
Abstract
Studies have strongly suggested a disturbed regulation of dopaminergic neurotransmission in attention-deficit/hyperactivity disorder (ADHD) and Parkinson’s disease (PD). A genetic and phenotypic overlap between both disorders is discussed. A well-studied risk gene for PD is the gene coding for α-synuclein (SNCA). α-Synuclein, a protein located primarily in the presynaptic vesicles, has been suggested to play a role in the modulation of dopamine transporter (DAT) function. DAT is the target of psychostimulants for the treatment of ADHD and plays a key role in regulating the dopamine concentrations in the synaptic cleft. In our sample consisting of German families with children affected by ADHD, we tested for association of allelic variants of two functionally relevant polymorphisms of the α-synuclein gene (NACP-Rep1: 156 families, 232 children; rs356219: 195 families, 284 children) with ADHD. Transmission disequilibrium test analysis revealed no over-transmission for NACP-Rep1 (OR 1, pnom = 1 padj = 1) and rs356219 (OR 1.28; pnom = 0288) in affected siblings. However, a subanalysis on trios with index children showed a nominal association of rs356219 with ADHD (OR 1.43, pnom = 0.020), which survived Bonferroni correction (padj = 0.039); again, no association for NACP-Rep1 (OR 0.8, p = 0.317, padj = 0.634) was found. In conclusion, we found in our pilot study a trend for an association of the rs356219 genotype in SNCA that may affect α-synuclein function and contribute to the aetiology of ADHD. In light of the small sample size of our study, the link between PD and ADHD through dopamine-related neurobiology warrants further investigations. Future studies on SNCA in large ADHD samples should focus on specified symptoms and traits, e.g. attentional capacities or emotional dysregulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The aetiology of attention-deficit/hyperactivity disorder (ADHD) still is subject of extensive research efforts. However, a large body of evidence suggests that ADHD is a highly heritable and multifactorial neurodevelopmental disorder. Multiple genes with small effect sizes as well as environmental factors contribute to the development and course of the disorder (Banaschewski et al. 2017; Thapar and Cooper 2016).
Previous studies have reported abnormalities in various neurotransmitter systems underlying the symptomatology of ADHD (for a review, see Faraone et al. 2015). In particular, disturbance of the dopaminergic neurotransmission has been linked to dysfunction of brain structures that modulate locomotion, executive function, working memory, emotional regulation, and reward processing. Though genetic studies have shown association between ADHD and genes involved in dopaminergic neurotransmission, study results remain heterogeneous and effect sizes are small (Albayrak et al. 2008). The dopamine transporter (DAT) is a presynaptic located protein that plays a key role in regulating the dopamine concentrations in the synaptic cleft by removing dopamine from the synaptic cleft and returning it to the presynaptic neurons (Giros et al. 1996). The effect of psychostimulants such as amphetamine and methylphenidate is mediated mainly by inhibition of DAT (Walitza et al. 2014). Finally, neuroimaging studies demonstrated an increased density of DAT in the striatum of ADHD patients using positron emission tomography (Fusar-Poli et al. 2012) and a morphological abnormality of the substantia nigra (dopamine neurons from this region project to the striatum) in children with ADHD using transcranial sonography (Krauel et al. 2010; Romanos et al. 2010).
Parkinson’s disease (PD) is the second most common neurodegenerative disease in the elderly (Ascherio and Schwarzschild 2016). Pathologically, PD is characterized by a preferential loss of neuromelanin-containing dopaminergic neurons in the substantia nigra pars compacta, accompanied by the accumulation of intracellular proteinaceous α-synuclein-rich inclusions named Lewy bodies and a reduction in striatal dopamine (Sian et al. 1999).
ADHD (Thapar and Cooper 2016) and PD (Kalia and Lang 2015) have several disturbances of neurotransmitter systems in common that lead to motoric and emotional dysfunctions, cognitive and attentional problems. Due to the pivotal role of a disturbed dopaminergic neurotransmission in ADHD as well as in PD, mutual neurobiological underpinnings have been discussed (Mehler-Wex et al. 2006, Kehagia et al. 2014). Previous studies on a potential genetic overlap of ADHD and PD found heterogeneous results (Jarick et al. 2014, Geissler et al. 2017). Analysis of shared heritability in common disorders of the brain demonstrated that there is little or no correlation between PD and psychiatric brain disorders including ADHD (The Brainstorm Consortium 2018). However, sufficient research is not available. It might be therefore very interesting to focus on specific subgroups of gene variants.
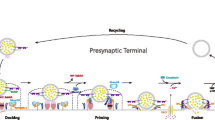
A well-studied risk gene for PD is the gene coding for α-synuclein (SNCA). Recent evidence suggests that dopaminergic neurotransmission is modulated by α-synuclein, which is located primarily in the presynaptic vesicles (Butler et al. 2017; Sidhu et al. 2004). Therefore, we carried out a family-based study to examine whether there is association of genetic variants of SNCA in ADHD. We selected two functional polymorphisms: a dinucleotide repeat polymorphism covering the 5′-upstream region (NACP-Rep1) and the single nucleotide polymorphism (SNP) rs356219 in the 3′-downstream region of SNCA. These variants have been reported to affect α-synuclein function by influencing SNCA transcription rate and mRNA stability or by altering the generation of alternative splice isoforms (Venda et al. 2010).
Materials and methods
Sample
Participants were recruited in children and parents, and phenotypically characterized by a team of experienced child and adolescent psychiatrists in the outpatient unit of the Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, University of Würzburg. Since recruitment started before publication of DSM-5, ADHD criteria according to DSM-IV were applied (American Psychiatric Association 2000). All patients agreed to participate in the study, and written informed consent was obtained from all participants. The study was approved by the Local Ethics Committee of the University of Würzburg.
Families were included if they had one or more children affected with ADHD to perform family-based association and genome-wide linkage studies. The index patient was required to be older than 8 years and to fulfil DSM-IV criteria for the combined subtype; other affected siblings in a family had to be older than 6 years.
Psychiatric diagnoses were based on the Schedule for Affective Disorders and Schizophrenia for School-Age Children—Present and Lifetime version (K-SADS-PL). Mothers received the unstructured introductory interview, the diagnostic screening interview, and if required Supplement Completion Checklist and upon fulfilment of screening criteria the appropriate diagnostic supplements. The child accomplished the screening interview of the K-SADS and in case of positive screening for mood or anxiety disorders the respective supplements of the K-SADS-PL. Additionally, the Child Behavior Checklist and a German Teachers’ Report on ADHD symptoms according to DSM-IV were assessed. ADHD combined subtype had to fulfil at least six criteria for inattention and at least six for hyperactive–impulsive symptoms, the inattentive subtype at least six symptoms of inattentiveness and less than six of hyperactivity–impulsivity, and the hyperactive–impulsive subtype at least six symptoms of hyperactivity–impulsivity and less than five symptoms of inattentiveness.
Exclusion criteria were IQ ≤ 75, confounding psychiatric disorders such as schizophrenia, any pervasive developmental disorder, Tourette’s disorder, primary mood or anxiety disorders, neurological disorders such as epilepsy, any acquired brain damage or foetal alcohol syndrome, premature deliveries, or maternal reports of severe pregnancy or birth complications.
Genotyping
DNA was extracted out of whole blood samples by standard salt precipitation. Procedures were described previously in detail (Renner et al. 2008). Genotyping of rs356219 and NACP-Rep1 was performed using the ABI PRISM SNaPshot Multiplex kit (Applied Biosystems, Germany) according to manufacturer instructions, followed by capillary electrophoresis on an ABI 3100 Genetic Analyzer. To assure high genotyping quality, potentially ambiguous genotypes were removed from analysis.
Finally, in the overall sample including index children and their siblings, 284 children of 195 families were genotyped successfully for rs356219 and 232 children of 156 families for NACP-Rep1. In the subsample consisting only of the index children, 195 children were successfully genotyped for rs356219 and 156 children for NACP-Rep1.
Statistical analysis
For all statistical analyses, PLINK v 1.9 (Purcell et al. 2007) was applied. Genotyping results of rs356219 and NACP-Rep1 were checked for Mendelian inconsistencies and set missing if aberrations occurred. None of the studied polymorphisms showed deviation from Hardy–Weinberg equilibrium. Minimum minor allele frequencies (MAF) were set to 0.1. Association of rs356219 with ADHD was tested applying the transmission disequilibrium test with two-tailed p values. Diallelic TDT was performed on SNCA NACP-Rep1 alleles 259 and 261, since the others were rare (MAF < 0.1). Thus, the 263 repeat which was determined as a risk allele for PD in previous reports could not be analysed (Maraganore et al. 2006). Bonferroni correction for multiple testing was applied.
Results
Table 1 shows the distribution of alleles of SNCA genotypes. Transmission disequilibrium test analysis revealed no over-transmission for NACP-Rep1 in affected siblings as shown in Table 2.
Since inclusion criteria differed between index children and affected siblings concerning age and ADHD subtype, a subanalysis on trios with index children only was performed (Table 2). This demonstrated a nominal association of rs356219 with ADHD, but again no association for NACP-Rep1 was detected. Table 3 shows the demographic characteristics of the sample genotyped for rs356219 with details on ADHD subtypes and comorbidities.
Discussion
In the present pilot study, we tested the hypothesis whether genetic variants in SNCA (promoter and 3′-region) are associated with ADHD. We found no association between ADHD and the NACP-Rep1 as well as rs356219 polymorphism in the overall sample including affected siblings. In the analysis including index children only, again no association was found for NACP-Rep1. However, an over-transmission of rs356219 was detected.
This interesting finding may be explained by the differing inclusion criteria. The index children were required to be at least 8 years old and suffer from combined type ADHD, whereas affected siblings were older than 6 years and could be affected by all subtypes (Table 3). The lower limit was chosen in order to ensure the relative persistence of ADHD symptoms and to exclude children who may show phenocopies of the disorder during preschool age but do not fulfil diagnostic criteria for ADHD during subsequent developmental stages (Shelton et al. 2000; Barkley et al. 2002).
Due to the small sample size, subanalyses regarding, for example, subtype, severity of symptoms, or comorbidities, were not possible. Yet, the result indicates a differential influence of rs356219 in ADHD subgroups and it would be highly interesting to perform subanalyses regarding symptomatology especially on potential underlying endophenotypes like emotional dysfunction or motoric patterns. Our sample was part of the newest ADHD GWAS by Demontis et al. (2018) on more than 20.000 individuals affected by ADHD and more than 30.000 controls. In this GWAS, rs356219 was also covered, but showed no association. However, the authors reported differential correlations in the included samples and discussed heterogeneity by, for example, phenotyping of the samples as a potential underlying factor. Thus, our German family sample might reflect a subgroup with more profound quantitative traits in an overlap between ADHD and PD.
Certainly, the rather low sample size is a major limitation to our study and we consider our results as preliminary, especially since due to low frequency we were not able to analyse the 263 repeat of NACP-Rep1, which was reported as a risk allele previously (Maraganore et al. 2006). Further, it was not possible to examine the potential influence of rs356210 on ADHD core symptoms or traits like irritability. However, since this is the first study on SNCA in ADHD it has a pilot character and may facilitate subsequent studies with larger sample sizes and especially the possibility to differentiate according to specific symptoms.
In future studies, it would be important to investigate larger sample sizes. Additionally, the focus should be on a search for common candidate genes for ADHD and PD and studies should include also assessments of environmental factors. It might be also interesting to examine ADHD subsamples with pronounced motoric symptoms or even PD. Further studies could also analyse common genetic linkages not to the clinical phenotypes but to the underlying endophenotypes such as impulsivity, emotional dysfunctions, inattentiveness, or motoric patterns.
In conclusion, we found evidence for an association between SNCA variant rs356210 and α-synuclein function that may affect the aetiology of ADHD. Despite the small sample size, our findings underline the interesting potential link in dopamine-related neurobiology between PD and ADHD.
References
Albayrak Ö, Friedel S, Schimmelmann BG, Hinney A, Hebebrand J (2008) Genetic aspects in attention-deficit/hyperactivity disorder. J Neural Transm 115:305–315
American Psychiatric Association (2000) Diagnostic and statistical manual of mental disorders, 4th edn, Text Revision. American Psychiatric Association, Washington
Ascherio A, Schwarzschild MA (2016) The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol 15:1257–1272
Banaschewski T, Becker K, Döpfner M, Holtmann M, Rösler M, Romanos M (2017) Attention-deficit/hyperactivity disorder: a current review. Dtsch Arztebl Int 114:149–159
Barkley RA, Shelton TL, Crosswait C, Moorehouse M, Fletcher K, Barrett S, Jenkins L, Metevia L (2002) Preschool children with disruptive behavior: three-year outcome as a function of adaptive disability. Dev Psychopathol 14:45–67
Butler B, Sambo D, Khoshbouei H (2017) Alpha-synuclein moduldates dopamine neurotransmission. J Chem Anat 83:41–49
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, Baldursson G, Belliveau R, Bybjerg-Grauholm J, Bækvad-Hansen M, Cerrato F, Chambert K, Churchhouse C, Dumont A, Eriksson N, Gandal M, Goldstein JI, Grasby KL, Grove J, Gudmundsson OO, Hansen CS, Hauberg ME, Hollegaard MV, Howrigan DP, Huang H, Maller JB, Martin AR, Martin NG, Moran J, Pallesen J, Palmer DS, Pedersen CB, Pedersen MG, Poterba T, Poulsen JB, Ripke S, Robinson EB, Satterstrom FK, Stefansson H, Stevens C, Turley P, Walters GB, Won H, Wright MJ ADHD Working Group of the Psychiatric Genomics Consortium (PGC); Early Lifecourse & Genetic Epidemiology (EAGLE) Consortium; 23andMe Research Team, Andreassen OA, Asherson P, Burton CL, Boomsma DI, Cormand B, Dalsgaard S, Franke B, Gelernter J, Geschwind D, Hakonarson H, Haavik J, Kranzler HR, Kuntsi J, Langley K, Lesch KP, Middeldorp C, Reif A, Rohde LA, Roussos P, Schachar R, Sklar P, Sonuga-Barke EJS, Sullivan PF, Thapar A, Tung JY, Waldman ID, Medland SE, Stefansson K, Nordentoft M, Hougaard DM, Werge T, Mors O, Mortensen PB, Daly MJ, Faraone SV, Børglum AD, Neale BM (2018) Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. https://doi.org/10.1038/s41588-018-0269-7
Faraone SV, Asherson P, Banaschewski T, Biederman J, Buitelaar JK, Ramos-Quiroga JA, Rohde LA, Sonuga-Barke EJS, Tannock R, Franke B (2015) Attention-deficit/hyperactivity disorder. Nat Rev Dis Primers 1:15020
Fusar-Poli P, Rubia K, Rossi G, Sartori G, Balottin U (2012) Striatal dopamine transporter alterations in ADHD: pathophysiology or adaptation to psychostimulants? A meta-analysis. Am J Psychiatry 169:264–272
Geissler JM, International Parkinson Disease Genomics Consortium members, Romanos M, Gerlach M, Berg D, Schulte C (2017) No genetic association between attention-deficit/hyperactivity disorder (ADHD) and Parkinson’s disease in nine ADHD candidate SNPs. Atten Defic Hyperact Disord 9:121–127
Giros B, Jaber M, Jones SR, Wightman RM, Caron MG (1996) Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379:606–612
Jarick I, Volckmar AL, Pütter C, Pechlivanis S, Nguyen TT, Dauvermann MR, Beck S, Albayrak Ö, Scherag S, Gilsbach S, Cichon S, Hoffmann P, Degenhardt F, Nöthen MM, Schreiber S, Wichmann HE, Jöckel KH, Heinrich J, Tiesler CM, Faraone SV, Walitza S, Sinzig J, Freitag C, Meyer J, Herpertz-Dahlmann B, Lehmkuhl G, Renner TJ, Warnke A, Romanos M, Lesch KP, Reif A, Schimmelmann BG, Hebebrand J, Scherag A, Hinney A (2014) Genome-wide analysis of rare copy number variations reveals PARK2 as a candidate gene for attention-deficit/hyperactivity disorder. Mol Psychiatry 19:115–121
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet 386:896–912
Kehagia AA, Housden CR, Regenthal R, Barker RA, Müller U, Rowe J, Sahakian BJ, Robbins TW (2014) Targeting impulsivity in Parkinson’s disease using atomoxetine. Brain 137:1986–1997
Krauel K, Feldhaus HC, Simon A, Rehe C, Glaser M, Flechtner HH, Heinze HJ, Niehaus L (2010) Increased echogenicity of the substantia nigra in children and adolescents with attention-deficit/hyperactivity disorder. Biol Psychiat 68:352–358
Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Krüger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C; Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium (2006) Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA 296:661–670
Mehler-Wex C, Riederer P, Gerlach M (2006) Dopaminergic dysbalance in distinct basal ganglia neurocircuits: implications for the pathophysiology of Parkinson’s disease, schizophrenia and attention deficit hyperactivity disorder. Neurotox Res 10:167–179
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575
Renner TJ, Walitza S, Dempfle A, Eckert L, Romanos M, Gerlach M, Schäfer H, Warnke A, Lesch KP, Jacob C (2008) Allelic variants of SNAP25 in a family-based sample of ADHD. J Neural Transm 115:317–321
Romanos M, Weise D, Schliesser M, Schecklmann M, Löffler J, Warnke A, Gerlach M, Classen J, Mehler-Wex C (2010) Abnormality of substantia nigra in attention-defi-cit/hyperactivity disorder. J Psychiatry Neurosci 35:55–58
Shelton TL, Barkley RA, Crosswait C, Moorehouse M, Fletcher K, Barrett S, Jenkins L, Metevia L (2000) Multimethod psychoeducational intervention for preschool children with disruptive behavior: two-year post-treatment follow-up. J Abnorm Child Psychol 28:253–266
Sian J, Gerlach M, Youdim MB, Riederer P (1999) Parkinson’s disease: a major hypokinetic basal ganglia disorder. J Neural Transm 106:443–476
Sidhu A, Wersinger C, Vernier P (2004) α-Synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson’s disease. FEBS Lett 565:1–5
Thapar A, Cooper M (2016) Attention deficit hyperactivity disorder. Lancet 387:1240–1250
The Brainstorm Consortium (2018) Science 360, eaap8757. https://doi.org/10.1126/science.aap8757
Venda LL, Cragg SJ, Buchman VL, Wade-Martins R (2010) α-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci 33:559–568
Walitza S, Romanos M, Warnke A, Greenhill L, Gerlach M (2014) Psychostimulants and other drugs used in the treatment of attention-deficit/hyperactivity disorder (ADHD). In: Gerlach M, Warnke A, Greenhill L (eds) Psychiatric drugs in childhood and adolescents. Springer, Vienna
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gerlach, M., Sharma, M., Romanos, M. et al. Family-based association study on functional α-synuclein polymorphisms in attention-deficit/hyperactivity disorder. ADHD Atten Def Hyp Disord 11, 107–111 (2019). https://doi.org/10.1007/s12402-019-00286-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12402-019-00286-8