
Abstract
3,4-Methylenedioxymethamphetamine (MDMA, ‘ecstasy’) is a selective 5-HT neurotoxin in rat brain which has been shown to produce acute neuroinflammation characterized by activation of microglia and release of interleukin-1beta (IL-1β). We aimed to determine whether or not minocycline, a semi-synthetic tetracycline antibiotic capable of inhibiting microglial activation, could prevent the inflammatory response and reduce the toxicity induced by MDMA. Adult male Dark Agouti rats were given minocycline twice a day for 2 days (45 mg/kg on the first day and 90 mg/kg on the second day; 12-h apart, i.p.). MDMA (12.5 mg/kg; i.p.) was given after the third minocycline injection and animals were killed either 1 h later for the determination of NFκB binding activity, 3 h later for the determination of IL-1β, 24 h later for the determination of microglial activation or 7 days later for the determination of [3H]-paroxetine binding as a measure of 5-HT neurotoxicity. MDMA increased NFκB activation, IL-1β release and microglial activation both in the frontal cortex and in the hypothalamus and 7 days later produced a reduction in the density of 5-HT uptake sites in both these brain areas. Minocycline prevented the MDMA-induced increase in NFκB activation, IL-1β release and microglial activation in the frontal cortex and prevented the 5-HT neurotoxicity 7 days later. However, in the hypothalamus, in spite of preventing MDMA-induced microglial activation, minocycline failed to prevent MDMA-induced NFκB activation, IL-1β release and neurotoxicity. This suggests that the protective mechanism of minocycline against MDMA-induced neurotoxicity in frontal cortex involves inhibition of MDMA-induced NFκB activation possibly through a reduction in IL-1β signalling.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
3,4-Methylenedioxymethamphetamine (MDMA or ‘ecstasy’) is a recreational drug commonly used by young people, particularly at crowded and warm dance venues, despite having been shown to be a potent neurotoxin in the brain of rodents and non-human primates (Green et al. 2003). Immunohistochemical techniques have demonstrated an apparent loss of 5-HT nerve terminals (Jensen et al. 1993; O’Hearn et al. 1988; Molliver et al. 1990) and biochemical studies have reported a reduction of [3H]-paroxetine binding to the pre-synaptic 5-HT transporter (Hewitt and Green 1994; Colado et al. 1997), a decrease in tryptophan hydroxylase activity (Stone et al. 1987; O’Shea et al. 2006) and a depletion of 5-HT and 5-hydroxyindole acetic acid (5-HIAA) content (Colado et al. 1997; O’Shea et al. 1998). MDMA also induces a sustained hyperthermic response which appears to modulate the long-term neuronal damage caused by the drug (Colado et al. 1999; Sanchez et al. 2004).
It has recently been shown that MDMA administration induces signs of neuroinflammation in the brain of Dark Agouti rats which is reflected by an increase in interleukin-1β (IL-1β) production and microglial activation (Orio et al. 2004; O’Shea et al. 2005). The IL-1β response appears within 1–3 h following MDMA injection, is partially a consequence of MDMA hyperthermia and seems to be involved in the long-term 5-HT neurotoxicity since the i.c.v. injection of IL-1β in the rat brain enhances the long-lasting reduction in 5-HT transporters and 5-HT concentration induced by MDMA (O’Shea et al. 2005). Activation of microglial cells following MDMA is independent of hyperthermia and there is no direct evidence of the possible implication of microglial activation in MDMA-induced long-term neurotoxicity.
Minocycline, a semi-synthetic tetracycline derivative, has an anti-inflammatory property which is completely independent and distinct from its antimicrobial mechanism (Amin et al. 1996; Rifkin et al. 1993). Recent reports have raised expectations for this category of antibiotic in the treatment of a wide range of neurodegenerative diseases. Minocycline has been shown to provide protection against brain ischemia (Yrjänheikki et al. 1998, 1999), excitotoxicity (Tikka and Koistinaho 2001), β-amyloid neurotoxicity (Ryu et al. 2004), spinal cord injury (Stirling et al. 2004) and dopamine neurotoxicity caused by 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Du et al. 2001; He et al. 2001; Wu et al. 2002). Neuroprotection is related to its ability to inhibit microglial activation (Yrjänheikki et al. 1998) and the subsequent release of cytotoxic substances such as oxygen and nitrogen reactive species as well as to the property of inhibiting the formation of mature IL-1β and the activation of NADPH-oxidase and inducible nitric oxide synthase (iNOS), COX-2 expression and prostaglandin E2 production (Yrjänheikki et al. 1999; Du et al. 2001; Wu et al. 2002).
The inducible transcription factor nuclear factor κB (NFκB) has been shown to play a key role in the regulation of many genes involved in immune and inflammatory responses. This transcription factor, ubiquitously expressed in neurons and glial cells, is sequestered in the cytoplasm by interaction with a family of inhibitory proteins (IκBs) (Ghosh et al. 1998) but can be activated in response to a broad range of stimuli which typically include free radicals and pro-inflammatory cytokines such as IL-1β and tumour necrosis factor-α (TNF-α) (Bowie and O’Neill 2000; Mattson and Camandola 2001). The currently known subunit members of the NFκB family in mammals are p50, p65 (RelA), c-Rel, p52 and RelB, the p65/p50 dimer being the predominant form of NFκB activated in many types of cells. IκBs are phosphorylated by a signal-activated kinase complex known as I-κB kinase (IKK) (Ghosh and Karin 2002) which causes them to release NFκB. The liberated NFκB dimer translocates to the nucleus where it induces transcription of target genes that mediate the inflammatory response by binding to high-affinity κB elements in their promoters (Pahl 1999; Bowie and O’Neill 2000).
The aims of this study are to examine: (1) the neuroprotective effect of minocycline against MDMA-induced neurotoxicity and the ability of this tetracycline to inhibit IL-1β release and microglial activation in the frontal cortex and hypothalamus of rats following MDMA, (2) the effect of MDMA on NFκB binding activity to the nucleus and the ability of pyrrolidine dithiocarbamate (PDTC) to inhibit NFκB activation and to provide protection against MDMA-induced neurotoxicity and (3) the ability of minocycline to prevent NFκB binding activity.
Materials and Methods
Animals and Drug Administration
Adult male Dark Agouti rats (175–200 g, Harlan Iberica, Barcelona, Spain) were used. They were housed in groups of six in conditions of constant temperature (21 ± 2°C) and a 12-h light/dark cycle (lights on: 07:00 a.m.) and given free access to food and water. Two dosing regimens of minocycline were employed in the neuroprotection studies. In the high-dose regimen, minocycline was administered twice a day (12 h apart) during two consecutive days at a dose of 45 mg/kg during the first day and 90 mg/kg during the second day. The same experiment was repeated using a lower dose of minocycline of 45 mg/kg i.p. twice a day (12-h apart) during two consecutive days. Rats were injected with MDMA (12.5 mg/kg, i.p.) or saline (1 ml/kg, i.p.) immediately after the third injection of minocycline and killed by decapitation 7 days later to determine 5-HT neurotoxicity. Pyrrolidine dithiocarbamate (PDTC, 50 and 100 mg/kg, i.p.) was injected 10 min before MDMA or saline (1 ml/kg, i.p.). Biochemical and immunohistochemical studies were performed in hypothalamus and frontal cortex since MDMA increases IL-1β release and microglial activation in both brain areas (Orio et al. 2004; O’Shea et al. 2005).
Minocycline (Sigma–Aldrich, Madrid, Spain) was dissolved in 0.9% w/v NaCl (saline) containing 10% of Tween 80 and injected in a volume of 5 ml/kg. MDMA (Ultrafine Chemicals Ltd., Manchester, UK) and PDTC (Sigma–Aldrich, Madrid, Spain) were dissolved in saline and injected in a volume of 1 ml/kg. Doses are quoted in terms of the base. Controls for each group were carried out following the same schedule of drug administration.
All experimental procedures were performed in accordance with the guidelines of the Animal Welfare Committee of the Universidad Complutense de Madrid (following European Council Directives 86/609/CEE and 2003/65/CE).
Measurement of Rectal Temperature
Immediately before and up to 6 h after MDMA injection, temperature was measured by use of a digital readout thermocouple (BAT12 thermometer, Physitemp, NJ, USA) with a resolution of 0.1°C and accuracy of ±0.1°C attached to a RET-2 Rodent Sensor which was inserted 2.5 cm into the rectum of the rat, the animal being lightly restrained by holding it in the hand. A steady readout was obtained within 10 s of probe insertion.
IL-1β Immunoassay
Levels of IL-1β were determined using a commercially available sandwich ELISA system (rat IL-1β immunoassay; Quantikine-M R&D Systems, Minneapolis, MN, USA). According to the manufacturer, the kit provides a valid measure of the levels of mature 17-kDa IL-1β (the limit of sensitivity was 5 pg/ml) but underestimates the precursor form, 31-kDa IL-1β (non-biologically active). Samples were prepared by homogenization of frontal cortex and hypothalamus in six volumes of ice-cold buffer (pH 7.0) containing 50-mM Tris, 320-mM sucrose, 1-mM dithiothreitol and a number of protease inhibitors (leupeptin 10 μg/ml, soybean trypsin 10 μg/ml, aprotinin 2 μg/ml and 0.2% phenanthroline). Samples were centrifuged at 14,000×g for 10 min at 4°C. Protein was determined in the supernatant fluid (Lowry et al. 1951). Supernatant fluids were assayed in triplicate following the manufacturer’s guidelines. The quantification of IL-1β was performed using a standard curve of increasing concentrations of recombinant IL-1β (4–1000 pg/ml). The optical density of each well was determined using a microplate reader (ELX808IU, Ultra Microplate Reader; Bio-Tek Instruments, Inc., Winoski, VT, USA) set to 450 nm (correction wavelength set at 540 nm). Intra- and interassay variations were less than 5 and 15%, respectively.
Immunohistochemistry of Microglia
Rats were anaesthetized with pentobarbitone and perfused transcardially through the left ventricle with 200 ml of 0.2-M sodium phosphate buffer as a vascular rinse followed by 300 ml of fixative solution containing 4% paraformaldehyde in 0.1-M sodium phosphate buffer (pH 7.4). Brains were removed, fixed in the same solution of paraformaldehyde for 4 h at room temperature and cryoprotected by immersion at 4°C in 0.1-M sodium phosphate buffer containing 30% sucrose. Brains were then placed on cryostat stages, frozen to −30°C and sliced at 8 μm in the coronal plane through the whole hypothalamus and frontal cortex.
Cerebral sections were incubated in the cold for 10 min in acetone and then for 30 min in 0.1% Triton X-100 in PBS and 30 min in 3% bovine serum albumin. Sections were then washed six times with PBS for 5 min each time and then incubated for 1 h with the primary antibody mouse anti-rat CD11b (Serotec; Raleigh, NC, USA; clone MRC OX-42, 1:50 dilution in PBS) to identify microglial cells. Sections next underwent six 5-min washes with PBS and were then incubated for 1 h in darkness with secondary antibody CyTM 2-labelled goat anti-mouse IgG (Amersham, Barcelona, Spain; 1:10 diluted in PBS; green colour with fluorescence maximum at 506 nm). Brain sections were washed in darkness six times with PBS (5 min each wash) and then mounted with glycerol (1:1 dilution in PBS). Visualization was performed under a fluorescence microscope (Eclipse TE300; Nikon Corporation, Tokyo, Japan) using Plan Fluor 20×/0.45 or 40×/0.6 objectives and a B2A Nikon filter for Cy2 fluorescence. Image acquisition was carried out with a laser scanning confocal imaging system (MRC1024; Bio-Rad, Hemel Hempstead, UK).
Preparation of Nuclear Extracts
Frontal cortex was homogenized in 500 μl and hypothalamus in 250 μl of buffer A [10-mM HEPES pH 7.9, 1-mM EDTA, 1-mM EGTA, 10-mM KCl, 1-mM dithiothreitol (DTT), 0.5-mM phenylmethanesulfonyl fluoride (PMSF), 40-μg/ml aprotinin, 4-μg/ml leupeptin, 4-μg/ml N-alpha-tosyl-l-lysine chloromethyl ketone (TLCK), 5-mM FNa, 10-mM Na2MoO4, 1-mM NaVO4] and Nonidet P-40® was added to reach 0.1% (v/v). Nuclei were collected by centrifugation at 13,000×g for 15 min at 4°C. The pellets were resuspended in 100 μl (cortex) or 50 μl (hypothalamus) of buffer A supplemented with 0.4-M NaCl and 20% glycerol and gently shaken for 30 min at 4°C. Nuclear protein extracts were obtained by centrifugation at 13,000×g for 15 min at 4°C and aliquots of the supernatant were stored at −80°C. All steps of the fractionation were carried out at 4°C. Nuclear proteins were quantified by the method of Bradford (1976).
Electrophoretic Mobility Shift Assay (EMSA) for NFκB
We measured p65/p50 binding activity since it is known that most of the NFκB transcriptional activity is mediated through these complexes (Baldwin 1996). Oligonucleotides were synthesized in an oligonucleotide synthesizer (Amersham Pharmacia Biotech, Little Chalfont, UK). The oligonucleotide sequence corresponding to the consensus NFκB binding site (nucleotides −978 to −952) was 5′TGCTAGGGGGATTTTCCCTCTCTCTGT3′ (Xie et al. 1994). Oligonucleotides were annealed with their complementary sequence by incubation for 5 min at 85°C in 10-mM Tris–HCl pH 8.0, containing 50-mM NaCl, 10-mM MgCl2, 1-mM DTT. Aliquots of 600 ng of these annealed oligonucleotides were end-labelled with the Klenow enzyme (Amersham Pharmacia Biotech, Barcelona, Spain) fragment in the presence of 50 μCi of [α-32P]dCTP (Amersham) and the other unlabelled dNTPs in a final volume of 20 μl. Then 15 × 104 dpm of the DNA probe were used for each binding assay of nuclear extracts as follows: 25 μg of nuclear protein were incubated for 20 min at 4°C with the DNA and 1 μg of poly(dI-dC), 5% glycerol, 1-mM EDTA, 0.05-mM NaCl, 5-mM MgCl2, 1-mM DTT and 20-mM HEPES pH 8.0 in a final volume of 20 μl. The DNA–protein complexes were separated on native 6% bisacrylamide gels in 0.5% Tris–borate–EDTA buffer (Díaz-Guerra et al. 1996). Supershift assays were carried out after incubation of the nuclear extracts with the antibody against NFκB protein p65 (0.5 μg, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at 4°C, followed by EMSA. Gels were run at 150 V and dried in a vacuum drier system (Amersham) for 2 h at 80°C. Then gels were exposed to autoradiography films (HyperfilmTM, Amersham) for several days at −80°C. Different exposure times were checked to ensure the optimal image resolution. Autoradiographs were quantified with a densitometric system (Total Lab, Biorad) and expressed in arbitrary units.
Quantification of [3H]-Paroxetine Binding in Tissue Homogenates
[3H]-Paroxetine binding was measured in fresh hypothalamical and cortical tissue by the method described in detail by Hewitt and Green (1994). Briefly, tissue was homogenized in ice-cold Tris–HCl (50-mM, pH 7.4) containing NaCl (120-mM) and KCl (5-mM) using an Ultra-Turrax. The homogenate was centrifuged at 30,000×g for 10 min at 4°C. The supernatant was discarded and the wash procedure repeated twice more. The pellet was finally resuspended in the Tris buffer at a concentration of 10-mg tissue/ml. Aliquots of tissue (800 μl) were incubated with [3H]-paroxetine (1 nM) for 60 min at room temperature in the absence and presence of 5-HT (100 μM) for determination of total and non-specific binding, respectively. Assays were terminated by rapid filtration through glass fibre filters and radioactivity determined by scintillation spectrometry. Protein was determined by the method of Lowry et al. (1951).
Measurement of MDMA Concentration in Cortical Tissue
Brain concentrations of MDMA were determined 1 h after injection following a previously described method with minor modifications (Sanchez et al. 2001). This time point was chosen because the cerebral concentration of MDMA normally peaks 60 min after MDMA injection (Esteban et al. 2001). The cortical tissue was homogenized in ice-cold sodium carbonate–sodium bicarbonate buffer (pH 11.5) using an ultrasonicator. The homogenate was centrifuged at 27,000×g for 20 min at 4°C. The supernatant was applied to a 145-mg C8 end-capped SPE light column (International Sorbent Technology, Waters). The column was washed with methanol (2 ml) followed by distilled water (2 ml) before applying the sample (400 μl of supernatant + 350 μl of distilled water). The column was washed with water (2 ml) before selective elution of MDMA with methanol (1 ml).
An aliquot (20 μl) of the resulting eluate was injected into a Waters HPLC system which consisted of a pump (Waters 510) linked to a manual sample injector (loop 20 μl, Rheodyne), a stainless steel column (RP 18, 5 μm, 150 × 4.6 mm, XTerra) fitted with a pre-column (RP 18, 5 μm, 20 × 3.9 mm, XTerra), and a UV/visible detector (Waters 2487). The current produced was monitored using an integrator (Waters M745). The mobile phase consisted of 20-mM potassium dihydrogen phosphate (75%) and acetonitrile (25%), pH 2.5; the flow rate was set to 0.8 ml/min and UV absorption was measured at 235 nm.
Statistics
Data from ELISA, EMSA and binding studies were analysed using one-way ANOVA followed by the Newman–Keuls multiple-comparisons test when a significant F value was obtained. Data from brain MDMA concentration were analysed using Student’s test. Statistical analyses of the temperature measurements were performed using the statistical computer package BMDP/386 Dynamic (BMDP Statistical Solutions, Cork, Ireland). Data were analysed by ANOVA with repeated measures (program 2V) or, where missing values occurred, an unbalanced repeated measure model (program 5V). Both used treatment as the between-subjects factor and time as the repeated measure. ANOVA was performed on both pre- and post-treatment data.
Results
Effect of Minocycline on MDMA-Induced Loss of 5-HT Transporters and Hyperthermia
One-way ANOVA indicated that there was a significant effect of treatment in frontal cortex (F 3,34 = 25.64, P < 0.0001; Fig. 1a) and hypothalamus (F 3,16 = 6.20, P = 0.0076; Fig. 1b). Post hoc Newman–Keuls test revealed that MDMA produced a significant reduction in the density of 5-HT uptake sites in frontal cortex (Fig. 1a) and hypothalamus (Fig. 1b) 7 days after drug injection. The high-dose regimen of minocycline administration attenuated this effect in frontal cortex (Fig. 1a), the effect being not significant in the hypothalamus (Fig. 1b). There was no neuroprotective effect following the lower dose of minocycline (data not shown); therefore, the lower dose was not used in the rest of experiments. Minocycline did not alter the density of 5-HT uptake sites in saline-treated rats.

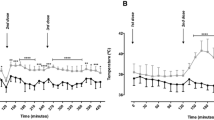
Effect of minocycline on MDMA-induced loss of 5-HT uptake sites in frontal cortex (a) and hypothalamus (b) and hyperthermia (c). Minocycline was injected i.p. every 12 h during two consecutive days at 45 mg/kg the first day and 90 mg/kg the second day. MDMA (12.5 mg/kg, i.p.) was injected immediately after the third injection of minocycline. Animals were killed 7 days after MDMA injection to perform [3H]-paroxetine binding in cortex and hypothalamus. Results are shown as mean ± SEM (n = 4–11). Different from saline group: * P < 0.05, *** P < 0.001. Different from MDMA-treated group: ΔΔ P < 0.01. Different from minocycline-treated group: f P < 0.05, ff P < 0.01. MDMA produced a marked increase in rectal temperature which peaked 30 min after drug administration and was sustained up to 6 h after treatment [F 1,22 = 24.07, P < 0.001]. Minocycline did not modify the hyperthermic response to MDMA [F 1,22 = 0.18, P = 0.65, n.s.] or did it modify the rectal temperature of saline-treated rats [F 1,16 = 0.0008, P = 0.97, n.s.]
MDMA produced a hyperthermic response which peaked between 30 and 60 min after treatment and was sustained for up to 6 h after MDMA injection. Minocycline had no effect on the hyperthermia of MDMA (Fig. 1c) or the rectal temperature of saline-treated animals (Fig. 1c).
Effect of Minocycline on Microglial Activation and IL-1β Release
Figure 2 shows OX-42 immunostaining in frontal cortex (upper panel) and hypothalamus (lower panel) of rats treated with minocycline and MDMA in combination or alone. There was increased OX-42 immunoreactivity in frontal cortex and hypothalamus 24 h after MDMA injection (Fig. 2b, f). Minocycline administration strongly reduced the number of OX-42 positive cells in both frontal cortex (Fig. 2c) and hypothalamus (Fig. 2g) 24 h after MDMA administration. Minocycline did not modify OX-42 immunostaining in saline-treated rats (Fig. 2d, h).

OX-42 immunostaining in frontal cortex (upper panel) and hypothalamus (lower panel) showing the effect of minocycline on MDMA-induced microglial activation: a, e saline and vehicle; b, f MDMA and vehicle; c, g minocycline and MDMA; d, h minocycline and saline. Minocycline was administered twice a day (12-h apart) during two consecutive days at 45 mg/kg i.p. (first day) and 90 mg/kg (second day). MDMA (12.5 mg/kg, i.p.) was administered immediately after the third minocycline injection. Rats were killed 24 h after MDMA administration. OX-42 immunostaining reveals the presence of activated microglia throughout the frontal cortex (b) and hypothalamus (f) 24 h after MDMA injection. Rats treated with minocycline and MDMA showed a decrease in the number of OX-42-stained cells both in frontal cortex (c) and hypothalamus (g) when compared with MDMA-treated rats (b, f). Minocycline did not change OX-42 immunostaining in saline-treated animals (d, h). Scale bar 25 μm
Regarding data of IL-1β release, one-way ANOVA indicated that there was a significant effect of treatment in frontal cortex (F 3,37 = 7.17, P = 0.0007; Fig. 3a) and hypothalamus (F 3,23 = 23.41, P < 0.0001; Fig. 3b). Post hoc Newman–Keuls test revealed that MDMA produced a substantial increase in IL-1β levels in frontal cortex (Fig. 3a) and hypothalamus (Fig. 3b) 3 h after drug administration. Minocycline significantly reduced this effect in frontal cortex but not in hypothalamus (Fig. 3a, b). Minocycline did not alter IL-1β levels in the brain of saline-treated rats (Fig. 3a, b).

Effect of minocycline on the MDMA-induced changes in interleukin-1beta (IL-1β) levels in frontal cortex (a) and hypothalamus (b). Minocycline was administered twice a day (12-h apart) during two consecutive days at 45 mg/kg i.p. (first day) and 90 mg/kg (second day). MDMA (12.5 mg/kg, i.p.) was administered immediately after the third minocycline injection. Rats were killed 3 h after MDMA administration. Results are shown as mean ± SEM (n = 4–13). Different from saline group: ** P < 0.01, *** P < 0.001. Different from MDMA-treated group: ΔΔΔ P < 0.001. Different from PDTC-treated group: fff P < 0.001
Changes Induced by MDMA in NFκB Activation
One-way ANOVA indicated that there was a significant effect of treatment in frontal cortex (F 4,18 = 7.70, P = 0.0017; Fig. 4a) and hypothalamus (F 4,20 = 7.32, P = 0.0015; Fig. 4b). Post hoc Newman–Keuls test revealed that shortly after MDMA administration there was an increase in NFκB (p50/p65 heterodimer) DNA-binding. This effect was evident 1 and 3 h after MDMA injection in both frontal cortex (Fig. 4a) and hypothalamus (Fig. 4b). No change was observed in either area 6 h after MDMA injection (Fig. 4a, b).

Time course of MDMA-induced NFκB DNA binding in frontal cortex (a) and hypothalamus (b). EMSA was used to determine the status of the NFκB complex 30 min, 1 h, 3 h and 6 h after MDMA administration. NFκB heterodimer p65/p50 (arrow) increased 1 and 3 h after MDMA injection both in frontal cortex and hypothalamus. Data presented as a percentage of saline-treated animals. Results are shown as mean ± SEM (n = 2–8). Different from saline-treated animals: * P < 0.05, ** P < 0.01, *** P < 0.001
Effect of Minocycline and PDTC on MDMA-Induced NFκB Activation
With respect to the minocycline study, one-way ANOVA indicated that there was a significant effect of treatment in frontal cortex (F 3,19 = 4.46, P = 0.0186; Fig. 5a) and hypothalamus (F 3,34 = 9.99, P < 0.0001; Fig. 5b). Post hoc Newman–Keuls test revealed that MDMA (12.5 mg/kg, i.p.) administration increased the DNA binding of the p65/p50 heterodimer of NFκB 1 h after drug injection in frontal cortex (Fig. 5a) and hypothalamus (Fig. 5b). Minocycline reduced the NFκB DNA binding induced by MDMA 1 h after drug administration in frontal cortex (Fig. 5a) but not in hypothalamus (Fig. 5b). Minocycline did not modify the NFκB binding activity in saline-treated animals (Fig. 5a, b).

Effect of minocycline on MDMA-induced NFκB DNA binding in frontal cortex (a) and hypothalamus (b). Animals pre-treated with minocycline showed an attenuation of MDMA-induced p65/p50 NFκB DNA binding in frontal cortex but not in hypothalamus as revealed by EMSA experiments. Animals were killed 1 h after MDMA injection. Data presented as percentage of saline-treated animals. Results are shown as mean ± SEM (n = 4–7 in cortex, n = 6–12 in hypothalamus). Different from saline group: * P < 0.05, ** P < 0.01, *** P < 0.001. Different from MDMA-treated group: Δ P < 0.05. Different from minocycline-treated group: f P < 0.05
Regarding the PDTC study, one-way ANOVA indicated that there was a significant effect of treatment in frontal cortex (F 3,18 = 3.51, P = 0.0415; Fig. 6a) and hypothalamus (F 3,29 = 10.30, P = 0.0001; Fig. 6b). Post hoc Newman–Keuls test revealed that MDMA (12.5 mg/kg, i.p.) administration increased the DNA binding of the p65/p50 heterodimer of NFκB 1 h after drug injection in frontal cortex (Fig. 6a) and hypothalamus (Fig. 6b). PDTC (100 mg/kg, i.p.) given immediately before MDMA (12.5 mg/kg, i.p.) prevented the effect of MDMA in frontal cortex (Fig. 6a) but not in hypothalamus (Fig. 6b). PDTC treatment did not modify the NFκB DNA binding in saline-treated animals (Fig. 6a, b).

Effect of PDTC on MDMA-induced NFκB DNA binding in frontal cortex (a) and hypothalamus (b). Animals pre-treated with PDTC (100 mg/kg, i.p.) showed an attenuation of MDMA-induced p65/p50 NFκB DNA binding in frontal cortex but not in hypothalamus as revealed by EMSA experiments. Animals were killed 1 h after MDMA injection. Data presented as percentage of saline-treated animals. Results are shown as mean ± SEM (n = 3–6 in cortex, n = 5–12 in hypothalamus). Different from saline group: * P < 0.05, ** P < 0.01, *** P < 0.001. Different from MDMA-treated group: Δ P < 0.05. Different from PDTC-treated group: ff P < 0.01
Effect of PDTC on MDMA-Induced Loss of 5-HT Transporters and Hyperthermia
One-way ANOVA indicated that there was a significant effect of treatment in frontal cortex (F 3,18 = 20.35, P < 0.0001; Fig. 7a) and hypothalamus (F 3,24 = 11.25, P < 0.0001; Fig. 7b). Post hoc Newman–Keuls test revealed that animals treated with PDTC (100 mg/kg, i.p.) immediately before MDMA administration (12.5 mg/kg, i.p.) showed attenuation in the loss of 5-HT uptake sites observed in MDMA-treated animals in frontal cortex (Fig. 7a) but not in hypothalamus (Fig. 7b). A lower dose of PDTC (50 mg/kg, i.p.) did not modify the loss of 5-HT uptake site density induced by MDMA in frontal cortex (data not shown).

Effect of PDTC on MDMA-induced loss of 5-HT uptake sites in frontal cortex (a) and hypothalamus (b) and hyperthermia (c). PDTC (100 mg/kg, i.p.) was injected immediately before MDMA administration (12.5 mg/kg, i.p.). Animals were killed 7 days after treatment. Results are shown as mean ± SEM (n = 4–8). Different from saline group: * P < 0.05, ** P < 0.01, *** P < 0.001. Different from MDMA-treated group: ΔΔ P < 0.01. Different from PDTC-treated group: ff P < 0.01. MDMA produced a marked increase in rectal temperature which peaked 30–60 min after drug administration and was sustained up to 6 h after treatment [F 1,10 = 72.7, P < 0.001]. Pre-treatment with PDTC (100 mg/kg, i.p.) did not modify the hyperthermic response to MDMA [F 1,13 = 2.40, P = 0.15, n.s.] or the rectal temperature of saline-treated rats [F 1,9 = 0.26, P = 0.62, n.s.]
MDMA (12.5 mg/kg, i.p.) administration to rats induced a rise in rectal temperature which peaked 30 min after injection (Fig. 7c). Pre-treatment with PDTC (100 mg/kg, i.p.) did not modify the MDMA-induced hyperthermia (Fig. 7c) or the rectal temperature of saline-treated animals (Fig. 7c).
Effect of Minocycline and PDTC on Cortical Levels of MDMA
In order to investigate the possible effect of minocycline and PDTC on the concentration of MDMA in the brain, rats were given each compound plus MDMA following the same protocol as that used in the experiments mentioned before. Rats were killed 1 h after MDMA injection. There was no difference between the MDMA levels found in the frontal cortex of rats treated with minocycline plus MDMA (27.50 ± 2.07 nmol/g tissue, n = 5) and those treated only with MDMA (31.78 ± 0.50 nmol/g tissue, n = 5). In a separate experiment the MDMA levels found in the cortex of rats treated with PDTC (100 mg/kg, i.p.) and MDMA (25.06 ± 2.11 nmol/g tissue, n = 4) were similar to those observed in rats treated with MDMA alone (20.43 ± 5.60 nmol/g tissue, n = 4).
Discussion
This study shows for the first time that the semi-synthetic tetracycline minocycline attenuates the MDMA-induced damage to 5-HT nerve endings in the frontal cortex of Dark Agouti rats and that the beneficial effect is associated with reduction of IL-1β release and microglia activation which remain in resting state when the animal is treated with minocycline. Minocycline exhibits region-specific actions in the CNS. In the hypothalamus, although minocycline treatment attenuates microglial activation, it does not reduce IL-1β release and it fails to afford protection against 5-HT neurotoxicity induced by MDMA.
IL-1β is generated as an inactive 31-kDa precursor protein (pro-IL-1β) (March et al. 1985) which is proteolytically processed into the 17-kDa mature IL-1β by a specific intracellular cysteine protease, the IL-1β converting enzyme (ICE) also termed caspase-1 (Thornberry et al. 1992). The enzyme is localized in the cytoplasm and on the external cell surface membranes where it forms a pore allowing the release of IL-1β from cells (Singer et al. 1995; Ferrari et al. 1996) to function as an intercellular messenger (Singer et al. 1995; Miossec et al. 1996). It has been demonstrated that minocycline attenuates the increased expression of procaspase-1 and inhibits caspase-1 upregulation induced by the R6/2 transgenic model of Huntington’s disease in the brain (Chen et al. 2000; Mievis et al. 2007), reduces the induction of caspase-1 mRNA following global brain ischemia (Yrjänheikki et al. 1998; Chen et al. 2000), and prevents diabetes-induced activation of caspase-1 in the early phase of diabetic retinopathy (Mohr 2004). Thus, it would to be expected that this tetracycline would be able to reduce IL-1β release in both brain areas. Nevertheless, we have recently observed regional heterogeneity in the release of IL-1β in response to MDMA (O’Shea et al. 2005). MDMA alters, in a region-specific manner, the mechanisms which regulate IL-1β production in the brain of Dark Agouti rats. Thus, MDMA shortly after administration increases pro-IL-1β synthesis and, in addition, facilitates the conversion of the inactive pro-IL-1β to the active form in the frontal cortex. This latter change is reflected by both an increase in caspase-1-like protease activity, and a higher protein expression of the p20 subunit, the active form of caspase-1. However, these effects are not observed in the hypothalamus since MDMA does not modify pro-IL-1β synthesis or caspase-1-protease cleavage in spite of also producing a rise in IL-1β release. These data indicate that the mechanisms leading to MDMA-induced externalization of IL-1β show region-specific differences and that in some brain areas such as hypothalamus IL-1β export is regulated independently of caspase-1-like protease activation. Thus, regional differences in the expression and elaboration of IL-1β may determine the susceptibility of brain regions to minocycline.
Our results show for the first time that MDMA produces NFκB pathway activation reflected as an increase in p65/p50 heterodimer DNA binding in frontal cortex and hypothalamus between 1 and 3 h after drug injection. The IL-1β release after MDMA injection could be one of the stimuli responsible for NFκB activation. There is abundant evidence that IL-1β acting through type-I IL-1 receptor (IL-1R1) can promote NFκB activity in brain cells via phosphorylation and degradation of IκBs (Auron 1998; Hu et al. 2005). Persistent IL-1β signalling causes activation of NFκB in human glial cells (Griffin and Moynagh 2006) and i.c.v administration of IL-1β induces NFκB activation in ependymal cells lining the lateral and third ventricle, choroid plexus, preoptic area and dentatus gyrus (Konsman et al. 2000). The effect of MDMA on IL-1β release (Orio et al. 2004) lasted longer than that on NFκB activation (this paper), which may indicate that activation of the transcription factor NFκB regulates the expression of a large number of different genes involved in inflammation which includes enzymes such as COX-2 and iNOS (Xie et al. 1994; Nadjar et al. 2005) and more proinflammatory cytokines such as IL-1β in a cyclic mechanism involving further IL-1β release and inflammation (Ahn and Aggarwal 2005).
In addition, suppression of the increase in cortical IL-1β levels by minocycline inhibits the MDMA-induced NFκB DNA binding in frontal cortex, whilst it has no effect in the hypothalamus where minocycline does not modify MDMA-induced IL-1β release.
Minocycline attenuates the MDMA-induced loss of 5-HT transporters in the frontal cortex and this protection is associated with the reduction of MDMA-induced IL-1β release and microglia activation and with the attenuation in the NFκB binding activity. In the hypothalamus, neither neuroprotection nor a reduction in IL-1β release or NFκB binding activity is observed although microglial activation was attenuated. It is worth noting that in the hypothalamus, a region with a readily releasable pool of mature IL-1β of neuronal origin (Lindberg et al. 2004), glial cells seem not to be the only source for the expression of this cytokine. There is evidence for the neuronal origin of immunoreactive IL-1β whose release is modulated by dopamine and corticotropin-releasing hormone in a concentration-dependent manner (Tringali et al. 1996, 1997) and it is well known that MDMA administration rapidly increases the extracellular concentration of dopamine in several brain areas.
Our findings on the effect of minocycline on MDMA neurotoxicity are at least partially consistent with those reported by Zhang et al. (2006). These authors showed that minocycline partially prevented the MDMA-induced 5-HT loss in frontal cortex and dopamine loss in the striatum of mice. Minocycline was also able to reduce microglial activation in the striatum. Nevertheless, the MDMA-induced microglial activation in mice seems not to be involved in the long-term loss of 5-HT occurring in frontal cortex as there is no microglial activation in this brain area following MDMA.
Reports on the effect of amphetamine derivatives on NFκB activation are scarce. Recently, Lai et al. (2009) have shown that methamphetamine, at doses producing dopamine neurotoxicity (three consecutive doses, 10 mg/kg each), enhances nuclear NFκB expression in striatal tissue 24 and 72 h after administration to mice. Mice also show an increased expression of TNF-α. Both factors seem to be involved in methamphetamine-induced damage as manipulations preventing the elevated TNF-α expression and activation of NFκB reduce methamphetamine-induced dopamine depletion.
NFκB has been found to be activated in response to a broad range of stimuli and conditions which include an increase in oxidative stress (Bowie and O’Neill 2000) and it is well known that MDMA and methamphetamine produce a rise in free radical content immediately after injection (Colado et al. 1997; Shankaran et al. 1999; Imam et al. 2001) which is involved in the neurotoxic effects induced by the amphetamines. In this study, the relatively specific inhibitor of NFκB activation PDTC attenuates the MDMA-induced increase in cortical NFκB DNA binding and partially prevents neurotoxicity in this same brain area. PDTC inhibits the activity of IκB-ubiquitin ligase (Hayakawa et al. 2003), an enzyme which degrades inhibitory IκB unit and frees NFκB for translocation and DNA binding. In addition, PDTC is also known to be a powerful thiol antioxidant (Bowie and O’Neill 2000; Tsuchihashi et al. 2003) and it scavenges haemin- and FeSO4-induced ROS in cultured neurons; therefore, this could be responsible for inhibition of the MDMA-induced NFκB activation leading to its neuroprotective actions. We cannot rule out the possibility that the antioxidant properties of PDTC may play a role in preventing MDMA-induced neurotoxicity.
What can also be stated unequivocally is that the neuroprotective effects of minocycline and PDTC against MDMA toxicity are not related to an effect on body temperature. The hyperthermic response immediately following MDMA was not modified in rats also given either minocycline or PDTC. In addition, neuroprotection appears to be unrelated to changes in MDMA metabolism since neither minocycline nor PDTC altered the concentration of MDMA in the brain.
Taken together, our results show that minocycline significantly attenuated the long-term loss of cortical 5-HT transporters induced by MDMA and acutely reduced microglial activation, IL-1β release and NFκB activation. The antioxidant PDTC also prevented the loss of the 5-HT marker in frontal cortex and NFκB binding activity. These results indicate that NFκB activation is involved in MDMA-induced neurotoxicity possibly through a mechanism involving IL-1β signalling.
Abbreviations
- IL-1β:
-
Interleukin-1β
- MDMA:
-
3,4-Methylenedioxymethamphetamine
- NFkB:
-
Nuclear factor kappaB
- PBS:
-
Phosphate buffered saline
- 5-HIAA:
-
5-Hydroxyindole acetic acid
References
Ahn KS, Aggarwal BB (2005) Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci 1056:218–233
Amin AR, Attur MG, Thakker GD, Patel PD, Vyas PR, Patel RN, Patel IR, Abramson SB (1996) A novel mechanism of action of tetracyclines: effects on nitric oxide synthases. Proc Natl Acad Sci 93:14014–14019
Auron PE (1998) The interleukin 1 receptor: ligand interactions and signal transduction. Cytokine Growth Factor Rev 9:221–237
Baldwin AS Jr (1996) The NF-kappa B and I kappa B proteins: new discoveries and insights. Ann Rev Immunol 14:649–683
Bowie A, O’Neill LA (2000) Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol 59:13–23
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6:797–801
Colado MI, O’Shea E, Granados R, Murray TK, Green AR (1997) In vivo evidence for free radical involvement in the degeneration of rat brain 5-HT following administration of MDMA (‘ecstasy’) and p-chloroamphetamine but not the degeneration following fenfluramine. Br J Pharmacol 121:889–900
Colado MI, Esteban B, O’Shea E, Granados R, Green AR (1999) Studies on the neuroprotective effect of pentobarbitone on MDMA-induced neurodegeneration. Psychopharmacology 142:421–425
Díaz-Guerra MJ, Velasco M, Martín-Sanz P, Bosca L (1996) Evidence for common mechanisms in the transcriptional control of type II nitric oxide synthase in isolated hepatocytes. Requirement of NF-kappaB activation after stimulation with bacterial cell wall products and phorbol esters. J Biol Chem 271:30114–30120
Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM (2001) Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci USA 98:14669–14674
Esteban B, O’Shea E, Camarero J, Sanchez V, Green AR, Colado MI (2001) 3,4-Methylenedioxymethamphetamine induces monoamine release, but not toxicity, when administered centrally at a concentration occurring following a peripherally injected neurotoxic dose. Psychopharmacology (Berl) 154:251–260
Ferrari D, Villalba M, Chiozzi P, Falzoni S, Ricciardi-Castagnoli P, Di Virgilio F (1996) Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J Immunol 156:1531–1539
Ghosh S, Karin M (2002) Missing pieces in the NF-kappaB puzzle. Cell 109(Suppl):S81–S96
Ghosh S, May MJ, Kopp EB (1998) NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 16:225–260
Green AR, Mechan AO, Elliott JM, O’Shea E, Colado MI (2003) The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”). Pharmacol Rev 55:463–508
Griffin BD, Moynagh PN (2006) Persistent interleukin-1beta signaling causes long term activation of NFkappaB in a promoter-specific manner in human glial cells. J Biol Chem 281:10316–10326
Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K (2003) Evidence that reactive oxygen species do not mediate NF-kappaB activation. EMBO J 22:3356–3366
He Y, Appel S, Le W (2001) Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res 909:187–193
Hewitt KE, Green AR (1994) Chlormethiazole, dizocilpine and haloperidol prevent the degeneration of serotonergic nerve terminals induced by administration of MDMA (‘ecstasy’) to rats. Neuropharmacology 33:1589–1595
Hu X, Nesic-Taylor O, Qiu J, Rea HC, Fabian R, Rassin DK, Perez-Polo JR (2005) Activation of nuclear factor-kappaB signaling pathway by interleukin-1 after hypoxia/ischemia in neonatal rat hippocampus and cortex. J Neurochem 93:26–37
Imam SZ, Newport GD, Itzhak Y, Cadet JL, Islam F Jr, Slikker W, Ali SF (2001) Peroxynitrite plays a role in methamphetamine-induced dopaminergic neurotoxicity: evidence from mice lacking neuronal nitric oxide synthase gene or overexpressing copper-zinc superoxide dismutase. J Neurochem 76:745–749
Jensen KF, Olin J, Haykal-Coates N, O’Callaghan J, Miller DB, de Olmos JS (1993) Mapping toxicant-induced nervous system damage with a cupric silver stain: a quantitative analysis of neural degeneration induced by 3,4-methylenedioxymethamphetamine. NIDA Res Monogr 136:133–149
Konsman JP, Tridon V, Dantzer R (2000) Diffusion and action of intracerebroventricularly injected interleukin-1 in the CNS. Neuroscience 101:957–967
Lai YT, Tsai YP, Cherng CG, Ke JJ, Ho MC, Tsai CW, Yu L (2009) Lipopolysaccharide mitigates methamphetamine-induced striatal dopamine depletion via modulating local TNF-alpha and dopamine transporter expression. J Neural Transm 116:405–415
Lindberg C, Eriksson C, Van Dam AM, Winblad B, Schultzberg M (2004) Neuronal expression of caspase-1 immunoreactivity in the rat central nervous system. J Neuroimmunol 146:99–113
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
March CJ, Mosley B, Larsen A, Cerretti DP, Braedt G, Price V, Gillis S, Henney CS, Kronheim SR, Grabstein K et al (1985) Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature 315:641–647
Mattson MP, Camandola S (2001) NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 107:247–254
Mievis S, Levivier M, Communi D, Vassart G, Brotchi J, Ledent C, Blum D (2007) Lack of minocycline efficiency in genetic models of Huntington’s disease. Neuromolecular Med 9:47–54
Miossec C, Decoen MC, Durand L, Fassy F, Diu-Hercend A (1996) Use of monoclonal antibodies to study interleukin-1 beta-converting enzyme expression: only precursor forms are detected in interleukin-1 beta-secreting cells. Eur J Immunol 26:1032–1042
Mohr S (2004) Potential new strategies to prevent the development of diabetic retinopathy. Expert Opin Investig Drugs 13:189–198
Molliver ME, Berger UV, Mamounas LA, Molliver DC, O’Hearn E, Wilson MA (1990) Neurotoxicity of MDMA and related compounds: anatomic studies. Ann N Y Acad Sci 600:649–661
Nadjar A, Tridon V, May MJ, Ghosh S, Dantzer R, Amédée T, Parnet P (2005) NFkappaB activates in vivo the synthesis of inducible Cox-2 in the brain. J Cereb Blood Flow Metab 25:1047–1059
O’Hearn E, Battaglia G, De Souza EB, Kuhar MJ, Molliver ME (1988) Methylenedioxyamphetamine (MDA) and methylenedioxymethamphetamine (MDMA) cause selective ablation of serotonergic axon terminals in forebrain: immunocytochemical evidence for neurotoxicity. J Neurosci 8:2788–2803
O’Shea E, Granados R, Esteban B, Colado MI, Green AR (1998) The relationship between the degree of neurodegeneration of rat brain 5-HT nerve terminals and the dose and frequency of administration of MDMA (‘ecstasy’). Neuropharmacology 37:919–926
O’Shea E, Sanchez V, Orio L, Escobedo I, Green AR, Colado MI (2005) 3,4-Methylenedioxymethamphetamine increases pro-interleukin-1beta production and caspase-1 protease activity in frontal cortex, but not in hypothalamus, of Dark Agouti rats: role of interleukin-1beta in neurotoxicity. Neuroscience 135:1095–1105
O’Shea E, Orio L, Escobedo I, Sanchez V, Camarero J, Green AR, Colado MI (2006) MDMA-induced neurotoxicity: long-term effects on 5-HT biosynthesis and the influence of ambient temperature. Br J Pharmacol 148:778–785
Orio L, O’Shea E, Sanchez V, Pradillo JM, Escobedo I, Camarero J, Moro MA, Green AR, Colado MI (2004) 3,4-Methylenedioxymethamphetamine increases interleukin-1beta levels and activates microglia in rat brain: studies on the relationship with acute hyperthermia and 5-HT depletion. J Neurochem 89:1445–1453
Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18:6853–6866
Rifkin BR, Vernillo AT, Golub LM (1993) Blocking periodontal disease progression by inhibiting tissue-destructive enzymes: a potential therapeutic role for tetracyclines and their chemically-modified analogs. J Periodontol 64:819–827
Ryu JK, Franciosi S, Sattayaprasert P, Kim SU, McLarnon JG (2004) Minocycline inhibits neuronal death and glial activation induced by beta-amyloid peptide in rat hippocampus. Glia 48:85–90
Sanchez V, Camarero J, Esteban B, Peter MJ, Green AR, Colado MI (2001) The mechanisms involved in the long-lasting neuroprotective effect of fluoxetine against MDMA (‘ecstasy’)-induced degeneration of 5-HT nerve endings in rat brain. Br J Pharmacol 134:46–57
Sanchez V, O’Shea E, Saadat KS, Elliott JM, Colado MI, Green AR (2004) Effect of repeated (‘binge’) dosing of MDMA to rats housed at normal and high temperature on neurotoxic damage to cerebral 5-HT and dopamine neurons. J Psychopharmacol. 18:412–416
Shankaran M, Yamamoto BK, Gudelsky GA (1999) Involvement of the serotonin transporter in the formation of hydroxyl radicals induced by 3,4-methylenedioxymethamphetamine. Eur J Pharmacol 385:103–110
Singer II, Scott S, Chin J, Bayne EK, Limjuco G, Weidner J, Miller DK, Chapman K, Kostura MJ (1995) The interleukin-1 beta-converting enzyme (ICE) is localized on the external cell surface membranes and in the cytoplasmic ground substance of human monocytes by immuno-electron microscopy. J Exp Med 182:1447–1459
Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W (2004) Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 24:2182–2190
Stone DM, Merchant KM, Hanson GR, Gibb JW (1987) Immediate and long-term effects of 3,4-methylenedioxymethamphetamine on serotonin pathways in brain of rat. Neuropharmacology 26:1677–1683
Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J et al (1992) A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356:768–774
Tikka TM, Koistinaho JE (2001) Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol 166:7527–7533
Tringali G, Mancuso C, Mirtella A, Pozzoli G, Parente L, Preziosi P, Navarra P (1996) Evidence for the neuronal origin of immunoreactive interleukin-1 beta released by rat hypothalamic explants. Neurosci Lett 219:143–146
Tringali G, Mirtella A, Mancuso C, Guerriero G, Preziosi P, Navarra P (1997) The release of immunoreactive interleukin-1 beta from rat hypothalamic explants is modulated by neurotransmitters and corticotropin-releasing hormone. Pharmacol Res 36:269–273
Tsuchihashi S, Tamaki T, Tanaka M, Kawamura A, Kaizu T, Ikeda A, Kakita A (2003) Pyrrolidine dithiocarbamate provides protection against hypothermic preservation and transplantation injury in the rat liver: the role of heme oxygenase-1. Surgery 133:556–567
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22:1763–1771
Xie QW, Kashiwabara Y, Nathan C (1994) Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 269:4705–4708
Yrjänheikki J, Keinänen R, Pellikka M, Hökfelt T, Koistinaho J (1998) Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA 95:15769–15774
Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho J (1999) A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA 96:13496–13500
Zhang L, Shirayama Y, Shimizu E, Iyo M, Hashimoto K (2006) Protective effects of minocycline on 3,4-methylenedioxymethamphetamine-induced neurotoxicity in serotonergic and dopaminergic neurons of mouse brain. Eur J Pharmacol 544:1–9
Acknowledgements
We thank Dr. Paloma Martin-Sanz for assistance with the EMSA procedure. This study was supported by MCYT (Grant no. SAF2007-65175), Plan Nacional sobre Drogas (Grant no. PR75/06-15077), Ministerio de Sanidad (Grant no. RTA-RD06/0001/006), UCM-CAM (Grant no. CCG07-UCM/SAL-2588). The authors received predoctoral funding from Ministerio de Educación y Ciencia (LO,ET) and Comunidad de Madrid (MI, NL).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Orio, L., Llopis, N., Torres, E. et al. A Study on the Mechanisms by Which Minocycline Protects Against MDMA (‘Ecstasy’)-Induced Neurotoxicity of 5-HT Cortical Neurons. Neurotox Res 18, 187–199 (2010). https://doi.org/10.1007/s12640-009-9120-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-009-9120-3