
Abstract
Poly (ADP-ribose) polymerase (PARP) inhibitors, a novel class of drugs that target tumors with DNA repair defects, have received tremendous enthusiasm. Early preclinical studies identified BRCA1 and BRCA2 tumors to be highly sensitive to PARP inhibitors as a result of homologous recombination defect. Based on this premise, PARP inhibitors have been tested in early phase clinical trials as a single agent in BRCA1 or BRCA2 mutation carriers and in combination with chemotherapy in triple-negative breast cancer patients. For high-risk populations, use of PARP inhibition as a prevention agent has been postulated, but no robust preclinical or clinical studies exist yet. We review the preclinical and clinical studies in treatment of breast cancer and rationale for use of PARP inhibitors as a prevention agent for high-risk populations. Of significance, PARP inhibitors vary significantly in mechanism of action, dosing intervals, and toxicities, which are highlighted in this review.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
DNA Repair and Cancer
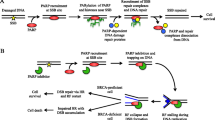
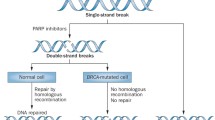
The human genome is constantly under genotoxic stress from various endogenous sources, such as oxygen radicals produced during metabolism, and exogenous sources, such as ultraviolet light. There are a large number of DNA repair systems that exist to accommodate a wide variety of DNA damage a cell encounters. The cellular response to DNA damage is variable, but highly regulated, and may result in survival of a normal cell, cell death, or mutagenesis depending on the magnitude of the insult and efficiency of repair [1]. There are multiple DNA repair pathways and these can be broadly divided into categories of direct repair, excision repair, and double-strand break repair. Double-strand DNA breaks are a highly lethal event and two major pathways, nonhomologous end joining and homologous recombination (HR), contribute to repair of these lesions [1]. The excision repair pathways, including base excision repair (BER), nucleotide excision repair (NER), and mismatch repair, use a “cut and patch” mechanism to excise the damaged or incorrect DNA strand and fill the resulting gap using the complementary DNA strand as a template [1]. Mechanistically, the overlapping and interacting nature of these pathways makes these repair systems highly complex. On the other hand, the pathway redundancy is important for effective DNA repair.
There is an integral connection between carcinogenesis and DNA repair. Maintenance of the integrity of the genome, which involves activity of DNA repair pathways, is essential for cell survival. Disruptions in these pathways can cause cells to accumulate DNA damage and, as a result, predispose them to mutations [2]. Mutations in genes that are involved in cell survival and growth, such as oncogenes and tumor-suppressor genes, can lead to development of cancer. Therefore, the molecular and genetic understanding of regulation of DNA repair pathways is important for the field of oncology [3]. One of the DNA repair proteins involved in the BER pathway, poly (ADP-ribose) polymerase (PARP), has been in the spotlight recently; PARP inhibitors, targeting DNA repair pathway defects, have shown significant promise in cancer treatment. We review existing preclinical and clinical studies of PARP inhibitors, focusing mainly on treatment of breast cancer along with their potential use in prevention of breast cancer.
Development of PARP Inhibitors
The current state of knowledge of PARP inhibitors is a result of over 40 years of research. PARPs are a family of enzymes involved in multiple cellular processes in addition to DNA repair; PARP1 is the best-characterized member and is one of the two DNA damage-activated nuclear PARPs [4]. It is comprised of three functional domains, including a DNA binding domain, an automodification domain, and a catalytic domain [5]. Following DNA damage, PARP1 is recruited and binds to the damaged DNA with a subsequent increase in catalytic activity that results in the formation of PARP using the substrate NAD+. These polymers are transferred to acceptor proteins and to PARP1 itself, which is important for recruitment of the BER machinery to the site of the DNA damage and relaxation of the chromatin structure to facilitate repair [6]. First-generation PARP inhibitors, such as 3AB, were simple analogs of nicotinamide and were shown to potentiate the effects of ionizing radiation and alkylating agents in both in vitro and in vivo studies [4]. This mechanism implicated a potential role of PARP inhibitors in the treatment of cancer, propelling the development of more potent and specific PARP inhibitors in the 1990s. Regarding treatment of cancer, two major strategies are being utilized for PARP inhibitors: 1) as sensitizers to DNA damaging chemotherapy or radiation, and 2) to exploit specific genetic alterations of certain cancers that leave them vulnerable to DNA damage, leading to cell death. The second treatment strategy is based on the principle of “chemical synthetic lethality” [7].
Preclinical Studies with PARP Inhibitors in Breast Cancer Models
In 2005, two pivotal articles suggested a novel application of PARP inhibitors in the treatment of cancer; BRCA1 and BRCA2 mutant cell lines, deficient in HR, were shown to be highly sensitive to PARP inhibitors as a result of this DNA repair defect [8••, 9••]. These studies suggest that deficiency in HR confers sensitivity to PARP inhibition, and this has been the premise of a novel treatment approach for patients with BRCA1- and BRCA2-deficient tumors. Although BRCA1 and BRCA2 proteins are best known for their important role in homologous recombination, BRCA1 has also been implicated as having additional roles in NER and BER [10, 11]. This suggests that DNA repair pathways other than HR may be responsible for conferring PARP inhibitor sensitivity as well.
Given the impressive preclinical results in BRCA1 or BRCA2 mutant cell lines, the immediate clinical application was to test the agents in the select group of BRCA1 and BRCA2 mutation carriers; however, they comprise only a minority of breast cancer cases [12, 13]. The “triple-negative” breast cancer (TNBC) subtype, lacking expression of estrogen and progesterone receptors, and lacking over-expression or amplification of the HER2/neu oncogene, represents approximately 10% to 15% of breast cancers and has an aggressive clinical course [14]. This subtype shares many pathologic and molecular features with BRCA1-associated breast cancers, including basal-like gene expression, high histologic grade, frequent p53 mutations, and increased genomic instability [15–17]. Preclinical work from our group shows that basal breast cancer cell lines, which include BRCA1 mutant and triple-negative breast tumors, but not luminal subtypes share defects in BER [11] and show increased sensitivity to PARP inhibition, cisplatin, and gemcitabine [18]. This supports the use of these chemotherapeutic agents in combination with PARP inhibitors in the clinical setting.
PARP Inhibitors for Treatment of Breast Cancer
Based on preclinical studies as described above, the majority of clinical studies in breast cancer have been confined to BRCA mutation-associated cancer and sporadic triple-negative breast cancer subtypes. The premise that HR defect, independent of hormone receptor-positive or negative phenotype, in BRCA-associated tumors confers PARP inhibitor sensitivity supports the rationale of including BRCA mutation-associated hormone receptor-positive breast cancers, as has been done in multiple ongoing clinical trials (Table 1).
Overview of Clinical Data
The current clinical trials with PARP inhibitors in breast cancer are being carried out in a variety of clinical settings including neoadjuvant, adjuvant, and metastatic (Table 1). PARP inhibitors currently in clinical investigation vary in multiple aspects including mechanism of action (reversible or irreversible inhibition), dosing intervals (continuous or intermittent), toxicities, and in combination with other chemotherapeutic agents (Tables 1 and 2). When clinical results are available from these ongoing studies, these factors will become important not only for interpretation of results, but also for further clinical development of a particular PARP inhibitor. The majority of clinical studies to date have been done with BSI-201 (now known as Iniparib [Sanofi-Aventis, Paris, France] and Olaparib [AstraZeneca, London, UK]). Other PARP inhibitors that are currently in clinical trial include ABT-888 (Veliparib [Abbott, Abbott Park, IL]) (Table 1), AG014699 (Pfizer, New York, NY) (Table 1), CEP-8983 (Cephalon, Frazer, PA), and MK-4827 (Merck, Ft. Washington, NJ).
BSI-201/Iniparib
Iniparib, an intravenous (IV) irreversible PARP inhibitor [5], dosed intermittently, has primarily been used in combination with gemcitabine and carboplatin in the clinical setting. Data with single agent BSI-201 in breast cancer are limited.
The first report of clinical results in treatment of sporadic triple-negative advanced breast cancer with BSI-201 was presented at the American Society of Clinical Oncology (ASCO) 2009 annual meeting by O’Shaughnessy et al. [19•]. In this randomized phase II trial, women with advanced breast cancer were treated with gemcitabine 1000 mg/m2 IV and carboplatin AUC of 2 IV on days 1 and 8, with or without the PARP inhibitor BSI-201 dosed at 5.6 mg/kg IV on days 1, 4, 8, and 11. A total of 123 women treated with up to two prior chemotherapy regimens for metastatic disease were enrolled. This study demonstrated improvements in the clinical benefit rate (21% vs 62%; P = 0.0002), overall response rate (16% vs 48%; P = 0.002), median progression-free survival (3.3 vs 6.9 months; hazard ratio [HR] = 0.342; P < 0.0001), and median overall survival (5.7 vs 9.2 months; HR = 0.348; P = 0.0005) among patients who received BSI-201. The improvement in overall survival with addition of BSI-201 was very impressive given that this was a metastatic breast cancer trial. At the 2009 San Antonio Breast Cancer Symposium, overall survival data from the same trial continued to show significant survival advantage among women treated with BSI-201 in an intention-to-treat analysis (7.7 vs 12.2 months; HR = 0.50; P = 0.005) [20]. Based on these notable results, this year, a randomized phase III registration study of gemcitabine and carboplatin, with or without BSI-201, was launched in a larger patient population with advanced breast cancer (n = 420) and has already completed accrual (Table 1). Our group is leading a phase II neoadjuvant triple-negative breast cancer study with gemcitabine, carboplatin, and BSI-201 in women with stage I-IIIA disease (Table 1).
Toxicities
In the randomized phase II trial with combination regimen of gemcitabine, carboplatin, and BSI-201 in patients with metastatic breast cancer, approximately 20% grade 3 or 4 hematologic toxicity was seen (Table 2). However, there was no difference in either hematologic or nonhematologic toxicity among patients who received BSI-201 compared with those who did not. Grade 3 or 4 nonhematologic toxicity was uncommon with this combination regimen with BSI-201 (Table 2).
Olaparib
Olaparib is an oral PARP inhibitor, dosed continuously, and has predominantly been used in the BRCA-mutation carrier population. Fong et al. [21••] reported a phase I dose escalation study of Olaparib in patients with advanced refractory solid tumors. This study was enriched with BRCA1 or BRCA2 mutation carriers, enrolling 22 patients with known BRCA1 or BRCA2 mutations out of a total of 60 patients. Only a small number of patients had breast cancer, including six with no BRCA mutations and three with documented BRCA2 mutations. No responses were observed in patients lacking known BRCA mutations. At the ASCO 2009 Annual Meeting, Tutt et al. [22] presented the first phase II results of olaparib for the treatment of women with BRCA1 and BRCA2 mutation-associated stage IIIB-IV breast cancer in which two sequential cohorts of 27 patients were enrolled at dose levels of 100 mg orally twice daily (po BID) and 400 mg po BID every 28 days, respectively. The objective response rate was 41% in the 400-mg po BID cohort and 22% in the 100-mg po BID cohort. About 50% of the patient population had triple-negative breast cancer and an additional 40% had hormone receptor-positive disease. This study clearly showed that the response to a PARP inhibitor is more dependent on germline mutations, such as BRCA1 or BRCA2, than the tumor’s phenotype, such as hormone receptor-positive or negative breast cancer. As stated above, most of the clinical studies have been done with single agent Olaparib to date. Ongoing studies include Olaparib in combination with carboplatin in patients with advanced BRCA1 or BRCA2 or sporadic triple-negative breast cancer (Table 1), and in combination with paclitaxel in patients with metastatic triple-negative breast cancer (Table 2).
Toxicities
Overall, Olaparib as a single agent has been fairly well tolerated, as highlighted in the two studies mentioned above by Fong et al. [21••] and Tutt et al. [22]. There were a few significant (grade 3 or 4) toxicities, including bone marrow suppression, nausea, fatigue, and dizziness (Table 2). When used in combination with paclitaxel in metastatic triple-negative breast cancer, neutropenia remains a significant toxicity even with addition of growth factors [23] (Table 2). This highlights the potential dose alterations that may be required, secondary to toxicities, when PARP inhibitors are combined with chemotherapeutics, and that there may be significant differences between different PARP inhibitors. Depending on the toxicity profile with combination agents, the choice of chemotherapeutics will likely be important as well.
Predictors of Response to PARP Inhibitors
A major area of needed investigation is to identify biological markers predictive of tumor sensitivity to PARP inhibitors. To date, pertinent to a minority of patients with breast cancer and other malignancies, BRCA mutations are predictive of response to PARP inhibitors. There are limited data with PTEN mutations [24] in vitro, but clinical studies with this patient population have not yet been undertaken.
Given that defects in DNA repair pathways are likely driving the sensitivity to PARP inhibitors, at least as single agents, the ultimate predictive biomarker would be a functional assay of DNA repair activity in the tumor. Such an assay could be applied to multiple malignancies and would be independent of specific underlying genetic or molecular defects. An important criterion for a functional assay is its ease of application to clinical samples on a large scale so it can be used for tissue samples acquired during clinical trials. Functional assays that specifically evaluate HR efficiency in vitro have been developed, but their application to fresh tumor samples may be difficult [25]. Our group has developed a cellular assay for BER activity that correlates with PARP sensitivity in triple-negative breast cancer cell lines using host-cell reactivation of a viral GFP reporter gene containing transcription-blocking oxidative DNA damage [11]. However, we have not yet successfully translated this to primary clinical samples. Several DNA damage response pathway genes are also being evaluated as biomarker candidates. γ- H2AX and RAD51 foci formation in response to DNA damage correlate with DNA strand breaks in in vitro studies [26, 27]; however, their clinical application may be limited. For a predictive biomarker in clinical trials, fresh tissue is usually collected prior to starting any drug treatment, but evaluation of these foci formation would require collection of tissue a few hours after delivery of DNA damaging agents, which is not practical for a large clinical trial. A novel assay to identify functional BRCA1 pathway defects using ex vivo irradiation of biopsied tissues appears promising as an assay that can profile individual tumors [28]; however, clinical application of this assay may again be quite challenging. PARP1 and PAR, a measure of PARP activity, have also been suggested as predictive biomarkers, but the data so far have been inconsistent and need further investigation.
Given the need for identifying robust biomarkers in tumors for PARP inhibitor sensitivity, ongoing clinical trials and their correlative studies including genomic analyses will hopefully identify gene expression signatures or underlying DNA alternations that will serve this need.
PARP Inhibitors for Prevention of Breast Cancer
A shift in focus on preventing the occurrence of breast cancer may ultimately have a much larger impact on reducing breast cancer mortality and morbidity. Successful chemoprevention agents such as selective estrogen receptor modulators (SERMs), specifically tamoxifen and raloxifene, reduce the incidence of hormone receptor-positive breast cancer in high-risk women, but these drugs do not prevent the development of hormone receptor-negative breast cancer [29–31]. In addition, for women identified to be at high risk due to a known cancer susceptibility gene mutation, such as BRCA1 or BRCA2, or strong family history of breast cancer, alternatives to surgical options for risk reduction are needed. Existing surgical options for this population include prophylactic bilateral mastectomy and bilateral salpingo-oophorectomy and provide significant risk reductions of 90% or more; however, studies have shown that majority of the mutation carriers would prefer an alternative prevention option [32].
Proposed Mechanism of Action of PARP Inhibitors for Breast Cancer Prevention
PARP inhibitors are unique in that they appear to selectively target malignant cells that have acquired at least one defect in a DNA repair pathway in a chemically “synthetic lethal” manner, and thus have little or no effect on normal DNA repair proficient cells, which is an important attribute for a potential chemoprevention agent. As a mechanism for cancer prevention for this class of agent, we propose that as premalignant cells undergo step-wise genetic alterations leading to breast carcinogenesis and acquire defects in DNA repair, they would be selectively eliminated by the PARP inhibitor, thus preventing the development of invasive breast cancer. In this scenario, inhibition of PARP may be able to delay the onset of or even prevent breast cancer entirely.
Preclinical and Clinical Studies of PARP Inhibitors in Breast Cancer Prevention
To date, there are no robust preclinical studies testing the efficacy of PARP inhibitors as a prevention agent. Testing of PARP inhibitors in a preclinical animal model of breast neoplasia would be an excellent way of elucidating the effects of this drug on targeted tissue (ie, breast or ovary), as well as normal tissue in a prevention setting. Genetically engineered mouse (GEM) models, specifically conditional mouse tumor models that mimic sporadic tumor onset and progression [33], similar to human disease, are ideal for testing such a chemoprevention agent.
PARP inhibitors have not yet been used clinically for cancer chemoprevention but are worth exploring for this indication. Women who may benefit from this novel class of chemoprevention agent include BRCA mutation carriers (estimated to be more than 300,000 in the United States alone) [34, 35] and breast cancer survivors with history of hormone receptor-negative breast cancer who are at a 10-fold elevated risk of developing a second hormone receptor-negative cancer [36]. Given the relatively short-term follow-up on safety and tolerability of PARP inhibitors, several questions need to be addressed before they can reach the level of acceptance of tamoxifen for long-term use. Clinical trials designed to prove that an agent prevents breast cancer require thousands of participants and multiple years of follow-up; the substantial cost of large, phase III chemoprevention trials has prompted investigators to initiate early stage, biomarker-based trials. These early stage trials of novel agents in breast cancer chemoprevention include the “presurgical window” in patients who have undergone a biopsy diagnosing breast cancer and are awaiting definitive surgery, primary prevention in patients at increased risk of developing breast cancer, and secondary prevention in breast cancer survivors. These trials evaluate breast cancer–associated biomarkers either directly in breast tissue, indirectly in serum, or by breast imaging. To test PARP inhibitor for breast cancer prevention, a randomized presurgical window trial has been proposed by Eastern Cooperative Oncology Group (ECOG), in which BRCA1 or BRCA2 mutation carriers will be treated with a PARP inhibitor for a short duration prior to prophylactic mastectomy. The main objective of this study will be to determine the effect of PARP inhibition on biomarker modulation in tissue and blood.
PARP Inhibition as a Prevention Agent: Challenges
Several general challenges for developing a chemoprevention agent and designing prevention trials exist. These include low tolerance of side effects from an agent to be administered to a high-risk population and the high cost of definitive trials powered to measure cancer incidence. In addition to these, development of PARP inhibitors in particular as prevention agents has unique challenges. PARP plays a role in DNA repair and maintenance of genomic integrity and one of the theoretical risks associated with long-term PARP inhibition might be increased mutation rate and cancer formation, particularly in BRCA mutation carriers. To date, in human clinical trials with PARP inhibitors, there has not been any increased cancer formation reported, but the duration of PARP inhibitor use and follow-up has been relatively short. Given that these prevention agents are being proposed for a high-risk population as opposed to a healthy population, the risks and benefits would have to be weighed accordingly. In terms of drug dosing for prevention, most agents have been tested with chronic daily dosing. An alternative dosing model may be pulse dosing, which may have a better tolerability and safety profile.
Conclusions
Significant results from preclinical studies and clinical trials highlight the potential promise that PARP inhibitors hold for treatment of a subset of breast cancer patients. These results have propelled PARP inhibitor development forward and several agents are actively in clinical trials. However, there is much that remains to be understood about the optimal use of these agents. Some of the challenges that we are facing today include identifying predictive biomarkers of response, which may be used for selecting a subset of patients who would benefit the most from treatment with this novel agent, determining optimal dose and schedule, and establishing efficacy as a single agent or in combination with other chemotherapeutics. Regarding use of these agents for prevention of breast cancer, there is lack of strong preclinical or clinical studies; however, based on the proposed mechanism as described earlier, PARP inhibitors have a great potential for reducing morbidity and mortality from breast cancer in a high-risk population. Long-term safety data need to be established for wide use of this agent in prevention setting. There are several ongoing clinical trials with strong translational components that should elucidate answers to some of these challenging questions. These trials will lead us to use PARP inhibitors optimally in a clinical setting, not only for treatment and prevention of breast cancer but also potentially for other malignancies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ford JM, Kastan MB: DNA damage response pathways and cancer. In Abeloff’s Clinical Oncology. Edited by Abeloff MD. Philadelphia: Churchill Livingstone; 2008:139–152.
Spry M, Scott T, Pierce H, D’Orazio JA: DNA repair pathways and hereditary cancer susceptibility syndromes. Front Biosci 2007, 12:4191–4207.
Rowe BP, Glazer PM: Emergence of rationally designed therapeutic strategies for breast cancer targeting DNA repair mechanisms. Breast Cancer Res 2010, 12:203.
Curtin NJ: PARP inhibitors for cancer therapy. Expert Rev Mol Med 2005, 7:1–20.
Rouleau M, Patel A, Hendzel MJ, et al.: PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010, 10:293–301.
Durkacz BW, Omidiji O, Gray DA, Shall S: (ADP-ribose) participates in DNA excision repair. Nature 1980, 283:593–596.
Simons A, Dafni N, Dotan I, et al.: Establishment of a chemical synthetic lethality screen in cultured human cells. Genome Res 2001, 11:266–273.
•• Bryant H, Schultz N, Thomas H, et al.: Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434:913–917. A pivotal preclinical study showing that BRCA2 deficient cells, as a result of homologous recombination deficiency, are very sensitive to PARP inhibitors.
•• Farmer H, McCabe N, Lord CJ, et al.: Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434:917–921. A pivotal preclinical study showing that BRCA1 or BRCA2 deficient cells, as a result of homologous recombination deficiency, are profoundly sensitive to PARP inhibitors, leading to apoptosis.
Hartman AR, Ford JM: BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nat Genet 2002, 32:180–184.
Alli E, Sharma VB, Sunderesakumar P, Ford JM: Defective repair of oxidative dna damage in triple-negative breast cancer confers sensitivity to inhibition of poly(ADP-ribose) polymerase. Cancer Res 2009, 69:3589–3596.
Narod S: Modifiers of risk of hereditary breast and ovarian cancer. Nat Rev Cancer 2002, 2:113–123.
Narod S, Foulkes W: BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 2004, 4:665–676.
Carey LA, Perou CM, Livasy CA, et al.: Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295:2492–2502.
Sorlie T, Tibshirani R, Parker J, et al.: Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A 2003, 100:8418–8423.
Cleator S, Heller W, Coombes RC: Triple-negative breast cancer: therapeutic options. Lancet Oncol 2007, 8:235–244.
Schneider BP, Winer EP, Foulkes WD, et al.: Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res 2008, 14:8010–8018.
Hastak K, Alli E, Ford JM.: Synergistic chemosensitivity of triple-negative breast cancer cell lines to PARP inhibition, gemcitabine and cisplatin. Cancer Res 2010 Aug 26 [Epub ahead of print].
• O’ Shaughnessy J, Osborne C, Pippen J, et al.: Efficacy of BSI-201, a poly (ADP-ribose) polymerase-1 (PARP1) inhibitor, in combination with gemcitabine/carboplatin (G/C) in patients with metastatic triple-negative breast cancer (TNBC): results of a randomized phase II trial [abstract 3]. Presented at 2009 American Society of Clinical Oncology Meeting. Orlando, FL; May 29 to June 2, 2009. This is the first report of clinical results from a phase II randomized trial with BSI-201 for treatment of sporadic triple-negative advanced breast cancer.
O’Shaughnessy J, Osborne C, Pippen J, et al.: Final Results of a Randomized Phase II Study Demonstrating Efficacy and Safety of BSI-201, a Poly (ADP-Ribose) Polymerase (PARP) Inhibitor, in Combination with Gemcitabine/Carboplatin (G/C) in Metastatic Triple Negative Breast Cancer (TNBC) [abstract 3122]. Presented at 32nd San Antonio Breast Cancer Symposium. San Antonio, TX; December 9–13, 2009.
•• Fong PC, Boss DS, Yap TA, et al.: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009, 361:123–134. This is the first phase I dose escalation study of olaparib in patients with advanced refractory solid tumors with patient population enriched for BRCA1 and BRCA2 mutation carriers.
Tutt A, Robson M, Garber JE, et al.: Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer [abstract CRA501]. Presented at 2009 American Society of Clinical Oncology Meeting. Orlando, FL; May 29 to June 2, 2009.
Dent R, Lindeman G, Clemons M, et al.: Safety and efficacy of the oral PARP inhibitor olaparib (AZD2281) in combination with paclitaxel for the first-or second-line treatment of patients with metastatic triple-negative breast cancer: Results from the safety cohort of a phase I/II multicenter trial [abstract 1018]. Presented at 2010 American Society of Clinical Oncology Meeting. Chicago, IL; June 4–8, 2010.
Mendes-Pereira AM, Martin SA, Brough R, et al.: Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med 2009, 1:315–322.
Weinstock DM, Nakanishi K, Helgadottir HR, Jasin M: Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol 2006, 409:524–540.
Bonner WM, Redon CE, Dickey JS, et al.: GammaH2AX and cancer. Nat Rev Cancer 2008, 8:957–967.
Banuelos CA, Banath JP, Kim JY, et al.: gammaH2AX expression in tumors exposed to cisplatin and fractionated irradiation. Clin Cancer Res 2009, 15:3344–3353.
Willers H, Taghian AG, Luo CM, et al.: Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol Cancer Res 2009, 7:1304–1309.
Li Y, Brown PH: Translational approaches for the prevention of estrogen receptor-negative breast cancer. Eur J Cancer Prev 2007, 16:203–215.
Vogel VG, Costantino JP, Wickerham DL, et al.: Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA 2006, 295:2727–2741.
Fisher B, Costantino JP, Wickerham DL, et al.: Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998, 90:1371–1388.
Metcalfe KA, Birenbaum-Carmeli D, Lubinski J, et al.: International variation in rates of uptake of preventive options in BRCA1 and BRCA2 mutation carriers. Int J Cancer 2008, 122:2017–2022.
Jonkers J, Berns A: Conditional mouse models of sporadic cancer. Nat Rev Cancer 2002, 2:251–265.
Whittemore AS, Gong G, John EM, et al.: Prevalence of BRCA1 mutation carriers among U.S. non-Hispanic Whites. Cancer Epidemiol Biomarkers Prev 2004, 13:2078–2083.
John EM, Miron A, Gong G, et al.: Prevalence of pathogenic BRCA1 mutation carriers in 5 US racial/ethnic groups. JAMA 2007, 298:2869–2876.
Kurian AW, McClure LA, John EM, et al.: Second primary breast cancer occurrence according to hormone receptor status. J Natl Cancer Inst 2009, 101:1058–1065.
Acknowledgment
James M. Ford is supported by research grants from the Breast Cancer Research Foundation and the Susan G. Komen for the Cure Foundation. Shaveta Vinayak is supported by research grants from the ASCO Cancer Foundation.
Disclosure
JMF has been a consultant for Lead Pharmaceuticals/BioMarin. He has received funding for a clinical trial from Sanofi-Aventis and an honorarium from the American Society for Clinical Oncology for an educational talk at the 2010 meeting.
SV reports no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vinayak, S., Ford, J.M. PARP Inhibitors for the Treatment and Prevention of Breast Cancer. Curr Breast Cancer Rep 2, 190–197 (2010). https://doi.org/10.1007/s12609-010-0026-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12609-010-0026-0