
Abstract
Shellfish depuration is a process that aims to eliminate pathogens from mollusk tissues. Seawater disinfection during the depuration process is important and ultraviolet (UV) light treatment is the most used method worldwide. Viral models are usually employed as surrogates of fastidious viruses in viability studies. The aim of this study was to employ methods based on green fluorescent protein (GFP) fluorescence and plaque forming units to detect, respectively, recombinant adenovirus (rAdV–GFP) and murine norovirus (MNV) artificially seeded in environmental matrices. These assays were applied to assess the inactivation of rAdV–GFP and MNV in seawater in recirculation shellfish depuration tanks with and without UV light treatment. Kinetics of rAdV GFP-expression was previously measured by UV-spectrophotometer. Flow cytometry (FC), fluorescence microscopy (FM), and plaque assay were used to determine virus titer and detection limits. The influence of the environmental matrix on the performance of the methods was prior determined using either drinking water or filtered seawater seeded with rAdV–GFP. Disinfection of seeded seawater was evaluated with and without UV treatment. The time of 24-h post-infection was established as ideal for fluorescence detection on rAdV–GFP infected cells. FC showed lower sensitivity, when compared to FM, which was similar to plaque assay. Seawater disinfection on depuration tanks was promising and rAdV–GFP declined 99.99 % after 24 and 48 h with and without UV treatment, respectively. MNV was completely inactivated after 24 h in both treatments. As conclusion, the depuration tanks were effective to inactivate rAdV–GFP and MNV and the UV disinfection treatment accelerated the process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Water quality has been affected by the presence of pathogenic microorganisms originated from incorrect disposal of treated, partially treated, or untreated sewage in watercourses. Enteric viruses are pathogenic microorganisms present in environmental waters and are the main causal agents of gastroenteritis worldwide (Figueras and Borrego 2010). According to Fong and Lipp (2005), there are hundreds of different types of human viruses present in human sewage, which, if improperly treated, can become a source of contamination in drinking and recreational waters. Based on that, it is important to choose a viral model to study persistence in environment as well efficiency of disinfection processes, such as chlorine, ozone, and UV treatment.
Viral disinfection studies must ensure that no virus remain viable after treatment. Thus, cultivable viruses in vitro need to be used as surrogates of fastidious viruses (Baert et al. 2009). Murine norovirus (MNV) has been widely used as a surrogate for human norovirus (Cannon et al. 2006), while recombinant human adenovirus (rAdV) that express green fluorescent protein (GFP) can be used as surrogate for studies of human adenoviruses. rAdV–GFP are defective in their replication, since the early gene E1 was replaced by the GFP gene. Early gene products, such as E1, are generally involved in viral gene transcription, DNA replication, host immune suppression, and inhibition of host cell apoptosis (Luo et al. 2007). Thus, rAdV replication is weakened in this condition, unless the replication occurs in permissive cell lines that express E1 gene products, such as human embryonic kidney (HEK) 293A cells (Graham et al. 1977). So, rAdV replication can be directly monitored by fluorescent methods based on GFP expression encoded by this gene incorporated into the viral DNA.
The plaque assay has long been considered as a standard method because of the inherent accuracy and reproducibility, but it is extensively time consuming (Gueret et al. 2002). HEK 293A cells infected with rAdV–GFP provide a novel reporter for assays of viral infectivity, allowing the use of flow cytometry and fluorescence microscopy as rapid and quantitative methods to monitor GFP expression in individual cells (Hitt et al. 2000).
Viral outbreaks related to consumption of contaminated shellfish have been identified in several countries in recent years, being detected multiple enteric viruses in shellfish and human feces, such as aichi virus, norovirus, astrovirus, enterovirus, and rotavirus (Le Guyader et al. 2008; Westrell et al. 2010). The most common cause of outbreaks is the consumption of shellfish grown in contaminated waters that are eaten raw or inadequately cooked (Fleming et al. 2006). So, oysters are mainly responsible for gastroenteritis outbreaks among the consumed shellfish (Potasman et al. 2002; Bosch and Le Guyader 2010; Bellou et al. 2013).
Human adenovirus has been used to evaluate the effectiveness of different treatments in environmental matrices, also being indicated by some authors as bioindicators of viral contamination (Albinana-Gimenez et al. 2009; Carratalà et al. 2013; Bofill-Mas et al. 2013). Furthermore, human norovirus is the main pathogen related to outbreaks by consumption of contaminated shellfish (Mesquita et al. 2011; Hall et al. 2011; Suffredini et al. 2012).
Shellfish grown in contaminated areas require a purification step prior to consumption. Depuration is a process by which the bivalves are allocated in clean seawater under conditions that maximize the natural filtration activity, resulting in expelling pathogens from their tissues and preventing recontamination (Lee et al. 2008a, b). Despite the efficiency of depuration process for bacteria removal (Corrêa et al. 2007; Lees et al. 2010; Love et al. 2010), there is no guarantee for absence of virus, once cases of viral diseases were reported after consumption of depurated shellfish that were according to bacterial standards (Bosch et al. 2005). According to Richards et al. (2010), bacteria are removed from shellfish faster than enteric virus. Thus, the rate of bacteria removal cannot accurately predict the rate of virus removal.
The seawater disinfection during depuration is important to avoid shellfish recontamination by microorganisms, especially in recirculation system, in which bivalves decontamination is dependent on this process. Chlorine, ultraviolet (UV) light, and ozone are the most used disinfectants during depuration processes. UV light treatment is efficient, of low cost, of easy maintenance, and does not interfere with the organoleptic properties from water or shellfishes, being the most used method worldwide.
The aim of this study was (i) to standardize methods based on GFP fluorescence detection, such as UV-spectrophotometer, flow cytometry, and fluorescence microscopy assays to detect rAdV–GFP in seeded environmental matrices (drinking and sea waters); (ii) to apply these assays to assess the inactivation of rAdV–GFP and MNV in seawater in a recirculation depuration tanks with and without UV light treatment.
Methods
Environmental Samples
Drinking water sample was harvested in a Water Treatment Plant located at Peri Lagoon, Florianópolis, Brazil, after regular treatment and just prior to chemical disinfection (without residual free chlorine). For standardization purposes, 2.0 × 106 PFU of rAdV–GFP was seeded in a total of 500 µL of this drinking water previously diluted 1:2 in DMEM 1X.
Filtered seawater sample came from Barra da Lagoa beach, Florianópolis, Brazil, and was concentrated by skimmed milk flocculation to 10 mL final volume, as previously described (Calgua et al. 2008). To analyze the seawater matrix influence and the viral recovery after flocculation, two flasks containing 10 L of filtered seawater were inoculated with 4.0 × 107 PFU of rAdV–GFP before and after the flocculation process, respectively. These concentrated samples were analyzed by flow cytometry and fluorescence microscopy assays.
These samples were not analyzed prior for the presence of wild strains of adenovirus because both techniques (flow cytometry and fluorescence microscopy) used in this study were purely based on fluorescence detection though they were able to detect only the artificially seeded rAdV–GFP.
Cytotoxicity Assay
To evaluate the cytotoxicity of the drinking water and the flocculated seawater on HEK 293A cells, the confluent monolayers were inoculated with 100 µL of two-fold scalar dilutions (1:2, 1:4, 1:8, 1:16, and 1:32) of each water matrix. Cells were incubated for 60 min at 37 °C in 5 % of CO2 and, after incubation, 650 µL of maintenance medium was added [DMEM 1X, containing 2 % FSB, 1 % HEPES, 1 % PSA (10 U/mL Penicillin, 10 μg/mL Streptomycin, 2 ng/mL Amphotericin)]. After 48 h, the cells were stained with Amido Black to check monolayer integrity.
Virus and Cell Line
Recombinant adenovirus (rAdV–GFP) serotype 5 was propagated in HEK 293A cells. HEK 293A cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM 1X), supplemented with 10 % of fetal bovine serum (FBS) and 1 % of HEPES. Virus stock was produced in 180-cm2 flasks by host cell infection using multiplicity of infection (MOI) of 5. After 24-h incubation, at which 100 % of cytopathic effect was reached, the flasks were freeze thawed three times, centrifuged at 2,500×g for 5 min, and the supernatant was recovered and stored at −80 °C. Virus stock titer was 4.0 × 107 PFU/mL.
MNV was propagated in murine macrophage cells (RAW264.7). RAW264.7 were maintained in DMEM 1X, supplemented with 10 % of fetal bovine serum (FBS), 1 % L-glutamin, 1 % non-essential amino acids, and 1.5 % of HEPES. Virus stock was produced in 180-cm2 flasks by host cell infection using MOI of 5. After 36–48-h incubation (enough for 100 % of cytopathic effect), the flasks were freeze thawed three times, centrifuged at 2,500×g for 5 min for cell debris removal, and the supernatant was stored at −80 °C. Virus stock titer was 2.0 × 107 PFU/mL.
Kinetics of GFP Expression
This assay was performed in order to determine which period post-infection (p.i.) was more appropriate to detect GFP fluorescence, ensuring optimal fluorescence quality as well to determine the first cycle of viral replication. For this, 3 × 105 of HEK 293A cells were incubated in 24-well plates for 24 h at 37 °C under 5 % CO2 atmosphere. Growth medium was discarded and the monolayers were inoculated with 100 µL of virus stock in distinct multiplicities of infection (MOI—rate of virus/cell) of rAdV–GFP (0.01; 0.1; 0.5; 1; and 10) in triplicates (MOI of 1 = 3 × 105 PFU). Cells were incubated for 60 min at 37 °C under 5 % CO2 atmosphere and, after that, 650 µL of fresh maintenance medium was added (DMEM 1X, 2 % FSB, 1 % HEPES, 1 % PSA). Every 2 h up to 60 h p.i. the plate was taken to the UV-spectrophotometer (Molecular Devices SpectraMax M2) for measuring therelative fluorescence of the attached cell monolayer (excitation/emission: 395/508 nm).
Viral Viability Assay
MNV Plaque Assay
MNV detection by plaque assay was performed as described by Gonzalez-Hernandez et al. (2012). Briefly, 2.0 × 106 RAW264.7 cells were incubated in a 6-well plate for 24 h at 37 °C under 5 % CO2 atmosphere. Growth medium was discarded and 250 µL of samples were inoculated in non-cytotoxic serial dilutions in duplicate. Cells were incubated for 60 min at 37 °C under 5 % CO2 atmosphere and, after incubation, the inocula were removed and cell monolayers were overlaid with 2.5 mL of 3 % of agar (low melting) (Sea Plate [Lonza]) in MEM 2X containing 2 % FSB, 1 % L-glutamin, 1 % non-essential amino acids, 1.5 % HEPES, and 1 % PSA. After 48 h p.i., cells were stained with 0.3 % neutral red solution and plaques were counted. The viral titer was determined as (average plaque counted × reciprocal dilution)/inoculum (mL). The results are expressed as Plaque Forming Unit per mL (PFU/mL).
rAdV–GFP Flow Cytometry (FC)
For flow cytometry assay, 3 × 105 of HEK 293A cells in confluent monolayers were incubated in 24-well plates during 24 h at 37 °C under 5 % CO2 atmosphere. Growth medium was discarded and 100 µL of decimal dilutions of virus stock, and spiked drinking water (without residual free chlorine) and concentrated seawater were inoculated in non-cytotoxic dilutions in triplicates. Cells were incubated for 60 min at 37 °C under 5 % CO2 atmosphere and, after that, 650 µL of fresh maintenance medium was added (DMEM 1X, containing 2 % FSB, 1 % HEPES, 1 % PSA). After 24 h p.i., the cells were harvested with tripsin, centrifuged at 2,000×g for 3 min, resuspended in 500 µL of PBS with 0.05 µM EDTA, and cell viability was determined on Countess® Automated Cell Counter (Invitrogen™). Data from 50,000 events were measured by flow cytometry (BD Biosciences FACSCanto™ II) and were analyzed using the free software program Weasel (version 3.1). The results were expressed as Green Fluorescence Units per mL (GFU/mL).
rAdV–GFP Fluorescence Microscopy (FM)
For fluorescence microscopy assay, 2.0 × 105 of HEK 293A cells were grown in 48-well plate for 24 h at 37 °C under 5 % CO2 atmosphere. Growth medium was discarded, and 100 µL of decimal dilutions of virus stock and spiked drinking water (without residual free chlorine) and concentrated seawater were inoculated in non-cytotoxic dilutions in triplicates. Cells were incubated for 60 min at 37 °C under 5 % CO2 atmosphere and, after incubation, 400 µL of fresh maintenance medium was added (DMEM 1X, containing 2 % FSB, 1 % HEPES, 1 % PSA). After 24 h p.i., chambers were mounted in the presence of 50 µL of mounting solution (5 % NaCl 5 M, 40 % PBS, 50 % ethanol, 5 % formaldehyde, 2.5 % DBCO) on the coverslip. Cells were then observed under an epifluorescence microscope with UV light (Olympus). The viral titer was determined as (average green cells counted × reciprocal dilution)/inoculum (mL). The results were expressed as Focus Forming Unit per mL (FFU/mL).
rAdV–GFP Plaque Assay
Plaque assay was performed for virus stock aiming to compare the results obtained by both fluorescence methods. The protocol was adapted from other study (Cromeans et al. 2008). Briefly, 5.0 × 105 of HEK 293A cells were incubated in 6-well plates for 24 h at 37 °C under 5 % CO2 atmosphere, when confluent monolayers were achieved. Growth medium was discarded, 300 µL of decimal dilutions of virus stock was added in duplicate to the cell monolayers, and the cells were incubated for 60 min at 37 °C under 5 % CO2 atmosphere. After this period, the inocula were removed and cell monolayers were overlaid with 2.5 mL of 0.6 % Bacto-agar in high-glucose DMEM 2X containing 4 % FBS, 0.1 mM sodium pyruvate, 26 mM MgCl2, and incubated at 37 °C during 7 days. After this period, the agar overlay was removed, cells were stained with 20 % Gram’s crystal violet, and plaques were counted macroscopically once the stain was removed. The viral titer was determined as (average plaque counted × reciprocal dilution)/inoculum (mL). The results were expressed as Plaque Forming Unit per mL (PFU/mL).
Depuration Tanks
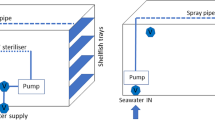
Two depuration tanks based on a closed system of seawater recirculation by a peristaltic pump (with a flow rate of 1,800 L/h), with a capacity of 300 L with a water flow of 1,800 L/h, were used (Fig. 1). One tank was coupled with a 36 W UV lamp (low-pressure lamp, monochromatic light output at 254 nm), with minimal dose of 44 mJ/cm2 delivered in each passage through these apparatus with an accumulated dose of 264 mJ/cm2 per hour. The second tank was not provided with UV lamp. The water in both tanks was maintained at 16 °C.

Recirculation depuration tank with UV light treatment
Seawater Depuration Assay
Both depuration tanks (with and without UV) were filled with 300 L of filtered seawater and spiked with rAdV–GFP and MNV at an approximate final concentration of 3.0 × 106 FFU/L and 3.0 × 106 PFU/L, respectively. Ten liters of each tank was collected at initial time (0 h) and after 24, 48, and 72 h of recirculation.
Samples of seawater were immediately concentrated by skimmed milk flocculation (Calgua et al. 2008). As concentration process control, in every time of sampling, ten liters of clean seawater was spiked with rAdV–GFP and MNV at the same concentration described above and concentrated by flocculation. After concentration, samples were immediately analyzed for viral viability. This experiment was made in triplicate.
Viral Stability in Seawater
To ensure that viral decay in depuration tanks was not due to matrix influence, flasks containing 10 L of filtered seawater were spiked with rAdV–GFP and MNV at the same concentration described above. One liter was collected at 0 h and another, after 24 h of agitation, and submitted to flocculation process. After concentration, samples were immediately analyzed for viral viability. This experiment was made in triplicate.
Statistical Analysis
The statistical analyses were performed using GraphPad Prism version 5.0. ANOVA test, Student’s t test, regression and linear correlation were performed, and all significant differences are quoted for P < 0.05.
Results
Kinetics of GFP Expression
The kinetics of GFP expression is shown in Fig. 2. The onset of cytopathic effect at MOI 10, 1.0, and 0.5 was observed 20 h p.i. After 40 h p.i., there were no attached cells at MOI 10 and at MOI 1.0 and 0.5; 100 % of cytopathic effect was observed. The statistical analysis showed that at MOI 10 and 1.0, there was a significant increase of fluorescence between 18 and 20 h p.i. (P < 0.05). Thus, the 24-h p.i. period was selected to perform flow cytometry and fluorescence microscopy assays, ensuring the achievement of the fluorescence peak for detection as well as the first cycle of viral replication.

Kinetics of GFP expression by UV-spectrophotometer in HEK293A cells infected with rAdV at MOIs of 0.01, 0.1, 0.5, 1.0, 10, and cellular control. Bars represent standard deviation of triplicate
Detection Limits
This study aimed to determine the rAdV–GFP detection limits by flow cytometry assay and fluorescence microscopy when compared with plaque assay. The use of the same virus stock and dilutions for all techniques was important to allow a direct comparison among the techniques. The 24-h p.i. period was used for flow cytometry and microscopy assays in accordance with the kinetics GFP expression results.
For FC assay, the detection limit of the virus stock was 2.15 × 105 GFU/mL. The titer of the viral working solution by FC assay was 1.43 × 109 GFU/mL (Fig. 3). Regression analysis showed that the correlation was linear, with an r 2 value of 0.9468, by plotting the normalized values in log10 of rAdV–GFP inoculated in decimal dilutions ranging from 4.0 × 102 to 4.0 × 107 PFU/mL versus the viral titer by flow cytometry assay obtained at each dilution (Fig. 4).

Detection limit of rAdV by flow cytometry, fluorescence microscopy, and plaque assay. Flow cytometry assays show detection limit of 10−4 dilution (2.15 × 105 GFU/mL), while fluorescence microscopy and plaque assay show 10−6 dilution (8.5 × 101 FFU/mL and 4.0 × 101 PFU/mL, respectively). Bars represent standard deviation of duplicates

Linear regression of plaque assay and flow cytometry assay. Normalized values in log10 of rAdV inoculated in plaque assay (PFU/mL) versus the viral titer obtained by flow cytometry assay (GFU/mL). The linear regression with an r 2 value of 0.9468 was obtained with the equation: y = 0.982x + 1.805
The detection limit of the virus stock by FM was 8.5 × 101 FFU/mL (Fig. 3). The titer of the viral working solution by FM assay was 8.5 × 107 FFU/mL. In order to compare, plaque assay showed virus stock titer of 4.0 × 107 PFU/mL and the limit of detection was 4.0 × 101 PFU/mL (Fig. 3).
For spiked environmental sample assays, the non-cytotoxic dilution established was 1:2 for drinking and seawater. No statistically significant differences were found between virus stock and virus diluted in drinking water and in flocculated seawater (P > 0.05) for FC and FM assays, showing that the matrices had no effect on method sensitivity, and for this reason the results are shown just for virus stock.
Viral Stability in Seawater
The rAdV–GFP and MNV had a non-significant (P > 0.05) reduction after 24 h of contact with seawater. rAdV–GFP had a 0.37 log decay and MNV was more unstable in seawater, with a 1.12 log reduction. The mean value and standard deviation of viruses log reduction are shown in Table 1.
Seawater Disinfection Assay
rAdV–GFP viability decay can be visualized in Fig. 5. In tank with UV treatment, a 4 log reduction was observed after 24 h of water circulation and it was not possible to detect viable virus after 48 h. For tank without UV, there was approximately 2 log reduction every 24 h of depuration, with no detection after 72 h. MNV has been totally inactivated from seawater in both depuration tanks after 24 h. The mean value and standard deviation of viral log reduction are shown in Table 2.

Fluorescence microscopy of HEK293A cells infected with rAdV–GFP at initial time (0 h) and after 24, 48, and 72 h of depuration in tanks with and without UV treatment. Numbers (1:10 and 1:4) represent the serial dilution. Bars = 200 μm
For this assay, the mean viral concentration method for recovery was 31 % for rAdV–GFP and 33 % for MNV.
Discussion
Recombinant adenovirus provides a versatile system for gene expression studies and therapeutic applications, including gene transfer in vitro, gene therapy, and vaccine therapy (Miller 1992; Graham and Prevec 1992; Nadeau and Kamen 2003). The possibility to transfer genes to a broad spectrum of cell types not depending on active cell division, as well as the high-titer preparations of adenoviruses made them the vectors of choice for these applications (Miller 1992; Graham and Prevec 1992; He et al. 1998; Nadeau and Kamen 2003). Although this usage is well established, we describe herein a novel application of rAdV–GFP in environmental virology field. The current study presented the employment of rAdV–GFP in environmental matrices, such as drinking water and seawater, as a way to study disinfection associated with viability decay.
Regarding the kinetics of GFP expression, at MOI 10 and 1.0, all the cells were killed by the virus after 40 h p.i., which explains the wide variation of relative fluorescence after this period. The 24-h p.i. period was chosen to perform the subsequent assays, ensuring the optimal fluorescence emission for detection, but mainly ensuring the first cycle of viral replication. It is already described that HAdV replication cycle is completed after 24–36 h in HeLa cells (Wold and Horwitz 2007), although viral replication can vary from cell to cell. This is important to not allow longer periods for viral replication which could overestimate the real titer of the virus where, at the end of one cycle, about 104 viral progeny particles per cell have been released after cell lysis (Green and Daesch 1961).
Fluorescence microscopy can be equally compared with plaque assay, since their detection limits were the same (10−6 dilution), as well a similar viral titer (Fig. 3). Nevertheless, the big difference regarding the incubation time (1 day for fluorescence microscopy versus 7 days for plaque assay) supports the choice of fluorescence microscopy assay.
A previous study (Li et al. 2010) reported the use of flow cytometry for rAdV detection after 1–5 days of incubation. They report that, after 3 days of incubation, fluorescent cells were detected at inoculation densities of 1 PFU/well, while after 1 day of incubation, it was only possible to detect fluorescence above 100 PFU/well, also showed in the present study (400 PFU/well). Therefore, it is remarkable that, when the incubation time is prolonged, the sensitivity of the assay increases but this fact masks the viral replication and the real viral titer.
The viral titer by FC (1.43 × 109 GFU/mL) was higher than by FM (8.5 × 107 FFU/mL). FM allowed to detect fluorescence in two decimal dilutions above than FC (10−6 and 10−4, respectively) (Fig. 3). Li et al. (2010) described a high detection limit of rAdV detection by FC after 24-h p.i.
The use of this viral model is economical and practical since its detection does not require the use of antibodies obligatorily required by methods as immunofluorescence microscopy and traditional flow cytometry, which has antibody labeling as principle. Therefore, with the use of rAdV–GFP, there is no chance of unspecific binding of antibodies (avoiding false-positives) and also decreasing the cell loss during extensive washes usually performed to remove antibodies. In addition, a previous study showed that detection of fluorescence by flow cytometry with rAdV was higher when compared with the use of anti-hexon or anti-E1A antibodies (Li et al. 2010). Furthermore, FM and FC are faster (1 day) than conventional methods like plaque assay (7–10 days) (Hitt et al. 2000). Although the FC showed higher detection limit (2.15 × 105 GFU/mL vs. 8.5 × 101 FFU/mL and 4.0 × 101 PFU/mL), the FM can be directly compared with the standard method of plaque assay.
The seawater disinfection is essential for an efficient mollusk depuration, especially in recirculation systems. Viral inactivation in water by UV light is well known. Calicivirus, like MNV, CaCV, and FCV, has high resistance to UV treatment, requiring a dose of approximately 40 mJ/cm2 for 4 log inactivation (Duizer et al. 2004; Lee et al. 2008a, b; Park et al. 2011). Regarding the adenovirus, Hijnen et al. (2006) showed several studies that evaluated the decay of different viruses in environmental water samples, indicating that adenovirus is the most resistant pathogen to inactivation by UV light, requiring doses higher than 170 mJ/cm2 to 99.99 % reduction of viable virus. Carratalà et al. (2013) described 6 log decay of adenovirus using high doses of UVB radiation in different water matrices, including seawater. UV light can not only degrade viral genome by inducing the formation of thymine dimers, but also can cause damage to viral capsid proteins, disabling them to infect cells (Eisched and Linden 2011; Wigginton et al. 2012).
Although the germicidal action of UV light in water is well known, few studies have evaluated the effectiveness of seawater treatment for viral inactivation in depuration tanks. A similar work by Corrêa et al. (2012) showed a decay of adenovirus genome copies of 4 log and 1 log after 72h in depuration tanks with and without UV, respectively. The same authors also showed highest susceptibility for MNV by qPCR, with 4 log reduction with UV and 2 log without UV, after 24-h depuration. In the present study it was proved, by viability assays, that 24-h UV treatment can inactivate a minimal of 4 log adenovirus and norovirus in seawater inside the depuration tanks, and the same result can be achieved by 48 h of depuration without UV. Viral decay in depuration tank without UV treatment cannot be sufficiently explained in this work, but it is possible that recirculating system can increase water oxygenation and also cause physical impacts to viruses particles, leading to inactivation.
Viral stability in seawater showed no significant reduction after 24 h for adenovirus and norovirus. Similar results were showed by Corrêa et al. (2012). Enriquez and Gerba (1995) observed a high stability of adenovirus in seawater, with approximately 1.5 log viral decay in 60 days. Lee et al. (2008a, b) showed that MNV showed instability in saline solution at a concentration of 0.5 M NaCl (similar amount present in seawaters) and the viral viability decreased about 0.5 log every 24 h.
The seawater concentration method with skimmed milk flocculation is well established and studies have reported an average genomic copies recovery of approximately 50 % for adenovirus and 80 % for MNV (Calgua et al. 2008, 2013a, b). In this study, we showed a recovery around 30 % of both infectious viruses.
Genome copies evaluation is not always proper for environmental samples since it does not show the real risk of contamination, since the virus can be damaged and still be detected. Therefore, whenever possible, it is very important to apply techniques that are preceded by cell infection in vitro to evaluate infectious viruses (Bosch et al. 2008; Baert et al. 2009). Thus, some viral models have been used, especially in the disinfection and stability studies in different matrices, such as human and MNV adenovirus. rAdV–GFP was used in this study as a surrogate of human adenovirus, and its detection was fast and efficient by fluorescence-based emission.
Conclusions
We demonstrated that FC and FM have proven to be feasible, reliable, non-time consuming and non-expensive to detect viable recombinant adenovirus in environmental matrices. Nevertheless, due to the lower detection limit of FC, FM showed to be more adequate and can be used for rAdV–GFP viability studies. Drinking water and seawater did not interfere in fluorescence detection, allowing the use of these water matrices and rAdV–GFP for further studies involving viral persistence in the environment as well as efficiency of disinfection processes. As conclusion, the depuration tanks were effective to inactivate rAdV–GFP and MNV and the UV radiation treatment accelerated the disinfection process. Additional studies shall be conducted in order to investigate the viral decay in seawater without UV treatment and to assess viral viability in shellfish after depuration. Also, the rAdV–GFP should be tested in different environmental matrices and using distinct disinfection methods.
References
Albinana-Gimenez, N., Miagostovich, M. P., Calgua, B., Huguet, J. M., Matia, L., & Girones, R. (2009). Analysis of adenoviruses and polyomaviruses quantified by qPCR as indicators of water quality in source and drinking-water treatment plants. Water Research, 43(7), 2011–2019.
Baert, L., Debevere, J., & Uyttendaele, M. (2009). The efficacy of preservation methods to inactivate foodborne viruses. International Journal of Food Microbiology, 131(2–3), 83–94.
Bellou, M., Kokkinos, P., & Vantarakis, A. (2013). Shellfish-borne viral outbreaks: A systematic review. Food and Environmental Virology, 5(1), 13–23.
Bofill-Mas, S., Rusiñol, M., Fernandez-Cassi, X., Carratalà, A., Hundesa, A., & Girones, R. (2013). Quantification of human and animal viruses to differentiate the origin of the fecal contamination present in environmental samples. BioMed Research International,. doi:10.1155/2013/192089.
Bosch, A., Abad, F. X., & Pintó, M. R. (2005). Human pathogenic viruses in the marine environment. In S. Belkin & R. R. Colwell (Eds.), Oceans and health: pathogens in the marine environment (pp. 109–131). New York: Springer.
Bosch, A., Guix, S., Sano, D., & Pintó, R. M. (2008). New tools for the study and direct surveillance of viral pathogens in water. Current Opinion in Biotechnology, 19(3), 295–301.
Bosch, A., & Le Guyader, S. F. (2010). Viruses in shellfish. Food and Environmental Virology, 2(3), 115–116.
Calgua, B., Fumian, T., Rusiñol, M., Rodriguez-Manzano, J., Mbayed, V. A., Bofill-Mas, S., et al. (2013a). Detection and quantification of classic and emerging viruses by skimmed-milk flocculation and PCR in river water from two geographical areas. Water Research, 47(8), 2797–2810.
Calgua, B., Mengewein, A., Grunert, A., Bofill-Mas, S., Clemente-Casares, P., Hundesa, A., et al. (2008). Development and application of a one-step low cost procedure to concentrate viruses from seawater samples. Journal of Virological Methods, 153(2), 79–83.
Calgua, B., Rodriguez-Manzano, J., Hundesa, A., Suñen, E., Calvo, M., Bofill-Mas, S., & Girones, R. (2013b). New methods for the concentration of viruses from urban sewage using quantitative PCR. Journal of Virological Methods, 187(2), 215–221.
Cannon, J. L., Papafragkou, E., Park, G. W., Osborne, J., Jaykus, L. A., & Vinjé, J. (2006). Surrogates for the study of norovírus stability and inactivation in the environment: A Comparison of murine norovírus and feline calicivirus. Journal of Food Protection, 69(11), 2761–2765.
Carratalà, A., Rusiñol, M., Rodriguez-Manzano, J., Guerrero-Latorre, L., Sommer, R., & Girones, R. (2013). Environmental effectors on the inactivation of human adenovíruses in water. Food and Environmental Virology, 5(4), 203–214.
Corrêa, A. A., Albarnaz, J. D., Moresco, V., Poli, C. R., Teixeira, A. L., Simões, C. M. O., & Barardi, C. R. M. (2007). Depuration dynamics of oysters (Crassostrea gigas) artificially contaminated by Salmonella enterica serovar Typhimurium. Marine Environmental Research, 63(5), 479–489.
Corrêa, A. A., Souza, D. S., Moresco, V., Kleemann, C. R., Garcia, L. A., & Barardi, C. R. (2012). Stability of human enteric viruses in seawater samples from mollusc depuration tanks coupled with ultraviolet irradiation. Journal of Applied Microbiology, 113(6), 1554–1563.
Cromeans, T. L., Lu, X., Erdman, D. D., Humphrey, C. D., & Hill, V. R. (2008). Development of plaque assays for adenoviruses 40 and 41. Journal of Virololical Methods, 151(1), 140–145.
Duizer, E., Bijkerk, P., Rockx, B., De Groot, A., Twisk, F., & Koopmans, M. (2004). Inactivation of caliciviruses. Applied and Environmental Microbiology, 70(8), 4538–4543.
Eisched, A. N., & Linden, K. G. (2011). Molecular indications of protein damage in adenoviruses after UV disinfection. Applied and Environmental Microbiology, 77(3), 1145–1147.
Enriquez, C. E., & Gerba, C. P. (1995). Concentration of enteric adenovírus 40 from tap, sea and waste water. Water Research, 29(11), 2554–2560.
Figueras, M. J., & Borrego, J. J. (2010). New perspectives in monitoring drinking water microbial quality. International Journal of Environmental Research and Public Health, 7(12), 4179–4202.
Fleming, L. E., Broad, K., Clement, A., Dewailly, E., Elmir, S., Knap, A., et al. (2006). Oceans and human health: Emerging public health risks in the marine environment. Marine Pollution Bulletin, 53(10–12), 545–560.
Fong, T. T., & Lipp, E. K. (2005). Enteric viruses of humans and animals in aquatic environments: health risks, detection, and potential water quality assessment tools. Microbiology and Molecular Biology Reviews, 69(2), 357–371.
Gonzalez-Hernandez, M. B., Cunha, J. B., & Wobus, C. E. (2012). Plaque assay for murine norovirus. Journal of Visualized Experiments,. doi:10.3791/4297.
Graham, F. L., & Prevec, L. (1992). Adenovirus-based expression vectors and recombinant vaccines. Biotechnology, 20(1), 363–390.
Graham, F. L., Smiley, J., Russell, W. C., & Nairn, R. (1977). Characteristics of a human cell line transformed by DNA from human adenovirus type 5. Journal of General Virology, 36(1), 59–74.
Green, M., & Daesch, G. E. (1961). Biochemical studies on adenovirus multiplication. II. Kinetics of nucleic acid and protein synthesis in suspension cultures. Virology, 13(1), 169–176.
Gueret, V., Negrete-Virgen, J. A., Lyddiatt, A., & Al-Rubeai, M. (2002). Rapid titration of adenoviral infectivity by flow cytometry in batch culture of infected HEK293 cells. Cytotechnology, 38(1–3), 87–97.
Hall, A. J., Vinjé, J., Lopman, B., Park, G. W., Yen, C., Gregoricus, N., et al. (2011). Updated norovírus outbreak management and disease prevention guidelines. Morbidity and Mortality Weekly Report, 60(RR03), 1–15.
He, T. C., Zhou, S., da Costa, L. T., Yu, J., Kinzler, K. W., & Vogelstein, B. (1998). A simplified system for generating recombinant adenoviruses. Proceedings of the National Academy of Science of USA, 95(5), 2509–2514.
Hijnen, W. A., Beerendonk, E. F., & Medema, G. J. (2006). Inactivation credit of UV radiation for viruses, bacteria and protozoan (oo)cysts in water: A review. Water Research, 40(1), 3–22.
Hitt, D. C., Booth, J. L., Dandapani, V., Pennington, L. R., Gimble, J. M., & Metcalf, J. (2000). A flow cytometric protocol for titering recombinant adenoviral vectors containing the green fluorescent protein. Molecular Biotechnology, 14(3), 197–203.
Le Guyader, F. S., Le Saux, J. C., Ambert-Balay, K., Krol, J., Serais, O., Parnaudeau, S., et al. (2008). Aichi virus, norovírus, astrovírus, enterovírus, and rotavírus involved in clinical cases from a french oyster-related gastroenteritis outbreak. Journal of Clinical Microbiology, 46(12), 4011–4017.
Lee, R., Lovatelli, A., & Ababouch, L. (2008). Bivalve depuration: fundamental and practical aspects. FAO Fisheries Technical Paper. No. 511. Rome: FAO.
Lee, J., Zoh, K., & Ko, G. (2008b). Inactivation and UV disinfection of murine norovírus with TiO2 under various environmental conditions. Applied and Environmental Microbiology, 74(7), 2111–2117.
Lees, D., Younger, A., & Doré, B. (2010). Depuration and relaying. In G. Rees, K. Pond, D. Kay, J. Bartram, & J. Santo Domingo (Eds.), Safe management of shellfish and harvest waters (pp. 145–181). London: IWA Publishing.
Li, D., He, M., & Jiang, S. C. (2010). Detection of infectious adenoviruses in environmental waters by fluorescence-activated cell sorting assay. Applied and Environmental Microbiology, 76(5), 1442–1448.
Love, D. C., Lovelace, G. L., & Sobsey, M. D. (2010). Removal of Escherichia coli, Enterococcus fecalis, coliphage MS2, poliovirus, and hepatitis A virus from oysters (Crassostrea virginica) and hard shell clams (Mercinaria mercinaria) by depuration. International Journal of Food Microbiology, 143(3), 211–217.
Luo, J., Deng, Z. L., Luo, X., Tang, N., Song, W. X., Chen, J., et al. (2007). A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nature Protocols, 2(5), 1236–1247.
Mesquita, J. R., Vaz, L., Cerqueira, S., Castilho, F., Santos, R., Monteiro, S., et al. (2011). Norovírus, hepatitis A virus and enterovírus presence in shellfish from high quality harvesting areas in Portugal. Food Microbiology, 28(5), 936–941.
Miller, A. D. (1992). Human gene therapy comes of age. Nature, 357(6378), 455–460.
Nadeau, I., & Kamen, A. (2003). Production of adenovirus vector for gene therapy. Biotechnology Advances, 20(7–8), 475–489.
Park, G. W., Linden, K. G., & Sobsey, M. D. (2011). Inactivation of murine norovirus, feline calicivirus and echovirus 12 as surrogates for human norovirus (NoV) and coliphage (F+) MS2 by ultraviolet light (254 nm) and the effect of cell association on UV inactivation. Letters of Applied Microbiology, 52(2), 162–167.
Potasman, I., Paz, A., & Odeh, M. (2002). Infectious outbreaks associated with bivalve shellfish consumption: a worldwide perspective. Clinical Infectious Diseases, 35(8), 921–928.
Richards, G. P., Mcleod, C., & Le Guyader, F. S. (2010). Processing strategies to inactivate enteric viruses in shellfish. Food and Environmental Virology, 2(3), 183–193.
Suffredini, E., Magnabosco, C., Civettini, M., Rossetti, E., Arcangeli, G., & Croci, L. (2012). Norovirus contamination in different shellfish species harvested in the same production areas. Journal of Applied Microbiology, 113(3), 686–692.
Westrell, T., Dusch, V., Ethelberg, S., Harris, J., Hjertqvist, M., Jourdan-da Silva, N., et al. (2010). Norovírus outbreaks linked to oyster consumption in the United Kingdom, Norway, France, Sweden and Denmark. Euro Surveillence, 15(12).
Wigginton, K. R., Pecson, B. M., Sigstam, T., Bosshard, F., & Kohn, T. (2012). Virus inactivation mechanisms: Impact of disinfectants on virus function and structural integrity. Environmental Science and Technology, 46(21), 12069–12078.
Wold, W. S. M., & Horwitz, M. S. (2007). Adenoviruses. In D. M. Knipe & P. M. Howley (Eds.), Fields virology (pp. 2396–2436). Philadelphia: Lippincott Williams & Wilkins.
Acknowledgments
We thank Ms. Carlos Guillermo Quiroz Carrillo for statistical assistance. Financial support for this research was provided by National Counsel of Technological and Scientific Development (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Conflict of interest
No conflict of interest declared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Garcia, L.A.T., Nascimento, M.A. & Barardi, C.R.M. Effect of UV Light on the Inactivation of Recombinant Human Adenovirus and Murine Norovirus Seeded in Seawater in Shellfish Depuration Tanks. Food Environ Virol 7, 67–75 (2015). https://doi.org/10.1007/s12560-014-9177-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12560-014-9177-x