
Abstract
To investigate the differences in gut bacterial community of Parabramis pekinensis at different growth stages, we collected wild P. pekinensis from the Jingjiang region of the Yangtze River, and detected the intestinal microflora structure using high-throughput sequencing technology. Results show that during stage I the dominant bacteria were Proteobacteria, Actinobacteria, and Firmicutes. During stage II, the proportion of Proteobacteria and Actinobacteria decreased, while the proportion of Firmicutes and Fusobacteria increased, especially Clostridium and Cetobacteria increased significantly. During stage III, Cetobacterium had a dominant position, while the proportion of Firmicutes decreased slightly. In stage IV, the male and female fish showed obvious differences. In the female gut, the proportion of Proteobacteria increased to the first place, while Fusobacteria decreased to the second place. In the male fish, the proportion of Fusobacteria dropped to the fifth, especially that of Cetobacterium decreased significantly, and that of Verrucomicrobia increased. In stage V, the proportion of Fusobacteria increased again to the first place, while Proteobacteria did not decrease significantly in the female gut. The gut bacterial community in males changed into a structure similar to stage I. In stage VI, the gut bacterial community in both females and males changed into a structure similar to stage I. There were significant differences in the intestinal microflora structure of P. pekinensis at different gonad development stages and sexes. To some extent, the changes in intestinal microflora structure reflect the changes in the nutritional requirements of P. pekinensis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, more attention has been paid to the role of intestinal flora in the physiological aspects of host feeding and nutrition. Owing to the breakthroughs in sequencing technology and the development of bioinformatics, research on intestinal flora has become very popular in recent years. This has greatly improved our understanding of the role of intestinal flora in host nutrition, metabolism, immunity, and many other physiological and biochemical functions (Parma et al. 2016; Ringø et al. 2016; Zarkasi et al. 2014). The intestinal flora plays an important role in fish nutrition (Carla et al. 2019). For example, the intestinal flora of Cyprinus carpio is involved in the synthesis of vitamin B1, vitamin B12, niacin, pantothenic acid, and biotin. There are multiple chitinolytic bacteria present in the digestive tract of fish that feed on crustaceans. There are also multiple xylan-decomposing bacteria feeding on algae present in the intestinal tract of fish. Additionally, the extracellular enzymes of Aeromonas hydrophila promote starch and protein digestion (Kashiwada et al. 1970). Inhibition of antibiotics can significantly reduce carboxymethyl cellulase activity in the intestinal tract of Ctenopharyngodon idella (Das and Tripathi 1991). Interestingly, fish with different diets also have different dominant intestinal flora. Clostridium, Citrobacter, and Leptothrix are closely related to cellulose digestion and are found in high abundance in the gut of herbivorous fish. Cetobacterium and Halomonas are related to protease production and are common in the intestinal tract of carnivorous fish (Liu et al. 2016). Abundance and diversity of intestinal flora showed an increasing trend in the order: carnivorous, omnivorous, and herbivorous (Lin et al. 2014; Larsen et al. 2014; Sou et al. 2015; Ward et al. 2009).
Growth stage is one of the main factors affecting the intestinal flora structure of fish. Studies on the relationship between fish growth stage and intestinal flora have mainly focused on the source and colonization of flora, and the characteristics of flora in the early development stage (Bakke et al. 2013). The intestinal flora of fish mainly originates from the fertilized eggs and the aquatic environment where they hatch, and then is established and gradually stabilized in the first feeding stage (Romero and Navarrete 2006). The structure of the intestinal flora is different at different growth stages, and the diversity of intestinal flora increases significantly with the development of fish (Parris et al 2016). Gonadal development is an important stage in the fish life cycle and is the basis of species reproduction. Gonadal development is affected by nutrition, water temperature, water flow stimulation, light, and many other conditions. Of these, the key condition for ensuring the development of gonads to maturity is the intake and accumulation of nutrients. In the middle and late stages of gonad development, fish need to store a large amount of nutrients in order to meet the reproduction requirements. Intestinal flora can promote the absorption of nutrients by the host, especially in the metabolism and transport of cholesterol (Rawls et al. 2004), and also stimulate the absorption of fatty acids in the intestinal epithelium, promoting the accumulation of lipid droplets (Semova et al. 2012). However, so far, there have been few studies on the structure of fish intestinal flora during gonad development.
Parabramis pekinensis is a species of Cyprinidae that is widely distributed throughout many major water systems in China. In recent years, human factors such as overfishing, habitat destruction, and construction of wading projects have led to a sharp decline in the population's resources. However, there are still few studies on the biological characteristics of P. pekinensis, such as its nutritional requirements and feeding characteristics, and its artificial propagation technology is still immature. Unlike other common herbivorous fishes, P. pekinensis does not stop feeding during reproduction; rather it tends to increase its food intake, which is of positive significance for the study of reinforcement breeding techniques for its parents. In this study, we analyzed the differences in the intestinal microbiota of P. pekinensis at different stages of gonad development by high-throughput sequencing and explored the nutritional requirements of P. pekinensis at different stages.
Materials and methods
Sampling and experimental design
Samples were collected from the wild in the Jingjiang section of the Yangtze River. The central sampling location is labeled with a "W" in Fig. 1. Samples were collected at three different sampling sites, all within 1 km of the central site. The average pH value of the water is 7.7 ± 0.16, and the average dissolved oxygen is 10.27 ± 0.35 mg/l. Based on the gonad development of P. pekinensis in natural environment, the gonad stages I and VI of P. pekinensis were mainly collected during August, and the remaining stages were mainly collected from April to June. See Table 1 for the specific water temperature during sample collection. The samples were stored on ice and sent to the laboratory.

The sampling point (★), Taizhou, Jiangsu Province, China
The 1- to 2-year-old fish without lesions or disease were selected as experimental subjects, and more than three batches of fish at the same gonad development stage were retained. The age of fish is judged by their scales (Lv et al. 2018). After measuring the body length, total length, and body weight of the experimental fish, the gonadal development stage was identified based on anatomy. The stages of gonadal development are divided into I-VI (Liu Y., 1993). The inclusions of the same sex and stage of development were mixed into the sample to be tested. The experiment was set up in three parallel directions, each containing 15 fish. See Table 1 for specific sample information. Intestinal flora was collected in an aseptic environment. The fish were placed on ice, the abdomen was sterilized using 75% ethanol and then cut, the intestines were straightened, and the intestinal contents were rinsed with sterile physiological saline and washed into sterile tubes. Intestinal inclusions collected from the same group of fish body samples were mixed into a sample for testing. Finally, the samples were labeled and stored at - 80 °C until DNA extraction.
DNA extraction, amplicon library preparation, and sequencing
Total DNA content was extracted from the intestinal samples using the QIAamp DNA Stool Mini Kit (Qiagen, Germantown, MD, USA) following the manufacturer’s protocol. Next, a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) was used to determine the concentration and purity of the extracted DNA. Polymerase chain reaction (PCR) was performed to amplify the V3-V4 region of bacterial 16S rRNA genes using the following primers: 341F, 5′-CCTAYGGGRBGCASCAG-3′; 806R, 5′-GGACTACNNGGGTATCTAAT-3′ (Hjort et al. 2014). The PCR products were visualized, purified (AxyPrep DNA Gel Extraction Kit, Axygen Biosciences, Union City, CA, USA), quantified (QuantiFluor™-ST, Promega, USA), and homogenized to form a DNA pool. Finally, the pooled products were sent to Novogene Co., Ltd. (Beijing, China) for paired-end sequencing using an Illumina HiSeq 2500 platform, following standard protocols. The sequencing data was deposited in the NCBI short read archive with the accession number PRJNA706781.
16S rRNA gene sequence and statistical analysis
After revealing the overlapping relationship between paired-end reads (Gregory et al. 2010), FLASH v1.2.7, Trimmomatic v0.33, and UCHIME v4.2 software were used to conduct quality filtering of the reads and the stitching effect, and finally, effective tags were obtained. The deblur (Amnon et al. 2017) method recommended by Qiime2 (Evan et al. 2019) was used for denoising analysis of sequences and generating feature tables. Species annotation was performed using the Silva 132 database (Release132, http://www.arb-silva.de) (Christian et al. 2013; Pelin et al. 2014). Alpha diversity index analysis (http://www.mothur.org/) included Chao1 (considering only the number of species) and Shannon (considering both the number of species and the abundance of each species), and t test in R language was used for significant difference analysis. β-diversity analysis was performed the sample principal coordinate analysis (PCoA). Species with significant differences in the abundance changes between groups and determined the significance of differences in community structure among different groups were analyzed by Linear discriminant analysis Effect Size (LEfSe) (Nicola et al. 2011).
Results
Amplicon sequence variants (ASV) analysis results
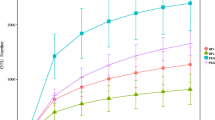
A total of 3,260,835 effective reads were detected in all samples, including 1,832,185 reads in the female intestinal flora and 1,428,650 reads in the male intestinal flora. A total of 2,023,868 high-quality reads were obtained through quality control and filtration, including 1,134,994 from the female intestinal flora and 888,874 from the male intestinal flora. After denoising and chimerization of tags, 753 ASVs were obtained in the female intestinal flora and 1156 ASVs in the male intestinal flora. After filtering the ASVs with abundance < 1‰ in the characteristic table, the common number of ASVs in the same sex at different stages of gonad development was analyzed (Fig. 2). A total of 85 ASVs were obtained from females, among which 59 ASVs were obtained from females at all stages, and the residual ASVs are unique to each gonadal development stage. A total of 119 ASVs were obtained from males, among which 32 ASVs were obtained at all stages, and 34 ASVs were obtained at stage I to V, while the stage VI own four unique ASVs. The rarefaction and Shannon curves indicated that the sequencing depth was sufficient (Online Resource 1), and the cumulative species curve indicated that the sample size was sufficient (Online Resource 2).

The common number of ASVs at different stages of gonadal development. The values on the picture are the number of ASVs contained in the sample. The number of common and unique ASVs between samples was displayed, and the coincidence of ASVs between samples was intuitively shown
General characteristics of intestinal microbiota
A total of 23 phyla and 348 genera were identified in all samples. The dominant flora were Proteobacteria (45.68%), Fusobacteria (20.75%), Firmicutes (13.86%), Actinobacteria (6.1%), and Chloroflexi (6.07%), accounting for 92.46% of the total bacteria. At the genus level, the dominant bacteria were Cetobacterium (20.72%), Klebsiella (20.02%), Rhizobiales, (4.65%), Rhodobacteraceae, (3.2%), and Clostridium (2.77%), which accounted for 51.36% of the total flora.
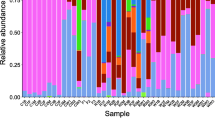
A total of 17 phyla were identified in the female samples, and 22 phyla were identified in the male samples (Fig. 3). At each gonadal development stage, the dominant bacterial species did not change significantly, and included Proteobacteria, Firmicutes, Actinobacteria, Fusobacteria, Chloroflexi, Verrucomicrobia, and Bacteroidetes. The lower level was primarily composed of several groups under Cetobacterium and Gammaproteobacteria (Online Resource 3).

Intestinal microflora analysis results of female (bar chart on the left) and male (bar chart the right) P. pekinensis (top 15 bacteria at the phylum level)
Difference of intestinal microflora in different gonadal development stages of P. pekinensis
The α-diversity and β-diversity indices of the intestinal inclusions of females and males were analyzed, respectively. The results showed that there were differences in the intestinal microflora structure between males and females at different stages of gonad development. There was a significant difference in the number of bacterial species between stages II and III in the microflora of the female intestinal tract (Fig. 4a, P = 0.023). There were significant differences in bacterial diversity between stage I and II, and between stage II and stage III (Fig. 4b, P = 0.015; P = 0.014), and the differences of bacterial diversity between stage II and VI were significant (Fig. 4b, P = 0.009). In terms of the microflora of the P. pekinensis intestinal tract, the species numbers of stage I and stage IV were significantly different (Fig. 4c, P = 0.023), and the species numbers of stage IV and stage VI were significantly different (Fig. 4c, P = 0.002). The diversity of bacteria in stage IV and VI was significantly different (Fig. 4d, P = 0.0003). PCoA also showed significant separation between the groups (Fig. 5a, female; Fig. 5b, male), which indicated that there were differences in the microbiota of P. pekinensis intestines between males and females at different gonad development stages.

Difference box chart; According to Chao1 and Shannon index analysis, a and b were female, while c and d were male (three mixed samples, statistically significant differences are indicated, P < 0.05)

PCoA of intestinal flora at every stage of gonadal development; a is the female, b is the male (analysis based on bray Curtis algorithm, the closer the samples were, the more similar the species composition was)
LEfSe (log10 ≥ 2.0) analysis showed that the specific microflora of stage I gonad development in female P. pekinensis were mainly Lactobacillales and Acidobacteriota (Fig. 6). The specific flora of stage II were Desulfobulbales, Acidimicrobiia, Gammaproteobacteria, and Acidobacteria. The specific flora of stage IV were Bdellovibrionota, Gammaproteobacteria, and Synechococcales. The specific flora of stage VI were Bacillales, Pseudomonadales. There was no significant difference in flora between stage III and V. The intestinal specific microflora of the stage I gonads in male P. pekinensis were Pseudomonadales, Proteobacteria, and Cyanobacteria (Fig. 7). The specific flora of stage II were Clostridia, α-proteobacteria, and Erysipelotrichales. The specific flora of stage III were Gammaproteobacteria and Fusobacteriales. The specific flora of stage IV were Planctomycetes, Verrucomicrobiae, and α-proteobacteria. The specific flora of stage V were Cytophagales, α-proteobacteria, Micrococcales, Acidimicrobiia, Acidobacteriota, and Verrucomicrobiae. The specific flora of stage VI were Nitrospirales, Clostridia, Gammaproteobacteria, Actinomycetales, Chloroflexales and Lactobacillales.

Analysis of different species of intestinal flora at different stages of gonadal development (for female), significantly discriminative taxa with absolute LDA score ≥ 2.0

Analysis of different species of intestinal flora at different stages of gonadal development (for male), significantly discriminative taxa with absolute LDA score ≥ 2.0
Discussion
The intestinal microbiota structure of P. pekinensis
Our study showed that Proteobacteria, Fusobacteria, Firmicutes, and Actinobacteria were the dominant microflora in P. pekinensis, which is typical of herbivorous fishes (Wu et al 2012; Ni et al 2014). However, typical bacterial species in the intestinal tract of omnivorous fishes (Kessel et al. 2011; Wang et al 2018), such as Chloroflexi, Verrucomicrobia, and Bacteroidetes were second only to the four microflora mentioned above, and the combined proportion of these three microflora was 10.86%, similar to the tendency of omnivores. In addition, the highest abundance (20.72%) of Cetobacterium, a typical bacterial species in the intestinal tract of carnivorous fish, also indicates a tendency to demand protein and fat (Yukgehnaish et al. 2020).
Hailong et al. (2021) studied the differences in the intestinal microflora of P. pekinensis among the Jingjiang section of the Yangtze River, the suburban rivers of Jingjiang, and circulating aquaculture. The results showed that 13 phyla and 201 genera were identified from the specimens of P. pekinensis intestines, which were dominated by Fusobacteria (39%), Firmicutes (29.79%), Proteobacteria (13.68%), Actinobacteria (12.05%), and Cyanobacteria (3.13%), accounting for 97.65% of the total bacteria. This is essentially the same as the results of this study. The reason for the difference in the dominant bacteria proportion in the two experiments may be the individual differences in age, size, and sex (Carla et al. 2019; Jiang et al. 2020). The experimental fish selected in the present study included female fish and male fish from 1 to 2 years old, while only the 2-year-old female fish were selected by Hailong et al. (2021), including the cultured population. In addition, it is possible that the analysis methods were different. In this study, the Amplicon Sequence Variants (ASV) method, rather than the operational taxonomic units (OTU) method, was used to analyze and annotate the sequences. ASVs are also commonly used for the analysis of microbial communities (Evan et al. 2019; Knight et al. 2018). Compared with OTU clustering, they both have advantages and disadvantages. The OTU clustering method can effectively overcome sequencing errors (i.e., discarding some sequencing errors by selecting representative sequences), but this method reduces the accuracy of classification, and some sequences below the set threshold cannot be accurately distinguished. ASV does not cluster sequences based on the distance threshold, which is equivalent to clustering at the level of 100% similarity, but at the same time increases the risk of single genomes splitting into separate clusters (Schloss 2021). Overall, compared with the OTU method, tags are merged and divided at a 97% similarity level, and the ASV method has a more comprehensive analysis, so more categories are identified.
It is worth noting that although all the fish collected in this experiment come from the water within 1 km and their living environment is basically the same, there are significant differences in water temperature due to different collection periods. This cannot be completely avoided in this experiment. We must follow the natural development of the P. pekinensis in the wild. However, temperature may also be a factor in the structural changes of P. pekinensis intestinal microflora, but this does not affect the purpose of this study. Many experiments have shown that water will not change the structure of the inherent intestinal flora of healthy fish, let alone the feeding characteristics of fish (Nayak 2010; Wang et al. 2018; Yukgehnaish et al. 2020). We have concentrated the collection period as much as possible to reduce the temperature difference. At the same time, we mixed the samples to eliminate individual differences. The objective of our experiment was to understand the structural changes of intestinal microflora of fish at different gonad development stages, and to infer the characteristics of the nutritional requirements of P. pekinensis.
Characteristics of intestinal flora at different stages of P. pekinensis gonadal development and speculation of their nutritional needs
The nutritional requirements of fish are closely related to their developmental stage, and the requirement for nutrients in late gonad development is much higher than that in the growth stage (Huang et al. 2009). The fertility and quality of sperm and ovum of parent fish can be directly affected by the nutritional storage of the parent fish. For example, feeding on a low-protein and high-fat diet can reduce the reproductive performance of Oncorhynchus mykiss (Watanabe et al. 1984). Dietary balance of essential amino acids can promote the synthesis of vitelloprotein in Sparus aurata (Tandler, et al. 1995). The egg viability of parent sea bass decreases with a decrease in dietary protein level (Joan, et al. 1994). The fecundity of Xiphophorus helleri parents also decreases with a decrease in dietary protein level (Chong, et al. 2004). Conversely, fish nutrient storage is related to the food they eat along with depending on the size of their digestive capacity. Many studies have confirmed the important function of intestinal flora in food digestion and nutrient absorption in fish. It is generally believed that the impact of fish intestinal flora on the host nutrition is via flora metabolism, which works in cooperation with the host itself. When the host cannot effectively digest substances itself, such as cellulose, aliens, biomass, etc., this material can instead become the energy source of intestinal flora, as it can effectively break down these substances. The resulting metabolites happen to be a digestible energy source for the host fish. The ability of herbivorous fish to digest cellulose is highly dependent on the help of intestinal flora (Kessel et al. 2011), and Pseudomonas fluorescens and Pseudomonas putida are the main decomposition forces of heteromorphic biomass (Austin et al., 1995). Therefore, it is very important to analyze the structure of intestinal flora in the study of fish nutrition, especially the feeding characteristics of herbivorous and omnivorous fish.
In stage I, Proteobacteria, Actinobacteria, Firmicutes, etc., were the dominant bacteria in the intestinal tracts of both female and male P. pekinensis, suggesting that the early stage of development of P. pekinensis may be mainly herbivorous. During stage II, the proportion of Proteobacteria decreased significantly, from 73.09 to 38.85% in females and from 52.09 to 20.73% in males. The proportion of Actinobacteria decreased while that of Firmicutes and Fusobacteria increased, especially Clostridium and Cetobacterium, indicating that P. pekinensis maybe no longer a typical herbivorous fish. They initially showed the need for fat and protein. Clostridium is also a common flora in fish intestines, and plays an important role in the synthesis of propionate, short-chain fatty acids, and butyrate (Eichmiller et al. 2016). In stage III, the highest proportion of the intestinal tract changed into Fusobacteria Cetobacterium (in females, this accounted for 68.32% and in males it accounted for 56.39%). Proteobacteria once again became the second-most dominant community, which may indicate that the need for protein and fiber of P. pekinensis are equally important during the rapid growth phase, while the decrease in Firmicutes indicates a slight decrease in the need for fat. At stage IV, Proteobacteria again became the most dominant flora, with Fusobacteria Cetobacterium ranking second in the female intestine and significantly decreasing in the male intestine (from 56.39 to 2.74%). This indicates that there are differences between male and female feeding habits at this stage. To ensure a reserve of nutrients, as well as reproductive performance, female fish require a high protein intake. Meanwhile the male fish turn from herbivores towards omnivores (Verrucomicrobia significantly increased). During stage V, Fusobacteria in female P. pekinensis again ranked first (43.45%), followed by Proteobacteria (37.9%), followed by Firmicutes (12.88%). This period was the breeding period for female fish, we found that their stomachs and intestines were always full through dissection. So, we hypothesized that female fish do not stop eating during reproduction, and their diet is still dominated by high-protein, cellulose, and fat-rich feed. The intestinal flora distribution of male fish was uniform at this stage; Proteobacteria accounted for the highest proportion, followed by Firmicutes, Chloroflexi, etc. Their stomachs and intestines were also mostly empty, indicating that there is no specific demand for food, which may also indicate a reduction in food intake during reproduction. In stage VI, the largest proportion of intestinal bacteria in male and female fishes changed into Proteobacteria again (78.55 and 67.94%, respectively), followed by Firmicutes and Actinobacteria, indicating that P. pekinensis at this stage had gradually changed into typical herbivorous fish.
In addition, LEfSe analysis found that Gammaproteobacteria appeared frequently as a specific strain in the middle and late gonad development, which may be caused by high chemical oil pollution in water. Scholars also found this phenomenon in the intestinal flora of southern flatfish (Yukgehnaish et al. 2020). Our sampling site is home to several shipyards, chemical plants, and a large gas station for ships, resulting in a certain amount of oil pollution in the water.
Admittedly, there are hundreds of species of bacteria in each phylum, and not all of them perform a single physiological function in the host. Therefore, the present study only discussed the nutritional preferences or general trends in the development of P. pekinensis at each stage from a macroscopic perspective, and provided a reference for the development of feeds for P. pekinensis. Their specific dietary requirements need to be verified further.
Availability of data and materials
The sequencing data were deposited in the NCBI Short Read Archive (SRA) with the accession number PRJNA706781.
References
Amnon A, Daniel M, Jose AN-M et al (2017) Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2(2):e00191-16
Austin B, Stuckey FL, Robertson PAW et al (1995) A probiotic strain of Vibrio alginolyticus effective in reducing diseases caused by Aeromonas salmonicida, Vibrio anguillarum and Vibrio ordalii. J Fish Dis 18(1):93–96
Bakke I, Skjermo J, Vo TA, Vadstein O (2013) Live feed is not a major determinant of the microbiota associated with cod larvae (Gadus morhua). Environ Microbiol Report 5(4):537–548
Carla PM, Fernando N-C, Paula S-M et al (2019) Sex, age, and bacteria: how the intestinal microbiota is modulated in a protandrous hermaphrodite fish. Front Microbiol 10:2512
Chong A, Ishak SD, Osman Z, Hashim R (2004) Effect of dietary protein level on the reproductive performance of female swordtails Xiphophorus helleri (Poeciliidae). Aquaculture 234(1–4):381–392
Christian Q, Elmar P, Pelin Y et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(D1):D590–D596
Das KM, Tripathi SD (1991) Studies on the digestive enzymes of grass carp, Ctenopharyngodon idella (Val.). Aquaculture 92:21–32
Eichmiller JJ, Hamilton MJ, Staley C et al (2016) Environment shapes the fecal microbiome of invasive carp species. Microbiome 4(1):44
Evan B, Ram RJ, Matthew RD et al (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37(8):852–857
Gregory CJ, Justin K, Jesse S et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336
Hailong G, Yaming F, Ya Z et al (2021) Differential study of the Parabramis pekinensis intestinal microbiota according to different habitats and different parts of the intestine. Annal Microbiol 71(1):1–11
Hjort HM, Hestbjerg HL, Jacob B et al (2014) High-resolution melt analysis for rapid comparison of bacterial community compositions. Appl Environ Microbiol 80(12):3568–3575
Huang X, Shi Z, Li W et al (2009) The aminoacids in different tissues of silver pomfret (Pampus argenteus) broodstock and their change with the gonad development. J Fish China 33(02):278–287 (in Chinese)
Jiang M, Xu M, Ying C et al (2020) The intestinal microbiota of lake anchovy varies according to sex, body size, and local habitat in Taihu lake. China Microbiologyopen 9(1):e00955
Joan C, Manuel C, Silvia Z et al (1994) Influence of nutritional composition of diet on sea bass, Dicentrarchus labrax L., reproductive performance and egg and larval quality. Aquaculture 128(3–4):345–361
Kashiwada K, Teshima S, Kanazawa A (1970) Studies on the production of B vitamins by intestinal bacteria of Fish-V evidence of the production of vitamin B12 by microorganisms in the intestinal canal of carp. Cyprinus Carpio Nippon Suisan Gakkaishi 36(4):421–424
Kessel MA, Dutilh BE, Neveling K et al (2011) Pyrosequencing of 16S rRNA gene amplicons to study the microbiota in the gastrointestinal tract of carp (Cyprinus carpio L.). AMB Express 1:41
Knight R, Vrbanac A, Taylor BC et al (2018) Best practices for analysing microbiomes. Nat Rev Microbiol 16(7):410–422
Larsen AM, Mohammed HH, Arias CR (2014) Characterization of the gut microbiota of three commercially valuable warmwater fish species. J Appl Microbiol 116(6):1396–1404
Lin Y, Jon A, Duane C et al (2014) Fish gut microbiota analysis differentiates physiology and behavior of invasive Asian carp and indigenous American fish. ISME J 8(3):541–551
Liu Y (1993) Reproduction physiology of Chinese cultured fish. Agriculture Press, Beijing (in Chinese)
Liu H, Guo X, Gooneratne R et al (2016) The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci Rep 6(1):24340
Lv D, Zhou Y, Ge Y et al (2018) Age structure and growth characteristics of Culter Ilishaeformis in Dianshan Lake. Acta Hydrobiol Sin 42(4):762–769 (in Chinese)
Nayak SK (2010) Role of gastrointestinal microbiota in fish. Aquac Res 41(11):1553–1573
Ni J, Yan Q, Yu Y, Zhang T (2014) Factors influencing the grass carp gut microbiome and its effect on metabolism. FEMS Microbiol Ecol 87:704–714
Nicola S, Jacques I, Levi W et al (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12(6):R60
Parma L, Candela M, Soverini M et al (2016) Next-generation sequencing characterization of the gut bacterial community of gilthead sea bream (Sparus aurata, L.) fed low fishmeal based diets with increasing soybean meal levels. Anim Feed Sci Technol 222:204–216
Parris DJ, Brooker RM, Morgan MA et al (2016) Whole gut microbiome composition of damselfish and cardinalfish before and after reef settlement. PeerJ 4:e2412
Pelin Y, Wegener PL, Pablo Y et al (2014) The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res 42:D643–D648
Rawls J, Samuel B, Gordon J (2004) Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc Natl Acad Sci 101(13):4596–4601
Ringø E, Zhou Z, Vecino JG et al (2016) Effect of dietary components on the gut microbiota of aquatic animals. A never-ending story? Aquac Nutr 22(2):219–282
Romero J, Navarrete P (2006) 16S rDNA-based analysis of dominant bacterial populations associated with early life stages of Coho Salmon (Oncorhynchus kisutch). Microb Ecol 51(4):422–430
Schloss PD (2021) Amplicon sequence variants artificially split bacterial genomes into separate clusters. mSphere 6(4):e0019121
Semova I, Carten JD et al (2012) Microbiota regulate intestinal absorption and metabolism of fatty acids in the Zebrafish. Cell Host Microbe 12(3):277–288
Sou M, Kamanda ND, Ulrich S (2015) Diet strongly influences the gut microbiota of surgeonfishes. Mol Ecol 24(3):656–672
Tandler A, Harel M, Koven WM, Kolkovski S (1995) Broodstock and larvae nutrition in gilthead seabream Sparus aurata-new findings on its mode of involvement in improving growth, survival and swimbladder inflation. Israeli J Aquac-Bamidgeh 47(3):95–111
Wang AR, Ran C, Ringø E, Zhou ZG (2018) Progress in fish gastrointestinal microbiota research. Rev Aquac 10(3):626–640
Ward NL, Steven B, Penn K et al (2009) Characterization of the intestinal microbiota of two Antarctic notothenioid fish species. Extremophiles 13(4):679–685
Watanabe T, Takeuchi T, Saito M, Nishimura K (1984) Effect of low protein-high calory of essential fatty acid deficiency diet on reproduction of rainbow trout. Nippon Suisan Gakkaishi 50(7):1207–1215
Wu S, Wang G, Angert ER et al (2012) Composition, diversity, and origin of the bacterial community in grass carp intestine. PLoS One 7:e30440
Yukgehnaish K, Kumar P, Sivachandran P et al (2020) Gut microbiota metagenomics in aquaculture: factors influencing gut microbiome and its physiological role in fish. Rev Aquac 12(3):1903–1927
Zarkasi KZ, Abell GCJ, Taylor RS et al (2014) Pyrosequencing-based characterization of gastrointestinal bacteria of Atlantic salmon (Salmo salar L.) within a commercial mariculture system. J Appl Microbiol 117(1):18–27
Acknowledgements
We would like to thank the members of the Taizhou Institute of Agricultural Sciences for their help. We also thank Novogene Co., Ltd. (Beijing, China) for providing sequencing services. We would like to thank Editage (www.editage.cn) for English language editing.
Funding
Jiangsu Agricultural Science and Technology Independent Innovation Fund “Study on the key technology of artificial larger-scale seedling breeding of Parabramis pekinensis” [No. CX(19)3011].
Author information
Authors and Affiliations
Contributions
HLG and YMF conceived and designed the study. HLG and ZJY collected the fish and prepared the samples. HLG drafted the manuscript YMF critically reviewed the manuscript and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethics approval
All procedures performed were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978) and approved by the Experimental Animal Ethics Committee of Taizhou Academy of Agricultural Sciences (Taizhou, China).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gu, H., Feng, Y. & Yang, Z. Differential study of the Parabramis pekinensis intestinal microbiota according to different gonad development stages. Fish Sci 88, 721–731 (2022). https://doi.org/10.1007/s12562-022-01631-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12562-022-01631-z