
Abstract
Colon cancer frequently metastasizes to the liver but the genetic and phenotypic properties of specific cancer cells able to implant and grow in this organ have not yet been established. The contribution of the patient’s genetic, physiologic and pathologic backgrounds to the incidence and development of hepatic colon cancer metastases is also presently misunderstood. At a transcriptional level, hepatic metastasis development is in part associated with marked changes in gene expression of colon cancer cells that may originate in the primary tumor. Other changes occur in the liver and are regulated by hepatic cells, which represent the new microenvironment for metastatic colon cancer cells. However, hepatic parenchymal and non-parenchymal cell functions are also affected by both tumor-derived factors and systemic host factors, which suggests that the hepatic metastasis microenvironment is a functional linkage between the hepatic pathophysiology of the colon cancer patient and the biology of its cancer cells. Therefore, together with metastasis-related gene profiles suggesting the existence of liver metastasis potential in primary tumors, new biomarkers of the prometastatic microenvironment supported by the liver reaction to colon cancer factors may be helpful for the individual assessment of hepatic metastasis risk in colon cancer patients. In addition, knowledge on hepatic metastasis gene regulation by the hepatic microenvironment may open multiple opportunities for therapeutic intervention during colon cancer metastasis at both subclinical and advanced stages.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The Cancer Microenvironment as a Functional Nexus between Cancer Cell Biology and Pathophysiology of the Cancer Patient
Pathophysiology of the Cancer Patient
When cancer cells disrupt normal physiological processes, the malignant disease moves from a pathogenic to a pathophysiologic level. Pathophysiology of the cancer patient refers to the specific alterations of body functions caused by cancer cells [1]. It includes the ensemble of biological and physical manifestations of the disease that correlate with underlying molecular abnormalities and cell disturbances in both cancer and host cells. Pathophysiologic parameters of the cancer patient represent the clinical signature of the disease. They are the basis for diagnosis and therapy and, at the same time, are useful references for translational research activities.
Pathophysiology of the cancer patient does not only depend on the cancer cells themselves. The cancer microenvironment, which is mainly constituted by tumor-activated host cells surrounding or even coexisting with cancer cells within the tumor, also remarkably contributes to the pathophysiology of the cancer patient [2]. Moreover, since abnormal cancer cells are frequently insensitive to many physiologic control factors from the host, the microenvironment of tumor-activated host cells can also act as an efficient translator of host signals to cancer cells. The microenvironment thus, represents an element with bidirectional properties having the capability to establish a functional linkage between cancer cell activities and systemic host factors, even if they come from anatomically remote organs.
Cancer microenvironment can induce and respond at the same time to tumor and host factors, acting as a relevant functional entity with an interface role between the biology of cancer cells and the complex pathophysiology of the cancer patient. For this reason, cancer microenvironment should be viewed as a constellation of tumor-activated host cells [3] that are functionally coupled not only to the tumor but also to the whole body. This suggests that specific alterations of body functions representing the pathophysiology of the cancer patient are contributed not only by cancer cell factors, but also by factors released from tumor-activated host cells at the cancer microenvironment (Fig. 1).

The cancer microenvironment as a functional nexus between cancer cell biology and pathophysiology of the cancer patient. Cancer cell activities can instruct surrounding tissues (I) and even distant organs (II) to undergo changes that promote cancer progression and metastasis. Cancer microenvironment can in turn induce and respond at the same time to cancer and host factors, acting as a functional entity with an interface role between the biology of cancer cells and the pathophysiology of the cancer patient (III-IV). Understanding the mechanisms by which cancer cells interact with their surroundings, both locally in the tumor organ and systemically in the body as a whole, may have implications for cancer prevention and therapy
During the pathogenic process of cancer metastasis, biological features of specific target organs also influence the microenvironment of metastatic cancer cells [4]. However, because dissemination of cancer cells usually occurs at advanced stages of cancer progression, target organs are already altered by previous factors released from primary tumor. Hence, the cancer metastasis microenvironment may not only depend on the anatomy and biology of the target organ [4], but also on pathophysiological features previously generated in the target organ as a consequence of primary tumor development and any other tumor-unrelated preexisting disease. For this reason, measurement of body tissue dysfunctions induced by primary tumor growth, and particularly measurement of the pathophysiological parameters that promote cancer progression is becoming increasingly relevant at a clinical level, not only for better understanding cancer biology and progression, but also for predicting metastasis risk and selecting the best treatment options for every patient.
Subclinical Events of Developing Cancer Pathophysiology
When a primary tumor is still microscopic, its systemic effects are negligible and it is difficult to recognize them clinically on the basis of pathophysiological parameters. However, early in the subclinical stage, primary tumors have already regional effects, which precede the later systemic effects. In particular, subclinical effects include those resulting from compression, destruction and replacement of normal tissues around the tumor. Physical microenvironment is increasingly recognized as having a major influence on cellular phenotype of host cells that are responsive to stretching [5]. Recent data have emphasized the importance of mechano-signal transduction pathways (mechanotransduction) in tumor progression [6]. In these cases, external forces are transmitted via cell surface receptors to cytoskeletal and signaling proteins inside the cell, driving phenotypic changes. Next, cancer cell growth can also affect its surrounding microcirculation, leading to ischemic areas whose subsequent reperfusion can increase environmental release of reactive oxygen intermediates (ROIs), promoting tissue damage, inflammation and altered gene expression to both host and cancer cells [7]. These tissue disturbances are further fostered by tumor-derived soluble factors also acting on normal tissues and promoting recruitment of stromagenic and preangiogenic cells. Host cell enrichment within cancer tissue largely varies but its growth as a tumor-specific stroma, adept at the tumor, is also promoted by tumor-derived factors [8], and it therefore represents an indirect biomarker of tumor functionality. Moreover, while tumor stroma is under cancer cell control, it is composed of normal cells that can also respond to physiologic host factors, which may play a critical role as a functional nexus between cancer cell biology and patient’s pathophysiology.
Another pathophysiological implication of the cancer microenvironment depends on the progressive development of an intratumoral microcirculation connected to lymphatic and blood vessel circulatory systems [9]. Once again, this has bidirectional implications: 1) by facilitating systemic spread of circulating cancer cells and tumor-derived soluble factors; and 2) by allowing other circulating host cells, soluble factors and hormones from the body to gain access into the tumor and to act on both cancer and tumor-activated stromal cells. Therefore, cancer microenvironment can remarkably contribute to the pathophysiology of the cancer patient even at a subclinical stage and should not be clinically ignored.
The Liver Prometastatic Reaction in the Pathophysiological Context of Patients with Colon Cancer
Hepatic Colon Cancer Metastasis as a “Prometastatic Model” of Cancer Pathophysiology
Experimental evidences support the contribution of cancer cell factors to cancer microenvironment regulation and, vice versa, of cancer microenvironment factors to cancer cell regulation [2, 3]. Nevertheless, the linkage between cancer patient’s pathophysiology and cancer cell biology is not yet fully elucidated, and valid models with translational potential are needed. The hepatic metastasis occurrence subsequent to colon cancer development may be a good model in that regard. First, both the innate genetics and the very diverse functional profiles that are operating in the liver for each patient may contribute to the diathesis, or predisposition level of the liver not only for hepatocarcinogenesis [10], but also for colon cancer cell invasion and colonization. At present, innate physiological conditions that either prevent or facilitate hepatic metastasis have not yet been clinically established. However, there are various pathophysiological conditions in the liver that lead to functional changes promoting metastasis [11]. Some of them are very common and include hepatic regeneration, fibrosis, inflammation, immune suppression and endocrino-metabolic alterations. However, at present, how altered hepatic microenvironments are in turn inducing the prometastatic gene expression phenotype of cancer cells in patients with colon cancer is still unclear.
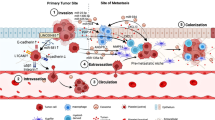
Second, both prometastatic circumstances operating at the microenvironment of colon cancer and remote effects of cancer cell factors on target organ microenvironment support the hepatic metastasis potential of colon cancer (Fig. 2). Examples of these tumor-dependent actions are: specific gene expression profiles at the primary tumor, inducing colon cancer cell receptors for prometastatic hepatic soluble factors; and colon cancer cell secretion of soluble factors and shedding of membrane-bound proteins that drain into portal vein circulation, inducing remote activation of hepatic cells and premetastatic niches for circulating cancer cells [12, 13]. However, how such metastatic cancer cell phenotypes are selected and regulated by the microenvironment at the primary tumor and the liver of colon cancer patients is at present unclear.

Prometastatic events at the microenvironment of primary colon cancer. The remote effects of colon cancer cell factors on target organ microenvironment results in premetastatic niche formation in turn supporting activation of metastatic colon cancer cell in the liver
Third, the metastatic process has been extensively studied using experimental models in mice, and many gene products associated with colon cancer cells' ability to metastasize to the liver have already been reported [14–16]. However, pathophysiological implications of the molecular genetics of colon cancer at the prometastatic microenvironment of the liver remain unclear at a clinical level. Moreover, most of experimental metastasis models have been done in healthy young animal, ignoring the fact that majority of colon cancer metastasis occur in aged livers and that when metastatic cells arrive in the liver, it is already activated by earlier subclinical delivery of soluble factors and even circulating cancer cells from the primary tumor (Fig. 3).

Clinical stages of the hepatic tissue reaction to colon cancer growth and dissemination. Stage I: Hepatic tissue reaction to soluble factor secretion and membrane-bound protein shedding from the primary colon cancer. Stage II) Hepatic tissue reaction to circulating colon cancer cells trafficking through the hepatic microcirculation. Stage III) Hepatic tissue reaction during the sublobular growth of avascular colon cancer micrometastases. Stage IV) Hepatic tissue reaction during panlobular and lobar growth of angiogenic colon cancer metastases. Stage I and II occur in patients with primary tumor. Stages III and IV occur once the primary tumor has been surgically removed
The major cause of death in colon cancer patients is related to liver metastasis, and therefore, identification of both hepatic microenvironment-specific biomarkers for metastasis risk prediction and liver-dependent metastasis gene products are urgent and relevant for diagnostic development and cancer microenvironment pharmaceutics in colon cancer patients.
Based on these general principles of cancer microenvironment regulation along the developing cancer pathophysiology and opportunities offered by the hepatic colon cancer metastasis model, the main purpose of this review is 1) to analyze the liver prometastatic reaction in the pathophysiological context of patients with colon cancer; and 2), the functional profile of hepatic colon cancer metastasis at a gene expression level, with particular emphasis on the microenvironmental control of genes associated with colon cancer cells' ability to metastasize to the liver.
The Liver Prometastatic Reaction to Colon Cancer Factors
Besides regional lymph nodes, the liver is the first organ where superior mesenteric vein-draining colon cancers deliver their soluble molecules and circulating cancer cells. This is an occult event occurring prior to colon cancer detection, since it may start as soon as the tumor becomes angiogenic. Hepatic effects of colon cancer products may act until surgical removal of the primary tumor, and may result in subclinical liver functional changes hard to detect by medical and surgical oncology. However, it represents a key pathophysiological event of early stage colon cancer patients that may have implications for hepatic metastasis pathogenesis and regulation.
Under experimental conditions, both hepatic parenchymal and non-parenchymal cells react to colon cancer molecules and cells by creating a prometastatic microenvironment supporting the hepatic colonization process of colon cancer cells. The “liver prometastatic reaction” involves inflammatory activation of hepatic microcirculation with release of abnormal amounts of proinflammatory cytokines, chemokines and ROIs from hepatic sinusoidal endothelium and Kupffer cells. It also involves activation of mannose receptor-dependent immune suppression, myofibroblast activation of perisinusoidal stellate cells and portal tract fibroblasts leading to increased production of vascular endothelial growth factor (VEGF), soluble intercellular adhesion molecule (ICAM)-1 and transforming growth factor beta (TGFβ). In turn, this supports tumor angiogenesis and stromagenesis; and activation of cholangiocytes and hepatocytes with release of large amounts of nerve growth factor (NGF), C-reactive protein (CRP) and other members of the acute-phase protein family, and epithelial cell growth factors [11, 13]. Moreover, tumor-derived products are not the only hepatic factors that increases predisposition to colon cancer metastasis. There are some tumor-unrelated biological backgrounds, such as discoidin domain receptor-2 (DDR-2) deficiency and pathophysiological circumstances of the liver (see “Prometastatic Implications of Non-Neoplastic Liver Pathophysiology”), such as hepatic fibrosis, regeneration and chronic inflammation that also may mimic the prometastatic reaction, promoting hepatic colonization of colon cancer cells under experimental conditions.
Hepatic Metastasis Predisposition in Colon Cancer Patients
Architectural and Functional Aspects of Liver Tissue with Implications for Cancer Metastasis
The complex functions of the liver in biosynthesis, metabolism, clearance, and host defense are tightly dependent on its specialized microcirculation and parenchymal/non-parenchymal cell heterogeneity. Interestingly, the same architectural and functional aspects of the liver that guarantee hepatic homeostasis are also determining the malignant behavior of cancer cells through the various stages of the metastatic process [17]. First, the functional heterogeneity of the hepatic microcirculation at the lobular level and its unique sinusoidal cell composition influence the trafficking, retention of, and damage to circulating cancer cells reaching the liver through the portal vein or hepatic artery [18]. Second, the perivascular hepatic mesenchymal cells —including fibroblasts from perilobular portal tract and perisinusoidal hepatic stellate cells— become “tumor-activated myofibroblasts” providing a unique stromal support for metastatic cell growth and tumor angiogenesis [19]. Third, the phenotypic heterogeneity [20] of the hepatic parenchymal cell compartment represents an additional bio-resource for tumor-associated stromal cells and autocrine growth factors through epithelial to mesenchymal transition [13]. Fourth, the status of the hepatic regional immunity [21], including innate immunity activation and adaptive immunity suppression by tumor-derived factors, also contribute to cancer cell survival and growth [13]. Altogether, these liver-unique elements constitute a functional microenvironment representing the specific biological background encountered by metastatic colon cancer cells entering the liver.
Innate Prometastatic Conditions of the Hepatic Microenvironment
Organ-specific features endow the liver with a functional microenvironment that may provide prometastatic cues to circulating cancer cells. Among these architectural/functional aspects of the liver are the following:
-
The dual, slow and tortuous character of the hepatic microcirculation [22], which provides the hepatic “territory” with a great accessibility and efficient filtration and recognition capabilities for circulating cells, microbes and soluble molecules. This is due to the anastomotic arrangement of networking capillaries within the hepatic tissue and to the special role that organ-specific subtypes of endothelial cells, macrophages and pericytes play in blood flow control [23]. These hemodynamic features not only provide access to the liver but may also facilitate the mechanical arrest of cancer cell emboli carried in the blood from tumors located in other organs. Another feature of the hepatic biology that is relevant at the microvascular phase of hepatic metastasis is the rich profile of surface molecules provided to disseminating cancer cells by fenestrated hepatic sinusoidal endothelium cells (HSECs) and organ-specific macrophages i.e. Kupffer cells lining the hepatic capillaries. Major molecules on the surface of these hepatic scavenger cells include diverse terminal oligosaccharide moieties [24], cell adhesion proteins [25], endocytotic receptors [26] and toll-like receptors (TLRs) [27] normally involved in the recognition of pathogen-associated molecular patterns and endogenous toll-like receptor ligands termed “damage-associated molecular patterns”. More importantly, most of these surface molecules are regulated by inflammatory cytokines and account for the efficient hepatic uptake and clearance of circulating gut-derived nutrients, toxins, microbes, waste molecules, aged cells, and even cancer cells.
Not surprisingly, both tumor-derived soluble factors and circulating cancer cells delivered to the liver through the portal vein induce HSECs to adopt a proinflammatory phenotype that manifests as activation of mannose receptor-mediated endocytosis [28], induction of adhesion molecules [29], and release of proinflammatory mediators [30, 31] and ROIs [32]. Resident Kupffer cells similarly become activated and new ones are recruited from the circulating precursor cell compartment into the tumor-activated hepatic microvasculature. Thus, cytokine release from tumor-activated hepatic microvascular cells can be identified as an early, tumor-specific, inflammatory response to liver-invading cancer cells that influences the occurrence of experimental metastasis (Fig. 4). Moreover, factors that attenuate the tumor-induced host proinflammatory response (e.g., endostatin, COX-2 inhibitors, IL-1 receptor antagonist, IL-18 binding protein) or block adhesion receptors (e.g., VLA-4 antagonists) for cancer cells have a therapeutic effect in the prevention of experimental liver metastasis [33–36].
-
The liver contains heterogeneous populations of endoderm-derived parenchyma cells [37] (i.e. hepatocytes and cholangiocytes), and mesenchymal non-parenchymal cells [38]. These cell populations coordinately establish the functional tissue structure supporting the glandular and metabolic functions of the liver. At the same time, this functional coordination serves to restore hepatic microarchitecture during physiological renewal and regeneration in response to a variety of pathophysiological circumstances [39]. However, this powerful tissue-reconstitution machinery can also be co-opted during the development of hepatic metastases. In fact, the same hepatic parenchymal and non-parenchymal cells can also contribute to intratumoral stroma and blood vessel generation in response to tumor-derived factors, providing a favorable milieu for the survival and growth of disseminating cancer cells.
-
Perivascular hepatic mesenchymal cells —including fibroblasts from perilobular portal tract and perisinusoidal hepatic stellate cells— become activated myofibroblasts in response to liver insults of viral, metabolic, toxic and tumoral origin [40, 41]. These activated mesenchymal cells are the major source of extracellular matrix during liver injury and secrete all the components of the hepatic scar. In addition, they also have proangiogenic [42] and parenchymal cell-stimulating activities, contributing to hepatic regeneration [43, 44]. However, when this stromal reaction is induced by tumor-derived factors, it provides a unique stromal cell support for cancer cell invasion and growth and for the induction of tumor angiogenesis with resulting prometastatic effects [42].
-
The liver contains specialized subpopulations of the host defense system including resident macrophages, dendritic cells, mast cells, cytotoxic natural killer (NK) cells and T lymphocytes. These provide potent innate and adaptive immune responses [45]. However, the same defense machinery can also be the source of immune regulatory mediators, such as interleukin (IL)-10, prostanoids, soluble ICAM-1, and transforming growth factor (TGF)-β, that induce a regional immune suppression intended to limit the damage to the liver parenchyma, that can be caused by prolonged exposure to inflammatory and immune factors [46]. While this local immune suppression can provide a favorable milieu for liver transplantation [47] and increases oral tolerance [48], it may also contribute to the occurrence of autoimmunity, infectious diseases and cancer metastasis in this organ [49]. Not surprisingly, the hepatic microenvironment can be relatively tolerant to invading microorganisms such as malaria sporozoites [50] and fungi [51] and is also favorable to the development of hepatic metastases.

Altered cytokine pattern in the hepatic blood on the 3rd day after experimental intrasplenic injection of colon cancer cells in mice. Murine Co-26, MC-38 and 51B colon cancer cells were intrasplenically injected into syngeneic mice and hepatic venous blood samples were collected on day 3. Cytokine concentration was determined by ELISA and expressed as pg/ml per unit of hepatic weight. Control mice were intrasplenically injected with the same basal medium without cancer cells. *Differences were statistically significant as assessed by Student “t” test (P < 0.01). Some of represented data come from our previous publications in this research program [28–30, 33–35, 40, 41, 80]
It appears therefore that in addition to specific features of the hepatic microcirculation that facilitate the intra-hepatic lodgment of circulating cancer cells, the microenvironment and mechanisms that support key hepatic functions such as blood clearance, molecular scavenging, hepatic regeneration, wound healing, and immunity may also play a role in the retention or destruction of circulating cancer cells and ultimately may facilitate the ability of some colon cancer cells to develop metastases. In other words, the same mechanisms and responses of hepatic cells that guarantee hepatic homeostasis may influence metastatic behavior of liver-colonizing cancer cells [13]. For example, production of VEGF significantly varied among primary cultures from individual hepatocytes isolated from liver samples obtained during surgical treatment of metastases from colon cancer patients (Fig. 5). More importantly, there was a correlation between VEGF production and proliferation of HT-29 human colon carcinoma cells given conditioned media (CM) from those primary cultured hepatocytes, suggesting a cancer cell growth-stimulating role for tumor-activated hepatocytes. VEGF production and in vitro migration also correlated in primary cultured hepatic stellate cell-derived myofibroblasts given the same CM from hepatocytes, suggesting an additional proangiogenic role for hepatocytes [52].

In vitro VEGF secretion by primary cultured hepatocytes from patients with advanced colon cancer and hepatic metastases. Hepatocytes came from the tumor-unaffected liver tissue of patients with stage IV colon cancers, undergoing partial hepatectomy for hepatic metastasis surgery. All experimental procedures were done in compliance with Spanish laws and regulations and were approved by the Ethics Committee. Hepatocytes were isolated by a two-step collagenase perfusion as described previously [100]. After Percoll purification, freshly isolated hepatocytes were plated onto 60-mm dishes at a density of 3.5 × 106 cells/dish and cultured in HAM-F12/William’s-E medium supplemented with 5% fetal calf serum, insulin (4 mg/l), hydrocortisone (10ذ6 mM), and gentamycin (50 mg/l). Samples from culture supernatants were obtained on the 48th hour of culture. VEGF concentration was determined by ELISA. Data are expressed as average values of triplicated cultures
Altogether, the functional diversity in the microenvironment representing the biological background encountered by metastatic cancer cells entering the liver may affect cancer patients’ susceptibility to hepatic metastasis. Moreover, the several hepatic cell reactions to colon cancer cells and their secreted soluble molecules raise the possibility that hepatic microenvironment-specific biomarkers could be identified in patients with colon cancer that may provide prognostic tools for metastasis risk prediction, prevention, surgery and anti-tumor drug selection for liver metastasis.
Prometastatic Implications of Non-Neoplastic Liver Pathophysiology
It is generally accepted that alterations in the local microenvironment during inflammation, wound healing or organ growth can provide a favorable climate for cancer cell growth and metastasis [53–56]. This is well illustrated in models of non-neoplastic models of the hepatic pathophysiology as for example: liver regeneration following partial hepatectomy, liver fibrogenesis in discoidin domain receptor-2 (DDR2)-deficient mice and cirrhosis. These models of prometastatic microenvironment offer opportunities for identification of new molecular targets for anti-metastatic therapy. Their major features are summarized below.
Regenerating Liver Microenvironment and Metastasis
Surgical resection of hepatic tumors remains the first choice for curative treatment of hepatic colon cancer metastases, giving the patient the only chance for long-term survival [57, 58]. However, in up to 45% of tumors, extended liver resection is necessary to achieve clear resection margins, and the reason for unresectability is often that the remnant liver is of insufficient volume to support postoperative liver function. In these cases, a surgical procedure combining preoperative portal vein embolization, which reduces blood flow to the resected hepatic segments and increases blood supply to the remaining liver, can be used to reduce the risk of postoperative liver failure after major liver resection, thereby increasing the number of resectable patients [59–61]. However, partial hepatectomy for patients with liver metastases is associated with a tumor recurrence rate approaching 80%. There are cases in which accelerated hepatic tumor growth occurs even after portal vein embolization [62], suggesting that factors involved in liver regeneration may stimulate the reactivation of dormant hepatic micrometastases, and even the hepatic implantation of circulating cancer cells. Therefore, despite the known survival benefits for patients undergoing partial hepatectomy for colorectal metastases, questions have arisen regarding the potential effects of the regeneration process on the growth of any residual tumor deposits. Experimental excision of 70% of total liver mass induces rapid hyperplastic growth of remnant liver tissue that restores initial organ mass within 2 weeks post-surgery [63]. The same surgical procedure in syngeneic cancer cell-injected mice significantly increases implantation and growth of liver metastases and promotes outgrowth of dormant micrometastases [64–69]. However, when cancer cells are introduced after liver regeneration is complete, the tumor load is comparable to that of non-surgical controls. Thus, stimulation of tumor growth consequent to partial hepatectomy appears to depend on factors associated with active liver regeneration. Not surprisingly, cellular and molecular changes resulting from partial hepatectomy and the subsequent liver regeneration process can influence the kinetics of cancer cell growth and contribute to recurrence. Several groups have explored the mechanisms underlying the rapid tumor growth after portal vein embolization. Kokudo et al. [70] assessed the proliferative activity of intrahepatic metastases in the embolized liver after portal vein embolization in patients with colorectal carcinoma metastases and found a significantly increased tumor Ki-67 labeling index in the metastases of the portal vein embolization group as compared to those not treated by portal vein embolization. It was postulated that the tumor growth after portal vein embolization may be controlled by 3 factors namely, the malignant potential of the tumors, changes in the blood supply after portal vein embolization and changes in the levels of cytokines or growth factors induced by portal vein embolization [70]. For example, cytokines such as hepatocytes growth factor (HGF), IL-6 and insulin-like growth factor (IGF)-1 and prostaglandins have been implicated in promoting hepatic regeneration, while other factors such as TGFβ, glucagon, and glucocorticoids are known to be inhibitory. HGF is one of the most powerful hepatotropic factor identified to date. Animal models of portal vein branch ligation showed that HGF mRNA levels markedly increased in the non-ligated growing lobe, but was increased only slightly in the ligated shrinking lobe. Increased tissue levels of HGF may also increase plasma levels, but its ability to promote cancer cell "scattering" and invasion raises some concern about its therapeutic safety. Takahara et al. [71] reported that an engineered cytokine derived from HGF and the HGF-like factor, macrophage-stimulating protein, was as effective as HGF at preventing liver injury and at promoting hepatocyte regeneration, but it was therapeutically safer than HGF because it lacked proangiogenic and prometastatic activity during intrahepatic dissemination of orthotopically-injected hepatocarcinoma cells. Therefore, a better understanding of underlying microenvironmental changes in the liver resulting from hepatectomy and the subsequent liver regeneration process is essential. This may enable alternative strategies to minimize tumor recurrence and improve patient survival after hepatectomy.
The Fibrotic Microenvironment of the Liver and Metastasis
Liver damage leads to an inflammatory response and to the activation and proliferation of mesenchymal cell populations within the liver that remodel the extracellular matrix as part of an orchestrated wound-healing response [19]. Chronic damage results in a progressive accumulation of scarring proteins (fibrosis) that alters tissue structure and function with increasing severity, leading to cirrhosis and liver failure. Frequently, circulating colon cancer cells can colonize the altered microenvironment of the cirrhotic liver. However, at the moment it is unclear whether changes in the architecture and function of cirrhotic livers are preventing or promoting metastasis. Malignant cancers rarely metastasize to livers with cirrhosis and autopsies have confirmed that the rate of metastasis to cirrhotic liver is lower than that to normal liver, suggesting that cirrhosis may inhibit metastasis formation [72–76]. A possible explanation is that venous shunting in cirrhosis prevents cancer cells from reaching the liver, and that changes in the architecture of cirrhotic sinusoids may reduce metastasis [75]. Moreover, the activation of Kupffer cells in cirrhosis may also inhibit the formation of hepatic metastases due to their tumoricidal effects [77]. However, the mechanisms responsible for reduced tumor growth remain unclear and as suggested by Vanbockrijck and Kloppel [78], the lower incidence of metastasis may actually be due to a shorter life expectancy of patients with cirrhosis. Moreover, recent observations argue against the antimetastatic effects of cirrhosis. For example, using intravital videomicroscopy Qi et al. [79] showed that the hepatic sinusoids were narrower in cirrhotic livers and more cancer cells were retained in terminal portal veins. This was also consistent with the increased expression of vascular adhesion molecules by sinusoids of cirrhotic livers. Interestingly, using confocal microscopy and the fluorescent nitric oxide probe 4,5-diaminofluorescein diacetate, the same authors detected a significantly lower level of NO release in livers with cirrhosis both under basal conditions and after cancer cell arrest, and a lower percentage of apoptotic cancer cells could be observed in the sinusoids of cirrhotic than normal livers. Consistent with these findings, more mitotic and Ki-67-expressing cancer cells were detected in the cirrhotic livers. Taken together the results suggest that microenvironmental changes in the architecture, adhesion molecule expression and NO production levels may cause more cancer cells to arrest, survive and proliferate in the microvasculature of livers with cirrhosis [79].
Discoidin Domain Receptor-2 Deficiency and Hepatic Colon Cancer Metastasis Predisposition
The transdifferentiation of HSCs into myofibroblasts is a central event in the fibrogenic response to hepatic injury and cirrhosis [19], and play a major role during stroma development in hepatic metastasis [41, 42]. Activated HSCs display myofibroblastic features, such as α-smooth muscle actin (SMA) expression and induction of tyrosine kinase receptors such as platelet-derived growth factor (PDGF) receptor-β and discoidin domain receptor-2 (DDR2). DDR2 is a tyrosine kinase receptor for fibrillar collagen whose expression increases during the HSC activation associated with hepatic fibrosis [80]. Type I collagen-dependent upregulation of DDR2 expression establishes a positive feedback loop in activated HSCs, leading to further proliferation and enhanced release of matrix metalloproteinases. However, mice lacking the DDR2 gene (DDR2−/−) had an enhanced susceptibility to carbon-tetrachloride-induced hepatic fibrosis, suggesting that DDR2-dependent genes may also be anti-fibrogenic. Based on these findings, we hypothesized that tumor stroma formation by transdifferentiated HSCs may be enhanced by DDR2 deficiency, predisposing hepatic tissue to cancer metastasis. Interestingly, experimental hepatic metastasis of murine MC38 colon carcinoma cells significantly increased in DDR2−/− as compared to wild type mice [81]. Immunohistochemical analysis showed that hepatic metastases in DDR2−/− mice had a higher density of HSC-derived myofibroblasts, neoangiogenic vessels and proliferating cancer cells than those in DDR2+/+ littermates. Consistent with the in vivo findings, secretion of endothelial cell adhesion- and migration-stimulating factors, and of tumor cell proliferation enhancing factors increased in supernatants derived from primary cultures of DDR2−/− HSCs, as compared to wild-type HSCs. These secreted factors further increased in the supernatants of DDR2−/− HSC cultures pretreated with cancer cell-conditioned media. Moreover, under basal culture conditions DDR2 deficiency already involved altered HSC expression of key genes associated with immune response regulation: it decreased immune-stimulating factor IL-18, while it increased immune-suppressant factors such as IL-10 and TGFβ. It also involves a decreased expression of IGF-I gene, while expression level of the pro-metastatic genes VEGF, bone morphogenetic protein 7 (BMP7), and syndecan-1 increased. More importantly, IL-10, VEGF and TGFβ gene expression further increased in HSCs from DDR2−/− mice, compared to those from DDR2+/+ mice, when they were treated with MCA38 cancer cell-conditioned medium (Fig. 6). IL-10 is a Th2-type cytokine that attenuates immune stimulation of hepatic fibrogenesis [82] and CEA-expressing colorectal tumors can prevent cancer cell death by inducing hepatic IL-10, thereby inhibiting hepatic NO-dependent cancer cell death [83]. TGF-β1 is a strong immune suppressor, a pro-angiogenic factor, and promotes metastasis of colorectal cancer cells, thereby acting as an oncogene [84]. Therefore, HSCs has the ability to regulate the hepatic metastasis microenvironment via DDR2-dependent factors. However, DDR2 deficient mice may create a unique prometastatic microenvironment operating in the absence of DDR2-dependent factors preventing the expression of key factors fostering tumor-induced hepatic fibrogenesis, tumor cell adhesion and proliferation, and endothelial cell migration. DDR2 may act therefore as a hepatic metastasis suppressor factor operating in tumor-activated HSCs. However, DDR2 expression in the microenvironment of hepatic metastases from patients with colorectal cancer is low, which may increase risk of metastasis [81].

Quantitative real time PCR analysis on hepatic stellate cell (HSC) expression level of selected DDR2-dependent genes identified by comparative gene expression analysis using DNA microarrays. HSCs from DDR2−/− and DDR2+/+ mice were isolated and primary cultured for three days and their total RNAs were isolated and hybridized into DNA microarray for gene expression analysis as described previously [41, 42, 81]. Genes that had previously been related to relevant mechanisms in the hepatic metastasis process were analyzed by quantitative real time RT-PCR, comparing HSC from DDR2−/− and DDR2+/+ under both basal culture conditions and in the presence of media conditioned (CM) by MC38 colon cancer cells [81]. Results are expressed as relative values with respect to RPL13 mRNA expression levels. Under basal culture conditions, DDR2 deficiency already involved altered HSC expression of key genes associated to immune response regulation: it decreased immune-stimulating factor IL-18, while it increased immune-suppressant factors IL-10 and TGFbeta. It also involved an expression decrease of IGF-I gene, while expression level of pro-metastatic genes VEGF, BMP7, and syndecan-1 significantly increased. More importantly, IL-10, VEGF and TGFbeta gene expression further increased in HSCs from DDR2−/− mice, compared to those from DDR2+/+ mice, when they were treated with MCA38 cancer cell-CM. *P < 0.01 compared to cells treated with conditioned media from untreated DDR2+/+ HSCs
Contribution of the Hepatic Microenvironment to Colon Cancer Gene Regulation
Gene Expression Profile of Hepatic Colon Cancer Metastasis
The development of hepatic metastasis is associated with altered expression of many colon cancer genes and regulatory pathways at both primary and metastatic tumors [85]. Several approaches to explore gene expression profiles associated with hepatic metastasis in human colon cancer have been used. Some analyses were made on liver metastasis gene signatures from patients with advanced colon cancer, and tried to identify those genes that were expressed in liver metastasis and not in their matching primary tumors [86–88]. Because selecting only these genes may not be sufficient to assess their role in metastasis, they revalidated selected genes using primary colon cancers without liver metastasis for 3–5 years after curative surgery [87]. For example, Bertucci et al. [88] found 194 discriminating genes differently expressed in primary tumors with and without metastases, and were able to divide patients with significantly different 5-years survival. In contrast, by comparing the expression patterns of 6 tumors and corresponding metastases from the same patients Koehler et al. [89] found that expression level of most differentially expressed genes varied moderately, suggesting that many expression modifications occur during an early phase of the carcinogenesis process and only few alterations are associated with metastatic progression. According to Agrawal et al. [90], one of these genes was osteopontin whose expression significantly increased from invasive cancer to metastatic primary tumor and liver metastases.
Others have investigated specific gene expression patterns predicting the metastatic potential of primary tumor from colon cancer patients. In this case, the main purpose was to discriminate the genes differently expressed in primary tumors with and without metastases, and therefore encoding the metastatic potential of the primary tumor. D’Arrigo et al. [91] found 37 discriminating genes between 10 radically resected primary tumors from patients who did not develop recurrence within 5-year follow-up, and 10 primary tumors from patients with synchronous metastases, and suggested that 29 of these genes could be a distinct metastatic fingerprint that may predict the risk of distance relapse. Yamasaki et al. [92] investigated the existence of liver metastatic potential in primary colorectal tumors using metastasis-related genes and reported that the profile of metastasized primary tumors resembled one of a metastatic lesion apart from a primary lesion rather than one of a non-metastasized primary tumor. Moreover, the expression profile of these genes allowed the classification of tumors diagnosed as localized cancer into two classes, the localized and the metastasized class, according to their final metastatic status. The disease-free survival and overall survival were longer in the localized class than the metastasized class, suggesting that the metastatic potential was already encoded in the primary tumor and detectable, which may allow the prediction of liver metastasis in patients diagnosed with localized tumors.
Others studies have compared gene expression patterns between metastatic and non-metastatic stage-matched human colon cancers in order to establish gene signatures that differentiate metastatic from non-metastatic primary tumors, and to identify genetic markers of metastasis risk. Using this approach, Fritzmann et [93] established a signature of 115 genes that differentiated metastatic from non-metastatic primary tumors, and reported that TGFβ inhibitor BAMBI was highly expressed in approximately half of metastatic primary tumors and metastases but not in non-metastatic tumors. In addition, they observed an inverse correlation between level of BAMBI expression and metastasis-free survival time of patients. BAMBI inhibited TGFβ signaling and increased migration in colon cancer cells, and overexpression of BAMBI caused colon cancer cells to form tumors that metastasized more frequently to liver and lymph nodes than control cancer cells.
Hepatic Microenvironment-Dependent Colon Cancer Metastasis Genes
Despite the potential pro-metastatic role of the hepatic microenvironment, it is unknown which genes tumor-activated hepatic cells specifically regulate in colon cancer cells in order to support their intra-hepatic growth. In this context, we used a microenvironmental approach to the study of genes associated with colon cancer cells' ability to metastasize to the liver in patients with advanced colon cancer.
First we identified hepatic colon cancer metastasis genes not expressed in tumor-unaffected areas of the same liver, but expressed in the primary tumors of patients that developed metastases within five years of diagnosis. To this aim, RNAs from hepatic metastasis, tumor-unaffected hepatic tissue and peripheral blood mononuclear cells from same colon cancer patients were purified, and the specific gene clusters representing the transcriptome of colon cancer cells developing hepatic metastases in patients was determined by DNA microarray and RT-PCR [52].
Second, among the identified hepatic metastasis genes, we next selected those that overlapped with genes whose expression level changed in cultured HT-29 colon carcinoma cells exposed to the conditioned medium from tumor-activated hepatocytes and hepatic stellate cell-derived myofibroblasts (Fig. 7). This allows determining if both tumor and liver cells were mutually influencing the expression of metastasis-associated genes. We also determined genes co-expressed in hepatic metastases and tumor-unaffected liver tissue, but not in the primary tumors. From this gene category we then selected those expressed either by HT-29 cells treated with liver cell-conditioned media, or by liver cells receiving HT-29-conditioned media. Identified genes suggested that metastatic colon cancer have hepatomimetic properties, and that hepatic tissue expressed colon cancer-specific genes under the regulation of tumor-derived factors [94, 95].

Experimental design for the study of colon cancer cell response to soluble factors from tumor-activated human hepatocytes and hepatic stellate cell-derived myofibroblasts. (I) HT-29 human colorectal cancer cells were cultured until subconfluency, serum-free fresh medium exchanged and a 12-hour conditioned medium (CM) was prepared and (II) added 50% diluted in fresh medium to primary cultured hepatocytes and stellate cell-derived myofibroblasts obtained from hepatic tissue fragments resected for treatment purposes from patients with hydatid cyst, and approved by our Ethics Committee. Next (III), new conditioned media were obtained from untreated and HT-29-CM-treated hepatocytes and hepatic myofibroblasts and added 50% diluted in fresh medium to HT-29 colon cancer cells
A third approach was to determine genes of tumor-unaffected areas from livers bearing colon cancer metastasis, not expressed by colon cancer cells. This gene category represented the genetic background of the liver prometastatic reaction induced by colon cancer-derived factors, and therefore, included those genes operating at the colon cancer metastasis microenvironment.
From these studies, three microenvironment-related gene expression categories were identified (Table 1):
-
a)
Hepatic metastasis genes not expressed in tumor-unaffected liver. Some of these genes were also expressed in the primary tumors particularly in a group of patients that developed metastases within 5 years of diagnosis. They were also expressed in HT-29 cells treated with cultured liver cell-conditioned media and in liver cells treated with HT-29 cell-conditioned media. This hepatic metastasis gene class included some well-known molecules that consistent with our findings have already been related to colon cancer metastasis and some of them are even considered as potential colon cancer targets.
For example, S100P is expressed at greater levels in colon cancer than matched normal tissue [96] and acts via receptor for activated glycation end products (RAGE) in its role as a paracrine factor to stimulate ERK phosphorylation and NFκB pathway [97]. Moreover, knocking down S100P gene expression in colon cancer cells, using lentivirus-mediated RNA interference, resulted in significant inhibition of cancer cell growth, migration and invasion in vitro, as well as liver metastasis in vivo [98]. Cadherin-H1 (CDH1) gene encodes E-Cadherin, a primary cell-cell adhesion molecule for epithelial cells whose under expression in primary tumors, including colorectal tumors [99], may facilitate the mobility of cancer cells required for dissemination, while its overexpression by colon cancer cells at hepatic metastasis sites herein detected, is consistent with previous studies on E-Cadherin regulation in epithelial cancer cells [100, 101], and may indicate reactivation of an epithelial cell differentiation program needed for secondary tumor growth. Osteopontin (SPP1) is a hypoxia-regulated extracellular matrix protein that has been proposed as lead marker for tumor progression and metastasis in colon cancer and hepatocellular carcinoma [102, 103]. Its serum level also increases an early marker of hepatic metastasis from uveal melanoma [104] while it gene silencing decreases metastatic ability of mouse colon cancer cells [105]. Transforming growth factor beta-induced (betaig-h3/TGFBI) gene encodes expression of an extracellular matrix protein secreted by colon cancer cells whose expression enhances aggressiveness and metastatic properties of colon cancer cells, while its expression inhibition reduces metastasis [106]. Thioredoxin-1 (Trx-1) gene encodes a redox protein whose increased expression in colorectal cancer is associated with decreased patient survival [107] and poor prognosis in patients with hepatic colorectal metastasis [108]. Calcyclin (S100A6) gene encodes a calcium transporter protein over-expressed by numerous malignancies, whose increased expression in human colorectal adenocarcinoma has been associated with invasion and metastasis [109].
-
b)
Genes co-expressed in hepatic metastases and tumor-unaffected liver tissue. This gene group was neither expressed in the primary tumors nor in the normal liver. It included both liver-specific genes expressed by HT-29 cells treated with liver cell-conditioned media, and colon cancer-specific genes expressed by liver cells receiving HT-29-conditioned media. Once again, in this hepatic metastasis gene class we found some molecules whose oncogenic implications have already been detected in malignant tumors, colon cancer included. For example, macrophage migration inhibitory factor (MIF) expression in the gastrointestinal tract contributes to inflammation and immune response regulation, but also to carcinogenesis. MIF expression is increased in sporadic human colorectal adenomas, and exogenous MIF drives tumorigenic behavior of epithelial cells in vitro [110]. Serum levels of MIF are increased in early stage colon cancer and have been proposed as an early diagnostic marker in colorectal carcinomas [111]. Peroxiredoxin IV gene expression has been reported to increase in metastatic colon cancer [112]. It scavenges extracellular ROIs [113], which may protect liver-infiltrating colon cancer cells from intrasinusoidal oxidative stress [32]. Chromosome segregation 1-like/cellular apoptosis susceptibility (CSE1L) gene over-expression has also been reported in colon cancer [114] and proposed as a potential prognostic marker for metastatic colorectal cancer [115]. Haptoglobin gene encodes an acute phase hepatic protein whose seric levels increase in patients with infectious, inflammatory and neoplastic diseases. Interestingly, it has been reported as a serum biomarker for patients with familial adenomatous polyposis [116] and even with metastatic colorectal carcinoma [117]. TNFSF14 (tumor necrosis factor ligand superfamily member 14) gene encodes an immunoregulatory cytokine of the TNF ligand family which increases by 6-fold in the liver of Lewis lung carcinoma cell-injected mice [118], and which induces T cell proliferation and prevents TNFα-mediated apoptosis in hepatocyte [119].
The finding that the gene expression profiles of hepatic metastases had some similarities with the profiles seen in tumor-unaffected hepatic tissue, suggest that metastatic colon cancer have hepatomimetic properties. Conversely, we also found that hepatic tissue expressed colon cancer-specific genes under the regulation of tumor-derived factors. This suggests that mutual gene expression mimicking occurs when metastatic cells are exposed to the hepatic microenvironment and vice versa. The identification of distinct microenvironment-related hepatic metastasis genes in clinical specimens from primary tumors implicates them in the hepatotropism and metastatic development of colorectal carcinoma.
-
c)
Genes of tumor-unaffected liver tissue not expressed in hepatic metastases. This gene group was expressed in liver cells, but not in colon cancer cells, and represented the genetic background of the hepatic metastasis microenvironment generated by hepatic cells that have reacted to colon cancer-derived factors. In this gene class, we still could be able to detect some genes occurring in patients that developed hepatic metastases in less than 5 years from first diagnosis, while others were absent in primary tumors or in normal liver.
These results suggest that although some hepatic metastasis genes may already be expressed in the primary colon cancer and may be predictive of metastasis risk in the cancer patients, additional changes to gene expression take place in the liver under the control of tumor-activated hepatic cells. While this microenvironmental regulation of hepatic metastasis genes may occur at the earliest stages of metastasis, it is likely that some of the altered genes are still under microenvironmental control even at an advanced phase of the metastatic process and may therefore have implications for therapy. Interestingly, most of the genes from the three microenvironment-related gene expression categories had already been related to mechanisms of cancer progression and some of them in colon carcinoma.
Hepatic Cell Lineage-Specific Regulation of Metastasis Genes from Colon Cancer Cells
How the microenvironment of a highly susceptible metastasis site such as the liver dictates the ability of a tumor to metastasize is in general unclear. In our model, tumor-activated hepatocytes and hepatic stellate cell-derived myofibroblasts were major components of the metastasis stroma supporting colon carcinoma cell growth in the liver. Cultured HT-29 colon cancer cells contained 235 genes significantly upregulated by hepatocyte-CM and 67 genes by hepatic myofibroblast-CM (Fig. 8 and Table 2). Majority of genes upregulated by both hepatic cell types were different, but some overlapped with those included in the specific gene cluster representing the transcriptome of cancer cells developing hepatic metastases in patients with colon carcinoma. This suggests that some human colon cancer genes that are expressed during metastasis growth are controlled in the liver by hepatocytes, by hepatic myofibroblasts, or by both hepatic cell types at same time. More importantly, consistent with in vitro colon cancer cell proliferation-stimulating effects of tumor-activated hepatocytes and HSC-derived myofibroblasts, around 50% of hepatic cell-dependent colon cancer cell genes belonged to the cell cycle-regulation class, suggesting a role of these tumor-activated hepatic cells in the control of colon cancer cell proliferation [11, 13, 17].

Differential contributions of tumor-activated hepatocytes and hepatic myofibroblasts to gene expression by HT-29 human colon carcinoma cells. a Signature genes characterizing the transcriptional responses of HT-29 cancer cells to conditioned media (CM) from tumor-activated primary cultured human hepatocytes and hepatic stellate cell-derived myofibroblasts. The level of expression of each gene in each treated HT-29 cell samples, relative to the level of expression of that gene in untreated control samples, is presented in the form of a heat map, using a red-black-green color scale (green: below control sample; black: equal to control sample; red: above control sample). Shown in b are gene expression changes in HT-29 cancer cells cultured in tumor-activated hepatocyte-CM as compared to basal medium and in HT-29 cells cultured in tumor-activated hepatic myofibroblast-CM as compared to basal medium. Blue lines: genes mainly up/down regulated by tumor-activated hepatocytes. Yellow lines: genes mainly up/down regulated by tumor-activated hepatic myofibroblasts
Conclusions
The liver is the first organ where colon cancers deliver their soluble molecules and circulating cancer cells. The effect of colon cancer products results in the “liver prometastatic reaction”, a special microenvironment supporting the experimental hepatic colonization process of colon cancer cells [11]. It represents a key pathophysiological event of early stage colon cancer patients that has implications for hepatic metastasis pathogenesis and regulation. However, tumor-derived products are not the only increasing hepatic predisposition to colon cancer metastasis. There are some tumor-unrelated biological backgrounds, such as DDR-2 deficiency, and pathophysiological circumstances of the liver, such as hepatic fibrosis, regeneration and chronic inflammation that also can mimic the prometastatic reaction, promoting hepatic colonization of colon cancer cells under experimental conditions.
The majority of genome analyses made on liver metastasis gene signatures from patients with advanced colon cancer have just identified those genes that were expressed in liver metastasis and not in their matching primary tumors. Others studies have compared gene expression patterns between metastatic and nonmetastatic stage-matched human colon cancers in order to establish gene signatures that differentiate metastatic from nonmetastatic primary tumors, and to identify genetic markers of metastasis risk. However, despite the potential prometastatic role of the hepatic microenvironment, it is at present unclear which genes tumor-activated hepatic cells specifically regulate in colon cancer cells for supporting their intrahepatic growth.
We used a microenvironmental approach to the study of genes associated with colon cancer cells' ability to metastasize to the liver in patients with advanced colon cancer. First we determined hepatic metastasis genes not expressed in tumor-unaffected areas of the same liver but expressed in the primary tumors of patients that developed metastases within 5 years of diagnosis. Among identified hepatic metastasis genes, we next selected those that overlapped with genes whose expression level changed in HT-29 colon carcinoma cells given the conditioned medium from tumor-activated hepatocytes and hepatic stellate cell-derived myofibroblasts. Second, we determined genes co-expressed in hepatic metastases and tumor-unaffected liver tissue, but neither in the primary tumors nor in normal liver. From this gene category we then selected those expressed either by cultured HT-29 cells treated with liver cell-conditioned media, or by primary cultured liver cells receiving HT-29-conditioned media. Identified genes suggest that metastatic colon cancer have hepatomimetic properties, and that hepatic tissue expressed colon cancer-specific genes under the regulation of tumor-derived factors. Some of these genes, as for example MIF, Peroxiredoxin-IV, CSEIL or TNFSF14, have already been related to mechanisms of cancer progression, including colon carcinoma.
A third approach was to determine genes of tumor-unaffected areas from livers bearing colon cancer metastasis, not expressed by colon cancer cells. This gene category represented the genetic background of the liver prometastatic reaction induced by colon cancer-derived factors, and therefore, genes at the colon cancer metastasis microenvironment.
Thus, we identified microenvironment-related colon cancer genes whose expression level was regulated by hepatocytes, hepatic myofibroblasts or both. We also identified some genes representing the liver prometastatic reaction. We do not yet know which specific tumor and hepatic cell factors are controlling the mutual gene expression mimicking occurring when metastatic cells are exposed to the hepatic microenvironment. Neither is it known on the specific role of these genes in the pathogenesis of hepatic metastasis. However, identified genes provide a new target context for therapeutic innovation in the cancer metastasis microenvironment of colon cancer patients. On the one hand, by developing drugs able to block liver factors supporting colon cancer metastasis genes and therefore preventing effects of the “liver prometastatic reaction” in colon cancer patients. On the other hand, by developing drugs able to block those factors from colon cancer cells able to induce the prometastatic microenvironment of the liver.
References
Egeblad M, Nakasone ES, Werb Z (2010) Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 18:884–901
Witz IP, Levy-Nissenbaum O (2006) The tumor microenvironment in the post-PAGET era. Cancer Letters 246:1–10
Fidler IJ (2001) Seed and soil revisited: contribution of the organ microenvironment to cancer metastasis. Surg Oncol Clin N Am 10:257–269
Paget S (1889) The distribution of secondary growths in cancer of the breast. Lancet 133:571–573
Wang HB, Dembo M, Hanks SK, Wang Y (2001) Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc Natl Acad Sci USA 98:11295–11300
Schedin P, Keely PJ (2011) Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol 3:a003228
Jessup JM, Laguinge L, Lin S et al (2004) Carcinoembryonic antigen induction of IL-10 and IL-6 inhibits hepatic ischemic/reperfusion injury to colorectal carcinoma cells. Int J Cancer 111:332–337
Beacham DA, Cukierman E (2005) Stromagenesis: The changing face of fibroblastic microenvironments during tumor progression. Semin Cancer Biol 15:329–341
Baeriswyl V, Christofori G (2009) The angiogenic switch in carcinogenesis. Semin Cancer Biol 19:329–337
Budhu A, Forgues M, Ye QH et al (2006) Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 10:99–111
Vidal-Vanaclocha F (2008) The prometastatic microenvironment of the liver. Cancer Microenvironment 1:113–129
Kaplan RN, Rafii S, Lyden D (2006) Preparing the “soil”: the premetastatic niche. Cancer Res 66:11089–11093
Vidal-Vanaclocha F (2011). The Tumor Microenvironment at Different Stages of Hepatic Metastasis P. Brodt (ed.), Liver Metastasis: Biology and Clinical Management, Cancer Metastasis – Biology and Treatment 16, doi:10.1007/978-94-007-0292-9_3, C_ Springer Science + Business Media B.V.
Herath NI, Boyd AW (2010) The role of Eph receptors and ephrin ligands in colorectal cancer. Int J Cancer 126:2003–2011
Bellam N, Pasche B (2010) Tgf-beta signaling alterations and colon cancer. Cancer Treat Res 155:85–103
Jiang Y, Kimchi ET, Staveley-O'Carroll KF et al (2009) Assessment of K-ras mutation: a step toward personalized medicine for patients with colorectal cancer. Cancer 115:3609–3617
Vidal-Vanaclocha F. Architectural and Functional Aspects of the Liver with Implications for Cancer Metastasis. P. Brodt (ed.), Liver Metastasis: Biology and Clinical Management, Cancer Metastasis – Biology and Treatment 16, doi:10.1007/978-94-007-0292-9_2,C_ Springer Science + Business Media B.V. 2011.
Barbera-Guillem E, Smith I, Weiss L (1993) Cancer-cell traffic in the liver. II. Arrest, transit and death of B16F10 and M5076 cells in the sinusoids. Int J Cancer 53:298–301
Friedman SL (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134:1655–1669
Jungermann K (1986) Functional heterogeneity of periportal and perivenous hepatocytes. Enzyme 35:161–180
Parker GA, Picut CA (2005) Liver Immunobiology. Toxicol Pathol 33:52–62
McCuskey RS (1994) The hepatic microvascular system. In: Arias IM, Boyer JL, Fausto N et al (eds) The liver: biology and pathobiology. Raven Press, New York, pp 1089–1106
Oda M, Han JY, Yokomori H (2000) Local regulators of hepatic sinusoidal microcirculation: recent advances. Clin Hemorheol Microcirc 23:85–94
Barberá-Guillem E, Rocha M, Alvarez A et al (1991) Differences in the lectin-binding patterns of the periportal and perivenous endothelial domains in the liver sinusoids. Hepatology 14:131–139
Scoazec JY, Feldmann G (1994) The cell adhesion molecules of hepatic sinusoidal endothelial cells. J Hepatol 20:296–300
Seternes T, Sorensen K, Smedsrod B (2002) Scavenger endothelial cells of vertebrates: a nonperipheral leukocyte system for high-capacity elimination of waste macromolecules. Proc Natl Acad Sci USA 99:7594–7597
Seki E, Brenner DA (2008) Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48:322–335
Arteta B, Lasuen N, Lopategi A et al (2010) Colon carcinoma cell interaction with liver sinusoidal endothelium inhibits organ-specific anti-tumor immunity via IL-1-induced mannose receptor. Hepatology 51:2172–2182
Mendoza L, Carrascal T, de Luca M et al (2001) Hydrogen peroxide mediates vascular cell adhesion molecule-1 expression from IL-18-activated hepatic sinusoidal endothelium: implications for circulating cancer cell arrest in murine liver. Hepatology 34:298–310
Vidal-Vanaclocha F, Fantuzzi G, Mendoza L et al (2000) IL-18 regulates IL-1 beta-dependent hepatic melanoma metastasis via vascular adhesion molecule-1. Proc Natl Acad Sci USA 97:734–739
Khatib AM, Auguste P, Fallavollita L et al (2005) Characterization of the host proinflammatory response to tumor cells during the initial stages of liver metastasis. Am J Pathol 167:749–759
Anasagasti MJ, Alvarez A, Martin JJ et al (1997) Sinusoidal endothelium release of hydrogen peroxide enhances very late antigen-4-mediated melanoma cell adherence and tumor cytotoxicity during interleukin-1 promotion of hepatic melanoma metastasis in mice. Hepatology 25:840–846
Vidal-Vanaclocha F, Amézaga C, Asumendi A et al (1994) Interleukin-1 receptor blockade reduces the number and size of murine B16 melanoma hepatic metastases. Cancer Res 54:2667–2672
Carrascal T, Mendoza L, Vacarcel M et al (2003) Interleukin-18 binding protein reduces B16 melanoma hepatic metastasis by neutralizing the adhesiveness and growth factors of sinusoidal endothelial cell. Cancer Res 63:491–497
Vidal-Vanaclocha F, Alvarez A, Asumendi A et al (1996) Interleukin 1 (IL-1)-dependent melanoma hepatic metastasis in vivo; increased endothelial adherence by IL-1-induced mannose receptors and growth factor production in vitro. J Natl Cancer Inst 88:198–205
Zubia A, Mendoza L, Vivanco S et al (2005) Application of stereocontrolled stepwise [3 + 2] Cycloadditions to the preparation of inhibitors of alpha(4)beta(1)-integrin-mediated hepatic melanoma metastasis. Angew Chem Int Ed Engl 44:2903–2907
Fausto N (2004) Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology 39:1477–1487
Smedsrød B, Le Couteur D, Ikejima K et al (2009) Hepatic sinusoidal cells in health and disease: update from the 14th International Symposium. Liver Int 29:490–501
Malik R, Selden C, Hodgson H (2002) The role of non-parenchymal cells in liver growth. Sem Cell Dev Biol 13:425–431
Ramadori G, Saile B (2004) Portal tract fibrogenesis in the liver. Lab Invest 84:153–159
Olaso E, Santisteban A, Bidaurrazaga J et al (1997) Tumor-dependent activation of rodent hepatic stellate cells during experimental melanoma metastasis. Hepatology 26:634–642
Olaso E, Salado C, Egilegor E et al (2003) Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology 37:674–685
Libbrecht L, Cassiman D, Desmet V et al (2002) The correlation between portal myofibroblasts and development of intrahepatic bile ducts and arterial branches in human liver. Liver 22:252–258
Asai K, Tamakawa S, Yamamoto M et al (2006) Activated hepatic stellate cells overexpress p75NTR after partial hepatectomy and undergo apoptosis on nerve growth factor stimulation. Liver Int 26:595–603
Li Z, Diehl AM (2003) Innate immunity in the liver. Curr Opin Gastroenterol 19:565–571
Parker GA, Picut CA (2003) Liver Immunobiology. Toxicol Pathol 33:52–62
Starzl TE, Murase N, Abu-Elmagd K et al (2003) Tolerogenic immunosuppression for organ transplantation. Lancet 361:1502–1510
Safadi R, Alvarez CE, Ohta M et al (2005) Enhanced oral tolerance in transgenic mice with hepatocyte secretion of IL-10. J Immunol 175:3577–3583
Kern M, Popov A, Kurts C et al (2010) Taking off the brakes: T cell immunity in the liver. Trends Immunol 31:311–317
Frevert U, Engelmann S, Zougbédé S et al (2005) Intravital Observation of Plasmodium berghei Sporozoite Infection of the Liver. PLoS Biol 3:1034–1046
Anttila VJ, Elonen E, Nordling S et al (1997) Hepatosplenic candidiasis in patients with acute leukemia: incidence and prognostic implications. Clin Infect Dis 24:375–380
Beaskoetxea J, Telleria N, Del Villar A et al (2006) Identification of microenvironmentally-regulated genes of colon carcinoma cells from human hepatic metastasis. Proc Amer Assoc Cancer Res 47:4298
Murthy SM, Goldschmidt RA, Rao LN et al (1989) The influence of surgical trauma on experimental metastasis. Cancer 64:2035–2044
Murthy MS, Scanlon EF, Jelachich ML et al (1995) Growth and metastasis of human breast cancers in athymic nude mice. Clin Exp Metastasis 13:3–15
Bogden AE, Moreau J-P, Eden PA (1997) Proliferative response of human animal tumors to surgical wounding of normal tissues: Onset, duration and inhibition. Br J Cancer 75:1021–1027
Hofer SO, Shrayer D, Reichner JS et al (1998) Wound-induced tumor progression: A probable role in recurrence after tumor resection. Arch Surg 133:383–389
Nordlinger B, Guiguet M, Vaillant JC et al (1996) Surgical resection of colorectal carcinoma metastases to the liver: a prognostic scoring system to improve case selection, based on 1568 patients. Cancer 77:1254–1262
Jaeck D, Bachellier P, Guiguet M et al (1997) Long-term survival following resection of colorectal hepatic metastases. Br J Surg 84:977–980
Makuuchi M, Le Thai B, Takayasu K et al (1990) Preoperative portal embolizationto increase safety of major hepatectomy for hilar bile duct carcinoma: a preliminary report. Surgery 107:521–527
Elias D, Roche A, Vavasseur D et al (1992) Induction of hypertrophy of a small left hepatic lobe by preoperative right portal embolization, preceding extended right hepatectomy. Ann Chir 46:404–410
Azoulay D, Castaing D, Smail A et al (2000) Resection of non resectable liver metastases from colorectal cancer after percutaneous PVE. Ann Surg 231:480–486
Liu H, Zhu S (2009) Present status and future perspectives of preoperative portal vein embolization. Am J Surgery 197:686–690
Elias D, de Baere T, Roche A et al (1999) During liver regeneration following right portal embolization the growth rate of liver metastases is more rapid than that of the liver parenchyma. Br J Surg 86:784–788
Higgins GM, Anderson RM (1931) Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol 12:186–202
Ichihashi H, Mabuchi H, Suenaga M et al (1984) Liver regeneration andtumor growth in the rat after partial hepatectomy. Jpn J Surg 14:510–514
Morimoto H, Nio Y, Imai S et al (1992) Hepatectomy accelerates the growthof transplanted liver tumor in mice. Cancer Detec Prev 16:137–147
Panis Y, Ribeiro J, Chretien Y et al (1992) Dormant liver metastases: an experimental study. Br J Surg 79:221–223
Gutman M, Singh RK, Price JE et al (1994) Accelerated growth of human colon cancer cells in nude mice undergoing liver regeneration. Invasion Metastasis 14:362–371
Asaga T, Suzuki K, Umeda M et al (1991) The enhancement of tumor growth after partial hepatectomy and the effect of sera obtained from hepatectomized rats on tumor cell growth. Jpn J Surg 21:669–675
Kokudo N, Tada K, Seki M et al (2001) Proliferative activity of intrahepatic colorectal metastases after preoperative hemihepatic portal vein embolization. Hepatology 34:267–272
Takahara T, Xue F, Mazzone M et al (2008) Metron factor-1 prevents liver injury without promoting tumor growth and metastasis. Hepatology 47:2010–2025
Gervaz P, Pak-art R, Nivatvongs S et al (2003) Colorectal adenocarcinoma in cirrhotic patients. J Am Coll Surg 196:874–879
Melato M, Laurino L, Mucli E et al (1989) Relationship between cirrhosis, liver cancer, and hepatic metastases. An autopsy study. Cancer 64:455–459
Pereira-Lima JE, Lichtenfels E, Barbosa FS et al (2003) Prevalence study of metastases in cirrhotic livers. Hepatogastroenterology 50:1490–1495
Seymour K, Charnley RM (1999) Evidence that metastasis is less common in cirrhotic than normal liver: a systematic review of post-mortem case-control studies. Br J Surg 86:1237–1242
Uetsuji S, Yamamura M, Yamamichi K et al (1992) Absence of colorectal cancer metastasis to the cirrhotic liver. Am J Surg 164:176–177
Song E, Chen J, Ouyang N et al (2001) Kupffer cells of cirrhotic rat livers sensitize colon cancer cells to Fas-mediated apoptosis. Br J Cancer 84:1265–1271
Vanbockrijck M, Kloppel G (1992) Incidence and morphology of liver metastasis from extrahepatic malignancies to cirrhotic livers. Zentralbl Pathol 138:91–96
Qi K, Qiu H, Sun D et al (2004) Impact of cirrhosis on the development of experimental hepatic metastases by B16F1 melanoma cells in C57BL/6 mice. Hepatology 40:1144–1150
Olaso E, Ikeda K, Eng FJ et al (2001) DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J Clin Invest 108:1369–1378
Badiola I, Olaso E, Crende O, et al (2011). Discoidin domain receptor 2 deficiency predisposes hepatic tissue to colon carcinoma metastasis. Gut, in press.
Zhang LJ, Zheng WD, Chen YX et al (2007) Antifibrotic effects of interleukin-10 on experimental hepatic fibrosis. Hepatogastroenterology 54:2092–2098
Jessup JM, Samara R, Battle P et al (2004) Carcinoembryonic antigen promotes tumor cell survival in liver through an IL-10-dependent pathway. Clin Exp Metastasis 21:709–717
Zhang B, Halder SK, Kashikar ND et al (2010) Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology 138:969–980
Nadal C, Maurel J, Gascon P (2007) Is there a genetic signature for liver metastasis in colorectal cancer? World J Gastroenterol 13:5832–5844
Kwon HC, Kim SH, Roh MS et al (2004) Gene expression profiling in lymph node-positive and lymph node-negative colorectal cancer. Dis Colon Rectum 47:141–152
Ki DH, Jeung HC, Park CH et al (2007) Whole genome analysis for liver metastasis gene signatures in colorectal cancer. Int J Cancer 121:2005–2012
Bertucci F, Salas S, Eysteries S et al (2004) Gene expression profi ling of colon cancer by DNA microarrays and correlation with histoclinical parameters. Oncogene 23:1377–1391
Koehler A, Bataille F, Schmid C et al (2004) Gene expression profiling of colorectal cancer and metastases divides tumours according to their clinicopathological stage. J Pathol 204:65–74
Agrawal D, Chen T, Irby R et al (2002) Osteopontin identified as lead marker of colon cancer progression, using pooled sample expression profiling. J Natl Cancer Inst 94:513–521
D’Arrigo A, Belluco C, Ambrosi A et al (2005) Metastatic transcriptional pattern revealed by gene expression profiling in primary colorectal carcinoma. Int J Cancer 115:256–262
Yamasaki M, Takemasa I, Komori T et al (2007) The gene expression profile represents the molecular nature of liver metastasis in colorectal cancer. Int J Oncol 30:129–138
Fritzmann J, Morkel M, Besser D et al (2009) A colorectal cancer expression profile that includes transforming growth factor beta inhibitor BAMBI predicts metastatic potential. Gastroenterology 137:165–175
Del Villar A, Telleria N, Beaskoetxea J, et al (2008). Differential contribution of hepatocytes and hepatic myofibroblasts to the microenvironmental regulation of hepatic metastasis genes from colon carcinoma patients. Proc Amer Assoc Cancer Res 48:
Beaskoetxea J, Telleria N, del Villar A, et al (2009). Exposure of colon carcinoma cells to hepatic microenvironment and of hepatic cells to tumor factors promotes gene expression mimic contributing to organ tropism and metastasis development. Proc Amer Assoc Cancer Res 49:
Arumugam T, Simeone DM, Schmidt AM, Logsdon CD (2004) S100P stimulates cell proliferation and survival via receptor for activated glycation end products (RAGE). J Biol Chem 279:5059–5065
Fuentes MK, Nigavekar SS, Arumugam T et al (2007) RAGE activation by S100P in colon cancer stimulates growth, migration, and cell signaling pathways. Dis Colon Rectum 50:1230–1240
Jiang L, Lai YK, Zhang J et al (2011) Targeting S100P inhibits colon cancer growth and metastasis by lentivirus-mediated RNA interference and proteomic analysis. Mol Med. doi:10.2119/molmed.2011.00008
Gout S, Huot J (2008) Role of Cancer Microenvironment in Metastasis: Focus on Colon Cancer. CAMI 1:69–83
Richert L, Alexandre E, Lloyd T et al (2004) Tissue collection, transport and isolation procedures required to optimise human hepatocyte isolation from waste liver surgical resections. A multi-laboratory study. Liver Int 24:371–378
Kim CH, Kim J, Kahng H, Choi EC (2007) Change of E-cadherin by hepatocyte growth factor and effects on the prognosis of hypopharyngeal carcinoma. Ann Surg Oncol 14:1565–1574
Agrawal D, Chen T, Irby R et al (2002) Osteopontin identified as lead marker of colon cancer progression, using pooled sample expression profiling. J Natl Cancer Inst 94:513–521
Pan HW, Ou YH, Peng SY et al (2003) Overexpression of osteopontin is associated with intrahepatic metastasis, early recurrence, and poorer prognosis of surgically resected hepatocellular carcinoma. Cancer 98:119–127
Reiniger IW, Wolf A, Welge-Lussen U et al (2007) Osteopontin as a serologic marker for metastatic uveal melanoma: results of a pilot study. Am J Ophthalmol 143:705–707
Wai PY, Mi Z, Guo H et al (2005) Osteopontin silencing by small interfering RNA suppresses in vitro and in vivo CT26 murine colon adenocarcinoma metastasis. Carcinogenesis 26:741–751
Ma C, Rong Y, Radiloff DR et al (2008) Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev 22:308–321
Raffel J, Bhattacharyya AK, Gallegos A et al (2003) Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J Lab Clin Med 142:46–51
Noike T, Miwa S, Soeda J et al (2008) Increased expression of thioredoxin-1, vascular endothelial growth factor, and redox factor-1 is associated with poor prognosis in patients with liver metastasis from colorectal cancer. Hum Pathol 39:201–208
Komatsu K, Murata K, Kameyama M et al (2002) Expression of S100A6 and S100A4 in matched samples of human colorectal mucosa, primary colorectal adenocarcinomas and liver metastases. Oncology 63:192–200
Wilson JM, Coletta PL, Cuthbert RJ et al (2005) Macrophage migration inhibitory factor promotes intestinal tumorigenesis. Gastroenterology 129:1485–1503
Lee H, Rhee H, Kang HJ et al (2008) Macrophage migration inhibitory factor may be used as an early diagnostic marker in colorectal carcinomas. Am J Clin Pathol 129:772–779
Li M, Lin YM, Hasegawa S et al (2004) Genes associated with liver metastasis of colon cancer, identified by genome-wide cDNA microarray. Int J Oncol 24:305–312
Okado-Matsumoto A, Matsumoto A, Fujii J, Taniguchi N (2000) Peroxiredoxin IV is a secretable protein with heparin-binding properties under reduced conditions. J Biochem 127:493–501
Tsao TY, Tsai CS, Tung JN et al (2009) Function of CSE1L/CAS in the secretion of HT-29 human colorectal cells and its expression in human colon. Mol Cell Biochem 327:163–170
Stella Tsai CS, Chen HC et al (2010) Serum cellular apoptosis susceptibility protein is a potential prognostic marker for metastatic colorectal cancer. Am J Pathol 176:1619–1628
Quaresima B, Crugliano T, Gaspari M et al (2008) A proteomics approach to identify changes in protein profiles in serum of Familial Adenomatous Polyposis patients. Cancer Lett 272:40–52
Li ZG, Zhao L, Liu L, Ding YQ (2007) Monitoring changes of serum protein markers in metastatic colorectal carcinoma model. Zhonghua Bing Li Xue Za Zhi 36:48–52
Maeda S, Hikiba Y, Sakamoto K et al (2009) Ikappa B kinasebeta/nuclear factor-kappaB activation controls the development of liver metastasis by way of interleukin-6 expression. Hepatology 50:1851–1860
Matsui H, Hikichi Y, Tsuji I et al (2003) LIGHT, a member of the tumor necrosis factor ligand superfamily, prevents tumor necrosis factor-alpha-mediated human primary hepatocyte apoptosis, but not Fas-mediated apoptosis. J Biol Chem 277:50054–50061
Acknowledgements
This work was supported in part by grants from the Spanish Carlos III Health Institute, Madrid (ADE09/90041), the Spanish Ministry of Innovation and Science (SAF2009-12376), and the Burdinola Professorship on Molecular Medicine to F. Vidal-Vanaclocha.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vidal-Vanaclocha, F. The Liver Prometastatic Reaction of Cancer Patients: Implications for Microenvironment-Dependent Colon Cancer Gene Regulation. Cancer Microenvironment 4, 163–180 (2011). https://doi.org/10.1007/s12307-011-0084-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-011-0084-5