
Abstract
Machado-Joseph disease (SCA3) is the most frequent spinocerebellar ataxia worldwide and characterized by remarkable phenotypic heterogeneity. MRI-based studies in SCA3 focused in the cerebellum and connections, but little is known about cord damage in the disease and its clinical relevance. To evaluate the spinal cord damage in SCA3 through quantitative analysis of MRI scans. A group of 48 patients with SCA3 and 48 age and gender-matched healthy controls underwent MRI on a 3T scanner. We used T1-weighted 3D images to estimate the cervical spinal cord area (CA) and eccentricity (CE) at three C2/C3 levels based on a semi-automatic image segmentation protocol. The scale for assessment and rating of ataxia (SARA) was employed to quantify disease severity. The two groups—SCA3 and controls—were significantly different regarding CA (49.5 ± 7.3 vs 67.2 ± 6.3 mm2, p < 0.001) and CE values (0.79 ± 0.06 vs 0.75 ± 0.05, p = 0.005). In addition, CA presented a significant correlation with SARA scores in the patient group (p = 0.010). CE was not associated with SARA scores (p = 0.857). In the multiple variable regression, we found that disease duration was the only variable associated with CA (coefficient = −0.629, p = 0.025). SCA3 is characterized by cervical cord atrophy and antero-posterior flattening. In addition, the spinal cord areas did correlate with disease severity. This suggests that quantitative analyses of the spinal cord MRI might be a useful biomarker in SCA3.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Machado-Joseph disease or spinocerebellar ataxia 3 (SCA3) is the most common autosomal dominant ataxia and characterized by remarkable phenotypic heterogeneity. Although ataxia is the major clinical feature in patients with SCA3, other manifestations are also frequently found, such as dystonia, pyramidal signs, dysarthria, and peripheral neuropathy [1, 2]. The disease is caused by a trinucleotide repeat expansion (CAG) located in the 10th exon of the ATXN3 gene, which encodes the protein ataxin-3 [3]. The mutation results in a protein with an abnormal chain of polyglutamines that aggregates into nuclear inclusions and interferes with its normal function leading to neuronal toxicity [4].
Neurodegeneration in SCA3 involves several regions of the central and peripheral nervous system. Pathological studies revealed severe neuronal loss predominantly in the cerebellum, brainstem, and basal ganglia [5]. This is indeed confirmed by recent quantitative neuroimaging data [6–9]. Damage to these regions accounts for the main clinical features in the disease, such as ataxia and movement disorders. However, these patients also present pyramidal signs (predominantly in the legs) and urinary dysfunction, which suggest that spinal cord damage also takes place in SCA3. This hypothesis is further supported by pathological data that revealed marked neuronal depletion at the ventral horns and dorsal root ganglia, combined with degeneration of Clarke’s columns, corticospinal tracts, vestibulospinal tracts, as well as the gracile and cuneate fasciculi [10–12].
In this scenario, it would be important to quantify spinal cord damage in these patients in vivo and to determine its clinical impact. High-field MRI scanners and novel post-processing techniques now enable reliable quantification of spinal cord morphometry [13–16]. Such analyses have proven useful to investigate similar diseases such as Friedreich’s ataxia and Amyotrophic lateral sclerosis, but were not yet fully explored in SCA3 [14, 15]. In fact, there is a single MRI-based study of cervical cord atrophy, but with a rather small sample size and analyses restricted to area measurement [16]. We have thus designed a study with a larger cohort of patients that captures the wide phenotypic variability of the disease. In addition, our analyses assessed both area and eccentricity of the cord. Our ultimate objective was to investigate the potential usefulness of quantitative spinal cord MRI as a biomarker for SCA3.
Methods
Subject’s Selection
We selected 51 consecutive patients with molecular confirmation of SCA3 regularly followed in the Neurogenetics Outpatient Clinic at UNICAMP hospital. Three patients were excluded from MRI analyses because of motion artifacts in their scans. None of the remaining 48 subjects presented other concomitant neurological or medical disease that might affect the spinal cord.
We selected a group of 48 healthy controls with age and gender matched to the 48 remaining patients with SCA3. None of these controls had family history of ataxia and/or hereditary neuropathies.
This study was approved by our institutional ethics committee and written informed consent was obtained from all participants.
Clinical Evaluation
Patients underwent detailed neurological examination and the SARA scale was used to quantify disease severity [17]. Age of onset, disease duration, and length of expansion (CAG) were determined for each subject and used for further analyses.
We classified SCA3 patients into the 4 classical clinical subtypes: I—patients with early onset, prominent dystonia and large (CAG) expansions; II—patients with prominent ataxia and subtle pyramidal signs; III—patients with late onset, peripheral neuropathy, and short (CAG) expansions; and IV—patients with a pure Parkinsonian phenotype.
MRI Analysis
Acquisition
Patients and controls underwent MRI examination on a 3T Intera Achieva-PHILIPS scanner on the same day of clinical evaluation. The images were acquired using T1-weighted 3D images covering the whole brain and the cervical spinal cord with the following acquisition parameters: TE = 3.2 ms, TR = 7.1 ms, flip angle = 8 °, voxel size = 1.0 × 1.0 × 1.0 mm, FOV = 240 × 240. Sagittal T1- and T2-weighted sequences of the cervical spinal cord were also included for anatomic evaluation and to rule out any associated abnormalities.
Segmentation and Measurements
We used the in-house-developed software SpineSeg for the quantitative analyses of the cervical cord [18]. SpineSeg resamples the MR images to correct for variations in imaging angle and the neck position, segments cross-sections of the spinal cord semi-automatically, and fits an ellipse to the segmentation. The only interactive inputs are a few nodes that define an approximation of the spine and neck curvature and a single point inside the spinal cord. The boundary of the spinal cord is segmented by automatic tree pruning, which consistently segments the object with minimal interaction, and an ellipse is fitted to the segmented object boundary.
The measurements of the cervical spinal cord were determined from sections obtained perpendicular to the lower limits (base) of the 2nd and 3rd cervical vertebrae. We used the mean values obtained from the analysis of three sequential slices at this spinal level. The following measures were used for analysis: cross-sectional area (CA) and eccentricity (CE) of the cord. All measurements were performed by a single evaluator (CNF), who was experienced with the method and blind to the clinical status of the subjects.
Cerebellar Volume
We employed the software FreeSurfer v.5.3 to segment automatically the cerebellum and calculate its total volume for each patient [19].
Statistical Analysis
Clinical and genetic data of patients and controls are detailed with descriptive statistics. Group comparison was performed with student’s t test. Differences of CA and CE in the SCA3 clinical subtypes were assessed using ANOVA. A linear regression model was used to evaluate: (i) the effects of disease duration, age at onset, and (CAG) expansion on CA; and (ii) the association between SARA scores and CA taking into account disease duration and cerebellar volume as covariates. Level of significance was set at α = 0.05 for all comparisons. Statistical analyses were performed with SYSTAT software version 9.0.
Results
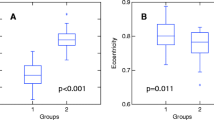
Demographic data and the spinal cord measurements of all subjects in the study are shown in Tables 1 and 2. Mean length of the expanded CAG repeat was 70.1 ± 5.4. Mean age at disease onset was 38.4 ± 12.6 y. The two groups—SCA3 and controls—were significantly different regarding CA (49.5 ± 7.3 vs 67.2 ± 6.3 mm2, p < 0.001) and CE (0.79 ± 0.06 vs 0.75 ± 0.05, p = 0.005), as we can see in Figs. 1 and 2. CA and CE were not significantly different between SCA3 clinical subtypes (p = 0.658 and 0.625, respectively). In the multiple variable regressions, we found that disease duration was the only variable associated with CA (Table 3). After covariation for disease duration and cerebellar volume, we found that CA was independently associated with SARA scores in the patient group (Table 3). CE was not associated with SARA scores (p = 0.857).

Box-and-whiskers plot showing the distribution of cord area (a) and eccentricity (b) in patients with SCA3 and controls

SpineSeg layout showing the segmentation of the cervical spinal cord in a patient with SCA3 (a) and a healthy control (b). T1 weighted sagittal images showing spinal cord atrophy in a patient with SCA3 (c) compared to a healthy control (d)
Discussion
There are several MRI-based studies in SCA3, but most of them focused in the cerebellum, brainstem, and basal ganglia [6–9]. Lukas et al. performed the first MRI-based study employing quantitative methods to investigate the spinal cord in SCA3 [16]. These authors scanned 14 patients in a 1.5T equipment and found significant atrophy at the cervical cord. However, such atrophy was not correlated with clinical parameters. In the present study, we confirmed cervical cord atrophy on a 3T scanner but found it to be significantly associated to ataxia severity and disease duration. Such difference might be explained because we recruited a much larger cohort and SARA instead of ICARS was employed as the clinical outcome measure. ICARS includes purely cerebellar items (such as dysarthria and the eye movement abnormalities) which might have biased the correlation analyses with the spinal cord parameters. Besides, a larger cohort allowed us to include an adequate number of patients with the different subtypes of the disease.
In our study, quantitative analyses revealed that patients with SCA3 have spinal cord atrophy combined with antero-posterior flattening. Damage to the ventral horns, dorsal root ganglia, and multiple tracts in the spinal cord are the pathological bases for these MRI findings [10–12]. Interestingly, the same pattern of atrophy and flattening was recently reported in a cohort of patients with Friedreich’s ataxia, which is known to harbor severe (but relatively restricted) damage to the posterior funiculi [14]. In contrast, degenerative diseases with lateral column involvement (e.g., amyotrophic lateral sclerosis) have atrophy, but no significant flattening [15]. Taken together, these data indicate that spinal damage in SCA3 resembles that found in Friedreich’s ataxia with predominant damage to posterior rather than lateral columns. This is in line with neurophysiological studies that identified reduction of sensory nerve amplitudes as a frequent finding in SCA3 [20, 21]. This hypothesis is further supported by previous reports that found hyperintense lesions in the dorsal, but not lateral funiculi of patients with SCA3 [20].
Conclusion
We have shown that SCA3 is characterized by cervical cord atrophy and flattening. In addition, the spinal cord areas did correlate with ataxia severity and disease duration. This suggests that quantitative analyses of spinal cord MRI might be a useful biomarker in SCA3. However, further prospective studies are needed to confirm its use as a prognostic parameter for these patients.
References
D’Abreu A, França Jr MC, Paulson HL, Lopes-Cendes I. Caring for Machado-Joseph disease: current understanding and how to help patients. Parkinsonism Relat Disord. 2010;16:2–7.
Maruyama H, Nakamura S, Matsuyama Z, Sakai T, Doyu M, Sobue G, et al. Molecular features of the CAG repeats and clinical manifestation of Machado–Joseph disease. Hum Mol Genet. 1995;4:807–12.
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, et al. CAG expansions in a novel gene for Machado–Joseph disease at chromosome 14q32.1. Nat Genet. 1994;8:221–8.
Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–8.
Rub U, Brunt ER, Deller T. New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado–Joseph disease). Curr Opin Neurol. 2008;21:111–6.
Lopes TM, D’Abreu A, França Jr MC, Yasuda CL, Betting LE, Samara AB, et al. Widespread neuronal damage and cognitive dysfunction in spinocerebellar ataxia type 3. J Neurol. 2013;260:2370–9.
Guimarães RP, D’Abreu A, Yasuda CL, França Jr MC, Silva BH, Cappabianco FA, et al. A multimodal evaluation of microstructural white matter damage in spinocerebellar ataxia type 3. Mov Disord. 2013;28:1125–32.
D’Abreu A, Franca Jr MC, Yasuda CL, Campos BA, Lopes-Cendes I, Cendes F. Neocortical atrophy in Machado–Joseph disease: a longitudinal neuroimaging study. J Neuroimaging. 2012;22:285–91.
Klockgether T, Skalej M, Wedekind D, Luft AR, Welte D, Schulz JB, et al. Autosomal dominant cerebellar ataxia type I. MRI-based volumetry of posterior fossa structures and basal ganglia in spinocerebellar ataxia types 1, 2 and 3. Brain. 1998;121:1687–93.
Rüb U, Seidel K, Ozerden I, et al. Consistent affection of the central somatosensory system in spinocerebellar ataxia type 2 and type 3 and its significance for clinical symptoms and rehabilitative therapy. Brain Res Rev. 2007;53:235–49.
Riess O, Rüb U, Pastore A, Bauer P, Schöls L. SCA3: neurological features, pathogenesis and animal models. Cerebellum. 2008;7:125–37.
Shimizu H, Yamada M, Toyoshima Y, et al. Involvement of Onuf’s nucleus in Machado-Joseph disease: a morphometric and immunohistochemical study. Acta Neuropathol. 2010;120:439–48.
Agosta F, Rocca MA, Valsasina P, et al. A longitudinal diffusion tension MRI study of the cervical cord and brain in amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry. 2009;80:53–5.
Chevis CF, da Silva CB, D’Abreu A, Lopes-Cendes I, Cendes F, França Jr MC, et al. Spinal cord atrophy correlates with disability in Friedreich’s ataxia. Cerebellum. 2013;12:43–7.
Branco LM, De Albuquerque M, De Andrade HM, Bergo FP, Nucci A, França Jr MC. Spinal cord atrophy correlates with disease duration and severity in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:93–7.
Lukas C, Hahn HK, Bellenberg B, Hellwig K, Globas C, Schimrigk SK, et al. Spinal cord atrophy in spinocerebellar ataxia type 3 and 6: impact on clinical disability. J Neurol. 2008;255:1244–9.
Braga-Neto P, Godeiro-Junior C, Dutra LA, Pedroso JL, Barsottini OG. Translation and validation into Brazilian version of the Scale of the Assessment and Rating of Ataxia (SARA). Arq Neuropsiquiatr. 2010;68:228–30.
Bergo FPG, França MC Jr., Chevis CF, Cendes F. SpineSeg: A 2012 Segmentation and Measurement Tool for Evaluation of Spinal Cord Atrophy. In: CISTI’2012 (7ª Conferencia Ibérica de Sistemas y Tecnologia de Información);; Madrid, Spain. IEEE; 2012. p. 400–403
Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–55.
França Jr MC, D’Abreu A¸ Nucci A, Cendes F, Lopes-Cendes I. Prospective study of peripheral neuropathy in Machado-Joseph disease. Muscle Nerve. 2009;40:1012–8.
Escorcio Bezerra ML, Pedroso JL, Pinheiro DS, Braga-Neto P, Povoas Barsottini OG, et al. Pattern of peripheral nerve involvement in Machado-Joseph disease: neuronopathy or distal axonopathy? A clinical and neurophysiological evaluation. Eur Neurol. 2013;69:129–33.
Acknowledgments
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and the Conselho Nacional de Pesquisa (CNPq). These are governmental Brazilian agencies that support research.
Conflict of Interest
The authors report no conflict of interests regarding this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fahl, C.N., Branco, L.M.T., Bergo, F.P.G. et al. Spinal Cord Damage in Machado-Joseph Disease. Cerebellum 14, 128–132 (2015). https://doi.org/10.1007/s12311-014-0619-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-014-0619-7