
Abstract
Introduction
Clopacin® (Acino Pharma AG) is a proprietary, besylate salt and lactose-free formulation of the widely-used anti-platelet treatment, clopidogrel. This study aimed to evaluate the bioequivalence of Clopacin® with the originator as reference drug, using a guideline-compliant trial design: open-labeled, randomized, single-dose (clopidogrel 75 mg tablet), two-period, crossover trial in 48 healthy male volunteers, with a 7 day wash-out period.
Methods
Plasma samples were collected at intervals and extracted before quantifying clopidogrel concentrations using a fully validated LC–MS/MS method. Bioequivalence of Clopacin® and the reference drug was established by comparison of the primary pharmacokinetic parameters, C max, AUC0–t, and AUC0–∞.
Results
The parameter values were similar for the two products (analysis of variance) and provided Clopacin/reference ratios (least squares means) of >90% and 90% confidence intervals (CIs 84.64–105.50%, 90.43–111.22%, 88.75–110.71%, respectively) that were well within the limits set for defining bioequivalence, according to international guidelines. The respective Clopacin® and reference drug values for mean time to maximal plasma clopidogrel concentration (t max) were 0.83 and 0.91 h, and for terminal elimination half-life were 3.99 and 3.51 h. The intra-subject coefficients of variability for maximal plasma clopidogrel concentration (C max), area under the plasma clopidogrel concentration versus time curve, at 48 h (AUC0–t) and extrapolated to infinity (AUC0–∞) were 32.2%, 30.2%, and 28.9% (least square means), respectively, and the respective power values were 99.5%, 97.1%, and 95.3%.
Conclusion
This bioequivalence study provided robust clopidogrel pharmacokinetic data that established the bioequivalence of Clopacin® and the reference originator drug.
Funding
Acino Pharma AG (formerly Cimex AG)
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Platelet aggregation plays a major role in the pathogenesis of atherosclerosis and thrombosis, and is initiated by the release of platelet aggregating substances, such as thromboxane A2 [1]. Antiplatelet drugs reduced the incidence of stroke, myocardial infarction and other occlusive vascular diseases [2]. Clopidogrel (CAS 113665-84-2) is one of the most commonly prescribed antiplatelet drugs [3]. It is a thienopyridine and acts via irreversible inhibition of the platelet P2Y12 adenosine diphosphate receptor [4, 5]. Its clinical utility, as an oral preparation, in preventing and treating cardiovascular disease has been established in several large scale clinical trials [6–10]. It is now established as a popular antithrombotic treatment option in the management of peripheral vascular disease [11] and acute coronary syndrome [12, 13].
The original clopidogrel product is now out of patent and several generic products are now additionally available [14]. Like the originator, these are all immediate-release formulations of orally-administered, systemically-active drugs. For such drug formulations, the European Medicines Agency (EMA) [15, 16] and United States Food and Drug Administration (FDA) [17] have published similar guidelines on the in vivo bioavailability studies that are required to establish bioequivalence of the generic and originator versions. Described here is a bioequivalence study that was part of a successful generic licensing application for Clopacin® (EU/1/09/532/001–007; Acino Pharma AG). The trial was conducted according to the EU guidelines that were current at the time [15]. The results of this study are discussed here with reference to the current, revised version of these guidelines [16]. The study was performed almost 10 years ago but is being reported now because of uncertainty among clinicians about switching to clopidogrel generic formulations, possibly based on doubts concerning the use of certain salt formulations [18] and/or on concerns about the validity of bioequivalence studies of clopidogrel [19].
Drug formulations of clopidogrel use salts of clopidogrel because the clopidogrel free base is unstable and because, as a salt, clopidogrel is more water-soluble [14]. The original product formulation contains the hydrogen-sulfate salt of clopidogrel while several alternative salt formulations are represented in generic formulations, including besylate (sometimes referred to as besilate), hydrochloride, resinate and napadisilate. Clopacin® contains clopidogrel besylate. Besylate is a sulfonic acid salt and a commonly used counter-ion in pharmaceutical preparations [20, 21]. Because of their pharmaceutical utility, the use of sulfonic acid salts in pharmaceutical development has been well-characterized, including in regard to the exclusion of any potential generation of toxic contaminants during drug manufacture [18]. Clopidogrel besylate has similarly good solubility in acidic media as other tested clopidogrel salts, including the hydrogen-sulfate salt, and is suitable for pharmaceutical production [22].
Generic medicinal products are currently defined in the European Union (EU) directive on medicinal products for human use [23] and by the FDA [24] as drugs that are bioequivalent to the reference originator drug. The EU and FDA define bioequivalence as sharing (1) the same qualitative and quantitative composition of active substance, (2) the same pharmaceutical form and (3) the same bioavailability of active substance, as demonstrated by the appropriate studies. These regulatory directives also state that different salt forms of the same active ingredient are considered to be the same active substance, unless the salt has significant effects on the efficacy and safety of the active substance [23, 24]. Thus, “pharmaceutical alternatives” (formulations containing different salt forms of the same active ingredient) and “pharmaceutical equivalents” (formulations containing the same salt form of the same active ingredient) are both considered generic, if they are shown to be bioequivalent to the originator [24].
Clopidogrel is a prodrug, being converted to its short-lived active metabolite, via two sequential, cytochrome-P450 isoenzyme (CYP)-dependent, oxidative steps and the generation of the major circulating metabolite SR26334 (2-oxo-clopidogrel) [25, 26]. Because of the rapid metabolism of clopidogrel, several early pharmacokinetic and bioequivalence studies of clopidogrel used measurement of SR26334 to obtain pharmacokinetic data [26–29]. Furthermore, Pawlowski and colleagues [19] have questioned the reliability of pharmacokinetic data based on clopidogrel measurement, with particular reference to the data of the first study to provide pharmacokinetic data based on measurement of clopidogrel [30]. Several more recent studies have provided pharmacokinetic data based on measurement of clopidogrel [31–35]. The bioequivalence study presented here also used measurement of clopidogrel, which is consistent with the current EMA [16] and FDA guidelines [17] that clearly state that, in the case of prodrugs, it is the pharmacokinetics of the parent compound, and not a metabolite, that is relevant for assessing bioequivalence.
Methods
The trial was conducted between March 10, 2006 and March 26, 2006, by the Contract Research Organisation, Lambda Therapeutic Research Ltd, at Premier House-l, Gandhinagar-Sarkhej Highway, Bodakdev, Ahmedabad-380 054, Gujarat, India (http://www.lambda-cro.com/). The trial was sponsored by Acino Pharma AG (formerly Cimex AG), Switzerland.
Ethics
Ethical approval of the study protocol, the informed consent form and other appendices was provided by an independent ethics committee (in Suraksha on 10 March 2006), which was kept informed of all the adverse events (AEs) occurring during the study. The study was compliant with the 1996 version of ICH guidelines for good clinical practice (GCP) and conducted according to a number of ethical guidelines for medical research on human subjects; those of the Declaration of Helsinki, of the World Medicine Association (WMA) (Tokyo, 2004) and of the Indian Council of Medical Research (1980).
The trial subjects were healthy volunteers, who were screened within 21 days prior to entering into the study after first providing their written informed consent.
Subjects
The target number of trial participants was 48. Healthy volunteers were enrolled.
Investigational Drugs
The test drug was the proprietary film-coated tablet formulation, Clopacin® (batch, 060281-75FT), which is manufactured and marketed by Acino Pharma AG (Aesch, Switzerland) (formerly Cimex AG, Liesberg, Switzerland), and contains clopidogrel 75 mg as a besylate salt. The reference, comparator drug was the film-coated tablet, originator product (batch, 501252), which is manufactured by Sanofi (formerly by Sanofi-Synthelabo), and contains clopidogrel 75 mg as the hydrogen-sulfate salt. Clopacin® contains the following excipients:—in the tablet core—macrogol 6000, microcrystalline cellulose (E460), crospovidone type A, hydrogenated castor oil;—in the film-coating—macrogol 6000, ethylcellulose (E462), titanium dioxide (E 171). The reference formulation contains the following excipients:—in tablet core—mannitol (E421), macrogol 6000, microcrystalline cellulose (E460), hydrogenated castor oil, low substituted hydroxypropylcellulose;—in film-coating—hypromellose (E464), lactose, triacetin (El518), titanium dioxide (E 171), red iron oxide (El72), carnauba waxed. So, Clopacin® is lactose-free and the reference product is not.
Trial Design and Procedure
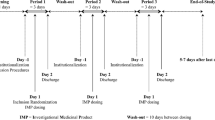
The study was an open-label (assessor blinded), balanced, randomized, two-treatment, two-period, two-sequence, single dose, crossover, comparative oral bioavailability study in healthy, adult, human male subjects under fasting conditions. The study design was compliant with the EMA’s guidance on drug bioavailability and bioequivalence studies, as was current at the time of the trial [15]. This comparative, crossover, bioequivalence study is compliant with the current EMA guideline [16]. Based on the sponsors in-house data derived from pilot studies, a sample size of 48 subjects was considered to be sufficient to establish bioequivalence between clopidogrel formulations under fasting conditions with adequate power.
Subjects were screened within 21 days prior to drug administration in period I. Possible drug abuse was tested via urine sample collected from each subject during the screening period. Subjects received one dose per study period and there was a wash-out period of 4 days between study periods I and II. A breath test for alcohol consumption was made on each subject immediately prior to each study period. Samples were collected for assessing the hematological status of each subject before starting study period II. During each study period, plasma samples were collected for measurement of clopidogrel concentrations for pharmacokinetic analysis.
Randomization
The order of receiving test and reference drugs during both periods of the trial were determined for each subject using a randomization schedule generated by the SAS statistical software, Version 9.1 (SAS Institute Inc., Cary, NC, USA).
Concomitant Medication
Subjects were instructed not to take any medicine at any time within 14 days prior to the investigational drug administration or during the trial.
Treatment and Trial Procedure
One tablet of clopidogrel free base 75 mg, which is the standard daily dose, was administered to each subject once in each period, and in the morning after 10 h of fasting. With the subject in a sitting position, one tablet of either of the investigational products was administered orally with 240 mL water. Subjects were instructed not to lie down for the next 90 min.
In each study period, venous blood samples were collected at the following intervals: immediately prior to drug administration and at 0.17, 0.33, 0.5, 0.67, 0.83, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 6, 8, 12, 16, 24, 36 and 48 h after drug administration. The time of collection of each blood sample was recorded and was taken as the time at the end sample collection—a tolerance of ±2 min was allowed in relation to the scheduled timing of sampling. The blood samples were collected into pre-labeled, heparinized vacutainers (Becton–Dickinson) placed in wet ice bath.
Analytical Procedures
The plasma samples of subjects were analyzed at the bioanalytical facility of Lambda Therapeutic Research Ltd (Ahmedabad, India). After liquid–liquid extraction of the plasma samples, using carbamazepine as internal standard, clopidogrel plasma concentrations were determined, using a chromatography/mass spectroscopy/mass spectroscopy (LC–MS/MS) (MDS SCIEX API 4000) method. The method had been validated according to International Guidelines [36] and the analyses were conducted according to Good Laboratory Practice guidelines. The methodology was similar to that used in other published studies of clopidogrel pharmacokinetics [37–39].
After injection of 20 µL of sample, chromatographic separation was performed on a micropore reversed-phase (C-18) column (150 × 4.60 mm), with an isocratic mobile phase (90% acetonitrile and 10% 2 mM ammonium acetate buffer, pH 3.0) and a flow rate of 1.2 mL/min. Clopidogrel and carbamazepine were monitored in the positive ion mode using multiple reaction monitoring transitions of m/z 322.10–212 and of m/z 237.20–194.10, respectively, with dwell times of 200 ms for each of the transitions. The retention times for clopidogrel and carbamazepine were about 2.5 min and 1.5 min, respectively. The chromatograms of the plasma extracts were free from significant interference at these retention times. An 8-point calibration curve was employed, with clopidogrel standard concentrations range of 10.36–4999.03 pg/mL. The goodness-of-fit (r 2) was mostly >0.99 and the lower limit of quantification of plasma clopidogrel was 10.36 pg/mL.
For the back-calculated concentrations of the calibration values of the standard curves, the precision varied from 2.5% to 5.8% and the accuracy from 95.8% to 102.2%. The respective values for inter-day precision and accuracy of quality control samples measured during the study were 8.4% and 101.1% (4474.93 pg/mL), 8.3% and 97.1% (2483.59 pg/mL) and 9.6% and 94.2% (30.70 pg/mL).
Pharmacokinetic Analysis
The timing of blood sample collection for the pharmacokinetic analysis was based on the previously reported pharmacokinetics of clopidogrel [31–35]. The sample clopidogrel concentration values were plotted on linear and semi-logarithmic scales to produced separate profiles of plasma clopidogrel concentration versus time for the test and reference drug, for each subject. The pharmacokinetic parameters were then derived for each subject from the individual concentration–time profiles of plasma clopidogrel, using a non-compartmental model and the WinNonlin Professional Software (Version-5.0.1, Pharsight Corporation, USA). The study protocol indicated that values of AUC0–∞ that have extrapolated areas that are >20% of total AUC0–∞ should be excluded from the pharmacokinetic analysis. Values below the lower limit of quantification of plasma clopidogrel were taken as zero.
Statistical Methods
Analysis of variance (ANOVA) was to be carried out for untransformed and natural log-transformed pharmacokinetic parameters, C max, AUC0–t and AUC0–∞. If the probability associated with their F-ratio was <0.05, this was considered as statistically significant. Least squares mean values for untransformed and the natural log-transformed pharmacokinetic parameters were computed.
The bioequivalence of the test and comparator drugs was determined according to European guidelines [16]. This was based on a statistical comparison of the parametric 90% confidence intervals (CI) of the primary outcome pharmacokinetic parameters—C max, AUC0–t, AUC0–∞. Two one-sided tests were applied to the ratios of least squares mean values of the two test drugs formulations, using root mean square error for untransformed and log-transformed data. Bioequivalence of test and comparator drugs was indicated if the 90% CI was within the acceptance range, which was 75–133% for C max and 80–125% for AUC0–t and AUC0–∞. Intra-subject variability and power of the three parameters were calculated for un-transformed and log-transformed pharmacokinetic parameters, using root mean square error values.
The wider limits of the 90% CI for C max were justified on account of the variability of plasma clopidogrel levels recorded in published studies [19]. This is in accordance with the guidelines [15, 16], which allow a higher tolerance for the mean C max ratio for drug products that have variable blood levels of the active substance and are not drugs with a narrow therapeutic index. Based on current clinical experience, clopidogrel is not considered to have a narrow therapeutic index [40].
Results
Patient Disposition
Fifty-two male subjects were screened. Two subjects tested positive for drug abuse in the urine analysis and were not enrolled in the trial. Another subject left of his own accord. Thus, 49 subjects were enrolled. The 49th subject to be enrolled did not enter study period I as the target of 48 subjects was reached and all these subjects complete the study period I. Subsequently, two more subjects withdrew from the trial, both before entering study period II, one on medical grounds and the other of his own will. Thus, 46 subjects completed the trial and plasma samples from 47 subjects (including those from the subject who withdrew on medical grounds) were analyzed. The mean demographic data for subjects completing each study period are shown in Table 1. A few deviations from the protocol were recorded in regard to collection of blood samples but these were judged to have had no significant impact on the study analyses and conclusions. There was 100% compliance recorded for oral intake of drug and this was reflected in the results of clopidogrel assay.
Pharmacokinetics
Presented in Fig. 1 are plots of the mean plasma clopidogrel concentrations against time over 48 h after administration of each of the two investigational drugs. The plasma concentrations of clopidogrel did not differ significantly between the reference drug and Clopacin®. The mean values of pharmacokinetic parameters derived from the plot of the untransformed data were very similar (Table 2). With both Clopacin® and the reference drug, the maximum clopidogrel plasma concentration was reached at a similar mean time of less than 1 h. The respective mean terminal elimination half-lives (t 1/2) were also similar (3.99 and 3.51 h, respectively). The statistical analyses of the primary pharmacokinetic parameters indicated the bioequivalence of Clopacin® and the reference drug (Table 3). The Clopacin®/reference drug ratio of the mean (geometric least-squares) of AUC0–∞, AUC0–t , and C max were all >90% and close to 100% and had 90% CI values (88.75–110.71%, 90.43–111.22%, 84.64–105.50%, respectively) that are well within the limits for bioequivalence (80–125% for the AUC parameters and 75–133% for C max). The ANOVA P values (Table 4) indicate the lack of any significant effect of the different product formulations on C max, AUC0–t, and AUC0–∞, as well as no significant period or sequence effect on these parameters (Table 4).

Mean plasma clopidogrel concentrations in subjects after oral administration of 75 mg clopidogrel, as Clopacin® or as reference drug
The bioequivalence study guideline that was current at the start of this trial [15] allowed exclusion from the analysis of AUC0–∞ values with >20% of AUC as extrapolation if, as in this study, it was stated in the trial protocol prior to starting the trial. This procedure is not encouraged in the current guideline [16], which states that the validity of the study should be questioned if >20% of the subjects’ AUC0–∞ values included >20% of AUC as extrapolation. Table 3 indicates that 19.5% (9/46) subjects had AUC0–∞ values for Clopacin® (n = 5) and/or reference drug (n = 5) excluded from the analysis because their values included >20% of AUC as extrapolation.
Safety Assessment
Each subject was exposed to 75 mg of clopidogrel once in each study period and had a washout period of 7 days between the two dosing days, in accordance with the randomization schedule.
Safety data are summarized in Table 5. Seven AEs were reported during the trial, with two AEs occurring prior to study period I, three during study period I, one during the washout period and one during study period II. All the AEs were mild and resolved. None caused death or were serious AEs but two were significant. The two significant AEs were recorded during study period I and were considered by the attendant physician to be possibly related to the investigational drug. Both were resolved. In one case, the subject had received Clopacin® and experienced vomiting and indigestion almost 12 h after drug intake. The AE was mild but required medication and was of 1.5 h duration. In the other case, the subject had received the reference drug and experienced diarrhea almost 20 h later. The AE was mild but required medication and was intermittent over a period of just over 2 days. The clinical laboratory values were all considered to be within clinically acceptable ranges.
Discussion
The bioequivalence study presented here was designed to conform to the European guideline on testing for bioequivalence [15] that was current at the time of the trial. The trial design is also fully compliant with the latest version of this guideline [16] and with the latest FDA guideline [17]. The current European guideline differs from the earlier version primarily by its specific focus on immediate-release drugs that act systemically, such as the clopidogrel formulations tested here. The specifics of the recommended trial design are precisely those used in this trial, namely a randomized, two-treatment, two-period, two-sequence, crossover study in healthy, adults of either single or mixed gender under fasting conditions. The statistical analyses applied in this trial were also fully compliant with the current guideline.
Bioequivalence of Clopacin® and the reference drug was established by comparison of the primary pharmacokinetic parameters, C max, AUC0–t and AUC0–∞. The Clopacin/reference ratio of the mean values of these parameters were all well-above 90% and the respective 90% CIs were well within the limits (80–125% for AUC parameters; 75–133% for C max) that were set for defining bioequivalence.
The 90% CI value for C max (84.64–105.50%) was actually well within 80–125%, which is inconsistent with the pre-trial assumption that clopidogrel drug products are ‘highly variable’ due to reported inter-individual variation in rates of clopidogrel metabolism [41]. In this trial, the calculated intra-subject coefficients of variation (CVs) for the pharmacokinetic parameters were close to or slightly above 30%. For C max, which had a CV of 32.2%, a value above 30% defines a ‘highly variable drug product’ according to the bioequivalence study guidelines [15, 16]. The high intra-subject variability (CV) for C max observed in this study is similar to other published data from pharmacokinetic studies of clopidogrel products [33–35]. However, much greater intra-subject variability in clopidogrel pharmacokinetics was reported in some published studies [19].
The safety data obtained in this study indicate that Clopacin® has a similar safety profile to that of the reference product, with no evidence of any significant safety issues. The pharmacokinetic data are robust, as revealed by the high power values (for detecting a 20% difference between test and reference drug) calculated for C max, AUC0–t and AUC0–∞ (log-transformed data). The power values were 99.5%, 97.1%, and 95.3%, respectively. The study also confirms the feasibility of using measurement of plasma concentrations of the prodrug, clopidogrel, to produce reliable pharmacokinetic data. According to current guidelines, this is essential for establishing the bioequivalence with the reference drug. Clopacin® is now approved and marketed in Europe [42, 43] and differs advantageously from the reference product in being a lactose-free formulation.
These data are important not only as a significant addition to the published data on clopidogrel generics drugs but because clopidogrel is a widely-used anti-platelet treatment and clinicians need to be assured of the availability of generic formulations of proven bioequivalence.
Conclusions
The pharmacokinetic data presented here establish that the Clopacin® formulation, which contains clopidogrel besylate salt and is lactose-free, is bioequivalent to the reference originator clopidogrel product, which contains clopidogrel hydrogen-sulfate and lactose. Also, both formulations were shown to have similar safety profiles.
References
Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340(2):115–26.
Anon X. Collaborative overview of randomised trials of antiplatelet therapy–I: prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. Antiplatelet Trialists’ Collaboration. BMJ. 1994;308(6921):81–106.
Tsoumani ME, Ntalas IV, Goudevenos JA, Tselepis AD. Evaluating the bioequivalence of clopidogrel generic formulations. Curr Med Res Opin. 2015;31(5):861–4.
Cattaneo M. The platelet P2Y receptors as targets for new antithrombotic drugs. J Thromb Haemost. 2003;1(6):1133–5.
Savi P, Herbert JM. Clopidogrel and ticlopidine: P2Y12 adenosine diphosphate-receptor antagonists for the prevention of atherothrombosis. Semin Thromb Hemost. 2005;31(2):174–83.
Chen ZM, Jiang LX, Chen YP, Xie JX, Pan HC, Peto R, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366(9497):1607–21.
Mehta SR, Yusuf S, Peters RJ, Bertrand ME, Lewis BS, Natarajan MK, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet. 2001;358(9281):527–33.
Sabatine MS, Cannon CP, Gibson CM, Lopez-Sendon JL, Montalescot G, Theroux P, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med. 2005;352(12):1179–89.
Steinhubl SR, Berger PB, Mann JT 3rd, Fry ET, DeLago A, Wilmer C, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA. 2002;288(19):2411–20.
Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345(7):494–502.
Rooke TW, Hirsch AT, Misra S, Sidawy AN, Beckman JA, Findeiss L, et al. Management of patients with peripheral artery disease (compilation of 2005 and 2011 ACCF/AHA Guideline Recommendations): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;61(14):1555–70.
Amsterdam EA, Wenger NK, Brindis RG, Casey DE Jr, Ganiats TG, Holmes DR Jr, et al. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130(25):e344–426.
O’Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos JA, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;127(4):e362–425.
Tsoumani ME, Kalantzi KI, Goudevenos IA, Tselepis AD. Clopidogrel generic formulations in the era of new antiplatelets: a systematic review. Curr Vasc Pharmacol. 2014;12(5):766–77.
EMA, CPMP/EWP/QWP/1401/98: Note for Guidance on the Investigation of Bioavailability and Bioequivalence—revision—came into operation in 2002. European Medical Agency 2001.
EMA, CPMP/EWP/QWP/1401/98 Rev. 1/Corr **: Guideline on the investigation of bioequivalence. European Medical Agency 2010.
FDA, Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA. Food and Drug Administration, Center for Drug Evaluation and Research (CDER) 2013.
Elder DP, Delaney E, Teasdale A, Eyley S, Reif VD, Jacq K, et al. The utility of sulfonate salts in drug development. J Pharm Sci. 2010;99(7):2948–61.
Pawlowska M, Duda J, Tejchman-Malecka B, Bogiel M, Marzec A, Sieradzki E. Usefulness of the parent compound determination in bioequivalence evaluation of clopidogrel generic products. Arzneimittelforschung. 2009;59(6):289–96.
Paulekuhn GS, Dressman JB, Saal C. Trends in active pharmaceutical ingredient salt selection based on analysis of the Orange Book database. J Med Chem. 2007;50(26):6665–72.
Seo JH, Park JB, Choi WK, Park S, Sung YJ, Oh E, et al. Improved oral absorption of cilostazol via sulfonate salt formation with mesylate and besylate. Drug Des Devel Ther. 2015;9:3961–8.
Zupancic V, Smrkolj M, Benkic P, Simonic I, Plevnik M, Ritlop G, et al. Preformulation investigation of some clopidogrel addition salts. Acta Chim Slov. 2010;57(2):376–85.
European Union, Directive 2001/83/EC of The European Parliament and of the Council: Community Code relating to Medicinal Products for Human Use (as amended). 2012 Article 10, 2(b).
FDA, (US, Food and Drug Administration), 21CFR320.1(c). 2015.
Savi P, Pereillo JM, Uzabiaga MF, Combalbert J, Picard C, Maffrand JP, et al. Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost. 2000;84(5):891–6.
Caplain H, Donat F, Gaud C, Necciari J. Pharmacokinetics of clopidogrel. Semin Thromb Hemost. 1999;25(Suppl 2):25–8.
Lagorce P, Perez Y, Ortiz J, Necciari J, Bressolle F. Assay method for the carboxylic acid metabolite of clopidogrel in human plasma by gas chromatography-mass spectrometry. J Chromatogr B Biomed Sci Appl. 1998;720(1–2):107–17.
Ksycinska H, Rudzki P, Bukowska-Kiliszek M. Determination of clopidogrel metabolite (SR26334) in human plasma by LC-MS. J Pharm Biomed Anal. 2006;41(2):533–9.
Souri E, Jalalizadeh H, Kebriaee-Zadeh A, Shekarchi M, Dalvandi A. Validated HPLC method for determination of carboxylic acid metabolite of clopidogrel in human plasma and its application to a pharmacokinetic study. Biomed Chromatogr. 2006;20(12):1309–14.
Lainesse A, Ozalp Y, Wong H, Alpan RS. Bioequivalence study of clopidogrel bisulfate film-coated tablets. Arzneimittelforschung. 2004;54(9A):600–4.
El Ahmady O, Ibrahim M, Hussein AM, Bustami RT. Bioequivalence of two oral formulations of clopidogrel tablets in healthy male volunteers. Int J Clin Pharmacol Ther. 2009;47(12):780–4.
Garces-Eisele J, Ruiz-Arguelles A, Estrada-Marin L, Reyes-Nunez V, Vazquez-Perez R, Guzman-Garcia O, et al. Pharmacogenetic selection of volunteers increases stringency of bioequivalence studies; the case of clopidogrel. Indian J Pharm Sci. 2014;76(4):281–6.
Kim SD, Kang W, Lee HW, Park DJ, Ahn JH, Kim MJ, et al. Bioequivalence and tolerability of two clopidogrel salt preparations, besylate and bisulfate: a randomized, open-label, crossover study in healthy Korean male subjects. Clin Ther. 2009;31(4):793–803.
Richter W, Erenmemisoglu A, Van der Meer MJ, Emritte N, Tuncay E, Koytchev R. Bioequivalence study of two different clopidogrel bisulfate film-coated tablets. Arzneimittelforschung. 2009;59(6):297–302.
Setiawati E, Yunaidi DA, Handayani LR, Santoso ID, Setiawati A, Tjandrawinata RR. Bioequivalence study of two clopidogrel film-coated tablet formulations in healthy volunteers. Arzneimittelforschung. 2011;61(12):681–4.
ICH, ICH harmonised tripartite guideline: Validation of analytical procedures: Text and methodology Q2(R1) (subsequently updated in 2005 as ICH guideline Q2B) 1996.
Brandt JT, Close SL, Iturria SJ, Payne CD, Farid NA, Ernest CS 2nd, et al. Common polymorphisms of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J Thromb Haemost. 2007;5(12):2429–36.
Kim BH, Kim JR, Lim KS, Shin HS, Yoon SH, Cho JY, et al. Comparative pharmacokinetics/pharmacodynamics of clopidogrel besylate and clopidogrel bisulfate in healthy Korean subjects. Clin Drug Investig. 2012;32(12):817–26.
Taubert D, Kastrati A, Harlfinger S, Gorchakova O, Lazar A, von Beckerath N, et al. Pharmacokinetics of clopidogrel after administration of a high loading dose. Thromb Haemost. 2004;92(2):311–6.
Steinhubl SR. Genotyping, clopidogrel metabolism, and the search for the therapeutic window of thienopyridines. Circulation. 2010;121(4):481–3.
Simon T, Verstuyft C, Mary-Krause M, Quteineh L, Drouet E, Meneveau N, et al. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med. 2009;360(4):363–75.
EMA, EMA website: http://www.ema.europa.eu/ema/.
Swissmedic, Swissmedic website: http://www.swissmedicinfo.ch/.
Acknowledgments
The trial was sponsored by Acino Pharma AG (formerly Cimex AG); the study was conducted by Lambda Therapeutic Research Ltd., Premier House-l, Gandhinagar-Sarkhej Highway, Bodakdev, Ahmedabad- 380 054, Gujarat, India, under contract from Acino Pharma AG (formerly Cimex AG. All the data reported here were provided to Acino Pharma AG (formerly Cimex AG) by Lambda Therapeutic Research Ltd., Premier House-l, Gandhinagar-Sarkhej Highway, Bodakdev, Ahmedabad- 380 054, Gujarat, India, under contract from Acino Pharma AG (formerly Cimex AG). The article processing charges for this publication were funded by Acino Pharma AG. The named author meets the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, takes responsibility for the integrity of the work as a whole, and has given final approval for the version to be published. The author had full access to all relevant aggregated study data required to understand and report research findings for this study and takes complete responsibility for the integrity of such data and the accuracy of the data analysis. The study protocol and clinical study report were submitted to the journal by Acino Pharma AG (formerly Cimex AG) in order to make them available to the journal Editor and the Reviewers. The manuscript was written by the author without any additional medical writing or editorial assistance.
Disclosures
Gerard P. McGregor receives consultancy fees from Acino Pharma AG and received a fee for writing this article.
Compliance with Ethics Guidelines
Ethical approval of the study protocol, the informed consent form and other appendices was provided by an independent ethics committee (in Suraksha on 10 March, 2006), which was kept informed of all the adverse events occurring during the study. The study was compliant with the 1996 version of ICH guidelines for good clinical practice (GCP) and conducted according to a number of ethical guidelines for medical research on human subjects; those of the Declaration of Helsinki, of the World Medicine Association (WMA) (Tokyo, 2004) and of the Indian Council of Medical Research (1980). Informed consent was obtained from all subjects for being included in the study.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
McGregor, G.P. Pivotal Bioequivalence Study of Clopacin®, a Generic Formulation of Clopidogrel 75 mg Film-Coated Tablets. Adv Ther 33, 186–198 (2016). https://doi.org/10.1007/s12325-016-0290-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-016-0290-0