (2006) Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res 1090:35–44
Acknowledgments
The study was supported by Italian Ministry of Health, Regione Piemonte (ET) and CARIGE (MT).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Tamagno, E., Guglielmotto, M., Monteleone, D. et al. Amyloid-β Production: Major Link Between Oxidative Stress and BACE1. Neurotox Res 22, 208–219 (2012). https://doi.org/10.1007/s12640-011-9283-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-011-9283-6