
Abstract
Background
Hormone therapy targeting the estrogen receptor (ER) pathway is the most common treatment used for ER-positive breast cancer. However, some patients experience de novo or acquired resistance, which becomes a critical problem. Activation of the insulin-like growth factor (IGF) pathway allows breast cancer cells to proliferate and is associated with the ER pathway. Little is known about the role of the IGF pathway in hormone therapy and resistance; therefore, we investigated whether the inhibition of this pathway may represent a novel therapeutic target for overcoming hormone therapy resistance in ER-positive breast cancers.
Methods
Crosstalk between the ER and IGF pathways was analyzed in breast cancer cell lines by inhibiting or stimulating either one or both pathways. We studied the effect of insulin-like growth factor one receptor (IGF1R) inhibition in aromatase inhibitor-resistant breast cancer cell lines and fulvestrant-resistant cell lines which were uniquely established in our laboratory.
Results
Under normal conditions, IGF signaling is controlled by ER signaling to promote cell growth. Temporary disruption of the estrogen supply results in attenuated ER signaling, and IGF-1 dramatically increased relative growth compared with normal conditions. In addition, IGF1R inhibitor strongly suppressd cell growth in hormone-resistant breast cancer cells where ER remains than cells where ER decreased or was almost lost.
Conclusions
Our study suggests that inhibition of the IGF pathway may be an effective strategy for ER-positive breast cancer therapy, even in hormone therapy-resistant cases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Approximately, 75% of all invasive breast cancers are estrogen receptor (ER)-positive and are treated using therapies targeting the ER pathway [1,2,3]. Hormone therapy is widely used in clinical adjuvant settings and in advanced cancer and has excellent outcomes [4]. However, some cancers acquire resistance and relapse during or after hormone therapy, resulting in poor prognosis [5]. Hence, there is an immediate requirement to identify an additional target and therapy to overcome resistance.
Estrogen and ER regulate mammary cell proliferation and apoptosis and are the dominant driver pathways, particularly in ER-positive breast cancers [6,7,8,9]. ER belongs to a nuclear receptor superfamily, functions as a ligand-activated transcription factor, and transactivates promoters of estrogen target gene expression. ER also exerts distinct cellular functions predominantly mediated by activation at key positions of ER, such as at serine 118 or 167, because of activating intracellular phosphorylation signaling pathways. To date, two major intracellular phosphorylation signaling pathways have been studied: MAPK and PI3K/Akt/mTOR [10,11,12,13]. Intracellular phosphorylation signaling pathways are mainly activated via membrane receptors, including epidermal growth factor receptor, human epidermal growth factor receptor two, and insulin-like growth factor (IGF) one receptor (IGF1R). The relationship between the ER and IGF pathways has been documented. Estrogen enhances the IGF pathway by inducing expression of IGF1R and its downstream signaling molecule insulin receptor substrate (IRS) [14, 15]. ER and IGF pathways respond to ligands, resulting in increased levels of cellular proliferation and enhanced signaling events [16, 17]. Activated IGF1R and ER show additive or synergistic effects when both ligands are added simultaneously [18]. The PI3K pathway is activated by IRS and leads to further phosphorylation of Akt/mTOR kinase.
Resistance to hormone therapy is clinically recognized as a significant problem and is highly correlated with involvement of intracellular phosphorylation pathways. Previously, our laboratory established several hormone-resistant cell line models: estrogen deprivation-resistant (EDR) and MCF-7-derived fulvestrant-resistant (MFR) cell lines [19, 20]. Our hormone-resistant cell lines support the involvement of intracellular phosphorylation pathways after acquired resistance to hormonal drugs. The importance of IGF and its signaling in ER-positive breast cancers is well-known [21]; however, little is known about the influence of therapy targeting the IGF pathway in hormone therapy-resistant breast cancer. Here, we explored the role of the IGF pathway in hormone therapy resistance and the role of the IGF pathway in ER-positive breast cancers and revealed that IGF pathway represents a possible therapeutic target for the treatment of ER-positive breast cancers. Our results indicate the usefulness of IGF-targeted therapy for some types of hormone therapy-resistant models and may provide an effective treatment option for hormone therapy-resistant breast cancers.
Materials and methods
Reagents
ADW-742 was purchased from Selleck Chemicals (Houston, TX, USA), fulvestrant was obtained from Sigma-Aldrich (St. Louis, MO, USA), and IGF-1 was acquired from R&D systems (Minneapolis, USA).
The antibodies for Western blotting were sourced as follows: total ERα (H-184) was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA), IGF1R β antibody (#3027) and β-tubulin were acquired from Cell Signaling Technology (Danvers, MA, USA), and phospho-S167 anti-ERα (ab31478) was obtained from AbCam (Cambridge, MA, USA). Secondary antibody conjugated with horseradish peroxidase was purchased from Bio-Rad Laboratories Inc. (Hercules, CA, USA).
Cells and cell culture
MCF-7-E10 breast cancer cells derived from MCF-7 cells were stably transfected with ERE-GFR reporter plasmids as reported previously [19]. Estrogen deprivation-resistant (EDR) cells (type one and type two cells) and fulvestrant-resistant cells (MFR) were established from MCF-7-E10 cells as described previously [19, 20]. Parent cells were maintained in RPMI1640 medium (Sigma-Aldrich) containing 5% fetal calf serum (FCS; Gibco BRL, Grand Island, NY, USA) and 1% penicillin/streptomycin (Gibco). MFR cells were maintained in fulvestrant-supplemented RPMI1640 medium (final concentration, 10 nM). Type one and type two EDR cells were cultured in phenol red-free RPMI1640 medium (Gibco BRL) supplemented with 5% dextran-coated charcoal-treated FCS and 1% penicillin/streptomycin. All cells were incubated at 37 °C in an atmosphere containing 5% CO2.
Cell proliferation assay
In inhibitor sensitivity assays, parent cell lines and MFR cells were maintained in RPMI1640 medium containing 5% FCS, seeded in 24-well culture plates, and grown to approximately 50% confluence. Each drug was added for 3 days, harvested, and counted using a Sysmex CDA-500 automated cell counter (Sysmex, Kobe, Japan). Type one and type two EDR cells were cultured in phenol red-free RPMI1640 medium.
Real-time RT-PCR
Total RNA was extracted using IsoGen lysis buffer (Nippon Gene, Toyama, Japan) according to the manufacturer’s instructions. Extracted RNA was converted to cDNA using a QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA, USA). Transcripts were detected using a Step One Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Relative copy numbers were calculated from a standard curve and normalized to housekeeping genes. The sequences of primers and probes were as follows: pS2, forward, 5′-TCC CCT GGT GCT TCT ATC CTA A-3′; reverse, 5′-ACT AAT CAC CGT GCT GGG GA-3′; PgR, forward, 5′-AGC TCA CAG CGT TTC TAT CA-3′; reverse, 5′-CGG GAC TGG ATA AAT GTA TTC-3′; and IGF1R, forward, 5′-GCA CCA ATG CTT CAG TTC CT-3′; reverse, 5′-CAG CGC ACA ATG TAG TAA CTC-3′.
Western blot analysis
Cell lysates were prepared using Lysis-M Reagent (Roche Diagnostics GmbH, Mannheim, Germany) supplemented with Phos STOP phosphatase inhibitor cocktail (Roche Diagnostics) according to the manufacturer’s instructions. Extracted proteins (5 µg) were separated using a 12% SDS–PAGE gel, and proteins were transferred to PVDF membrane. Protein expression was determined by Western blotting with specific antibodies, and expression signals were detected on an ImageQuant LAS 4000 image analyzer (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) using Immun-star HRP substrate (Bio-Rad).
Reporter plasmid construction and luciferase assays
Transient transfection of reporter plasmids was performed using Trans IT LT-1 Transfection Reagent (Mirus Bio LLC, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, cancer cells were grown to approximately 50% confluence in 24-well culture plates. Reporter plasmids (0.5 µg) were mixed with transfection reagent in serum-free medium and added to the culture medium. The vector pRL-TK (Promega Corporation) was also mixed with transfection reagent (internal transfection efficiency control) and incubated. After 24 h, cells were lysed and luciferase activity was determined using a Dual-Luciferase Reporter Assay System (Promega Corporation).
Statistical analyses
Student’s t test was used to assess the significance of differences between two groups performed in triplicate. Data were expressed as means ± SD. Probability value of < 0.05 was considered significant.
Results
IGF signaling plays an important role in luminal-type cell lines
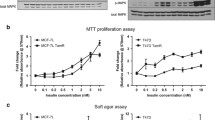
We explored the expression levels of IGF1R as a marker for each subtype of breast cancer cell line. IGF1R mRNA levels were higher in luminal-type cell lines (MCF-7 and T-47D cells) than in non-luminal-type cell lines (SK-BR-3 and MDA-MB-231) (Fig. 1a), and IGF1R protein expression corresponded to mRNA levels (Fig. 1b). We then evaluated the efficacy of the IGF1R-selective tyrosine kinase inhibitor, ADW-742, in the same cell lines. ADW-742 suppressed cell growth a greater extent to MCF-7 and T-47D cells than SK-BR-3 and MDA-MB-231 cells (Fig. 1c). These results and the well-known fact that luminal-type cell lines depend to a large extent on ER signaling suggest that growth suppression caused by IGF1R inhibition in breast cancer cells may be related to ER signaling.

Effect of IGF1R inhibitor in breast cancer cell lines. a IGF1R mRNA expression levels in each breast cancer cell line were analyzed using qPCR and the expression was normalized to that of RPL13A. Results are presented as mean ± standard deviation (SD), and P < 0.05 was considered significant. All experiments were performed independently in triplicates. b IGF1R protein expression levels were analyzed using Western blotting with β-tubulin as protein loading control. c Cell proliferation assay of breast cancer cell lines treated with IGF1R-selective tyrosine kinase inhibitor (ADW-742, 0.5 µM) was assessed relative to the negative control. Results are expressed as mean ± SD of the three independent experiments; *P < 0.05
Inhibition of IGF1R had little influence on ER signaling in normal luminal-type cell lines
To investigate the association between IGF1R inhibition and ER-related phenomena, we examined changes in ER activity in MCF-7 and T-47D cells using an IGF1R inhibitor. IGF1R inhibition suppressed cell proliferation in MCF-7 and T-47D cell lines (Fig. 1c), whereas no remarkable differences were observed in ER transcriptional activity in the estrogen response element (ERE)-luciferase assay (Fig. 2a). The mRNA levels of ER representative target genes, PgR and pS2, were unaltered (Fig. 2b). Furthermore, protein levels of total ER and phosphorylated Ser167 ER were unchanged (Fig. 2c), suggesting that growth suppression caused by IGF1R inhibition in breast cancer cell is independent of ER signaling.

Alteration of ER activity and target genes in MCF-7 and T-47D. a ER activity in luminal cells was measured using a luciferase reporter driven by the estrogen reporter element. b mRNA expression levels of ER target genes in luminal cell lines were analyzed using qPCR, and expression was normalized to that of RPL13A. Results are presented as mean ± SD. c Protein expression levels of total ER and phosphorylated Ser 167 ER were analyzed using Western blotting with β-tubulin as protein loading control. Cells were cultured in RPMI 1640 medium supplemented with 5% FCS
IGF1R is a target gene of ER
We further examined the relationship between IGF1R inhibition and ER signaling. MCF-7 cells highly expressed IGF1R mRNA when cultured in RPMI 1640 medium supplemented with 5% FCS. MCF-7 cells were then cultured in estrogen-depleted medium, with or without 1 nM estradiol. IGFR mRNA levels were decreased in cells cultured in estrogen-depleted medium, but were increased in the presence of estradiol. Moreover, this increase was suppressed by the addition of 100 nM fulvestrant (Fig. 3a). Protein levels corresponded to mRNA levels (Fig. 3b), showing that IGF1R gene expression was regulated by estrogen and ER levels.

Crosstalk between ER and IGF signaling. a mRNA expression levels of IGF1R were analyzed using qPCR and expression was normalized to that of RPL13A. MCF-7 cell lines were cultured in RPMI 1640 medium (MCF7) supplemented with 5% DCC-FCS for 3 days [MCF-7(E2-)], with 1 nM estradiol [MCF-7(E2-) + E2] and with 100 nM fulvestrant [MCF-7(E2-) + E2 + FUL]. Results are presented as mean ± SD of three independent experiments. b Protein expression levels of IGF1R were analyzed by Western blotting with β-tubulin used as protein loading control. c Cell proliferation assays of MCF-7 treated with IGF1 (20 µg/ml) were measured relative to the negative control using cells cultured in RPMI 1640 medium with 5% FCS (static state) and with 5% DCC-FCS for 3 days (estrogen-depleted state). The results are expressed as mean ± SD of three independent experiments; *P < 0.05
IGF1R functions as a compensatory pathway for ER signaling
Since IGF1R expression was shown to be controlled by ER signaling, we evaluated the contribution of IGF signaling via changes to ER signaling. The effects of IGF-1, an IGF1R ligand, were determined on cell proliferation, in static and estrogen-depletion states. In the estrogen-depleted state, IGF-1 dramatically increased relative growth compared within the static state (Fig. 3c), suggesting that IGF signaling significantly contributed to cell proliferation under conditions of reduced ER signal, and acted as a compensatory pathway for ER signaling.
Combined effect of IGF1R inhibition and suppression of ER signaling
We investigated the effect of IGF1R inhibition in combination with suppression of ER signaling, and examined changes in ER activity and representative target genes in MCF-7 cells. ER signaling was suppressed using two different methods: via administration of fulvestrant and by culturing cells in estrogen-depleted medium to mimic treatment with an aromatase inhibitor (AI). IGF1R inhibition suppressed cell proliferation in MCF-7 cells after culturing in estrogen-depleted medium for 3 days (Fig. 4a). Surprisingly, ER transcription activity was aberrantly activated (Fig. 4b), and ER target genes showed a similar tendency, although PgR showed no statistically significant difference (Fig. 4c). A combination of IGF1R inhibitor with fulvestrant suppressed cell proliferation to a greater extent than fulvestrant alone in MCF-7 cells (Fig. 4d). ER transcriptional activity did not differ between combined treatment and fulvestrant alone (Fig. 4e) and ER target genes were almost unchanged (Fig. 4f).

Combination effect of ER inhibition and IGF1R inhibitor. a Cell proliferation assay of MCF-7 cells cultured in RPMI 1640 medium with 5% DCC-FCS for 3 days and treated with IGF1R inhibitor (ADW-742, 0.5 µM) was measured relative to the negative control. b ER transcription activity in MCF-7 cells cultured in medium with 5% DCC-FCS and not treated (N.C.) or treated with IGF1R inhibitor (ADW-742, 0.5 µM) was measured using a luciferase reporter driven by the estrogen reporter element. c The mRNA expression levels of ER target genes in MCF-7 cultured in medium with 5% DCC-FCS and not treated (N.C.) or treated with IGF1R inhibitor (ADW-742, 0.5 µM) were analyzed using qPCR and expression normalized to that of RPL13A. The results are presented as mean ± SD. d Cell proliferation assay of MCF-7 treated with fulvestrant (500 pM) and a combination of fulvestrant (500 pM) with IGF1R inhibitor (ADW-742, 0.5 µM) was measured relative to the negative control. e ER transcription activity was measured in MCF-7 treated with fulvestrant (500 pM) alone and with a combination of fulvestrant (500 pM) and IGF1R inhibitor (ADW-742, 0.5 µM). f The mRNA expression levels of ER target genes in MCF-7 treated with under the same conditions were analyzed using qPCR and expression normalized to that of RPL13A
These results suggest that the combination of IGF1R inhibitor with suppression of ER signaling produced synergistic effects on cell proliferation. Moreover, the combination of IGF1R inhibitor with fulvestrant did not increase ER transcriptional activity or target genes.
IGF1R inhibitor potency in hormone-resistant cell lines
We investigated the effects of IGF1R inhibition following acquired hormone resistance. We previously reported two clones of AI-resistant breast cancer models obtained from MCF-7-E10 cells after long-term estrogen depletion (EDR cells). Type 1 cells showed increased ER expression levels, whereas type 2 showed decreased levels. Type 1 EDR cells were more sensitive to the IGF1R inhibitor than type 2 cells, but were similar to MCF-7-E10 cells (Fig. 5a). Type 1 EDR cells were sensitive to fulvestrant, whereas type 2 cells were not, as ER expression decreased and its function almost disappeared. Type 1 EDR cells showed a synergistic effect with combined IGF1R inhibitor and fulvestrant treatment, while type 2 cells showed no significant effect (Fig. 5b). Next, we examined MFR cells derived from MCF-7-E10 cells that had lost ER expression. Compared with MCF-7-E10 cells, MFR cells were not sensitive to the IGF1R inhibitor (Fig. 5c). In MFR cells, IGF1R mRNA levels were significantly decreased (Fig. 5d) and protein levels corresponded to mRNA levels (Fig. 5e). These results suggest that IGF1R inhibition may be effective after acquired hormone resistance when ER expression is maintained.

Effect of IGF1R inhibitor in endocrine-resistant cell lines. a Cell proliferation assay of estrogen deprivation-resistant cells (EDR) treated with IGF1R inhibitor (ADW-742, 0.5 µM) was measured relative to the negative control. The results are expressed as mean ± SD of three independent experiments; *P < 0.05. b Cell proliferation assay of EDR cells treated with fulvestrant (500 pM) alone and a combination of fulvestrant and IGF1R inhibitor (ADW-742, 0.5 µM). c Cell proliferation assay of MCF-7-derived fulvestrant-resistant (MFR) cells was measured. The results are expressed as mean ± SD of three independent experiments; *P < 0.05. d The mRNA expression levels of IGF1R were analyzed using qPCR and expression was normalized to that of RPL13A. e Protein expression levels of IGF1R were analyzed by Western blotting with β-tubulin as a protein loading control
Discussion
Hormone therapy is well-established for use in ER-positive breast cancer. However, in some cases, acquired resistance occurs after hormone therapy. This makes treatment extremely difficult; therefore, molecular targeting therapy is under development and is expected to help overcome hormone therapy resistance [22, 23]. Here, we examined the possibility of using drugs targeting the IGF pathway to overcome hormone therapy resistance in breast cancer cell lines.
We confirmed that IGF1R expression levels were higher in luminal cell lines and that they responded well to IGF1R inhibitors, thus indicating a role for IGF signaling in the ER signaling pathway. The promoter region of IGF1R gene included several specificity protein 1 (Sp1) binding sites which is suggested to bind with ER and regulate gene expression by estrogen [24]. Although inhibition of IGF1R did not influence ER signaling under normal conditions, stimulation of IGF1R dramatically promoted cell growth under conditions of estrogen depletion. These results indicate that IGF signaling has been controlled by ER signaling under normal conditions (Fig. 6a). Disruption of the estrogen supply resulted in a reduced dependence on ER signaling, indicating that IGF signaling acts as a major compensatory pathway to maintain cell survival (Fig. 6b). Thus, it is logical to approach treatment by targeting and blocking both pathways.

Models of crosstalk for ER-positive cells with varied ER signaling. a In normal conditions, IGF signaling is mediated by ER signaling, including ligand-dependent pathways and phosphorylation signaling pathways in ER-positive breast cancer cells. b Temporary disruption to ER signaling leads to IGF signaling playing a critical role for cell proliferation. c In cases of AI resistance with remaining ER signaling, IGF signaling still promotes cell proliferation controlled by ER signaling. d In cases of hormone therapy resistance with loss of ER signaling, cell proliferation does not depend on IGF signaling
To date, inhibition of ER signaling is achieved clinically via two main methods. One is the use of AIs, which block estrogen production from stromal cells. The other is the use of an estrogen receptor antagonist such as fulvestrant, which binds to the ERα and downregulates its expression. Next, we examined the combined effects of IGF1R inhibition with suppression of ER signaling using these two approaches. A synergistic effect on cell proliferation was shown. Surprisingly, ER transcriptional activity was drastically activated and ER target genes were elevated only when estrogen production was blocked. The differential increasing rates of ER activity and target genes were not always correspond well with each other, because the structure of promotor region in each ER target genes does not necessarily match with ERE-luciferase assay. This phenomenon of activation was not observed when cells were treated concomitantly with fulvestrant. This difference is likely attributable to the mechanism of ER signal inhibition. Fulvestrant binds to ER with a high affinity and leads to ubiquitin–proteasome degradation, decreasing the amount of ER. By contrast, AI acts by blocking estrogen production, thereby functional ER remains. ER-positive breast cancer cells highly depend on ER signaling, and once the major pathway is interrupted, they search for compensatory pathways, such as intracellular phosphorylation pathways, to survive. Activation of intracellular pathways occurs via the IGF pathway and other membrane receptor pathways. These pathways may be rescued when ER is diminished and deactivated.
We next explored the efficacy of IGF1R inhibition using hormone therapy-resistant cell lines to identify a treatment for hormone therapy-resistant cases. In our previous study, we reported the establishment of several endocrine-resistant cell lines, including type 1 and type 2 EDR cells and MFR cells. Type 1 EDR cells were sensitive to the IGF1R inhibitor, but the others were not. These results suggest that loss of ER due to long-term estrogen deprivation or long-term fulvestrant exposure leads to reduced IGF1R expression and, therefore, prevents IGF1R signaling (Fig. 6c, d).
Expression of ER in type 2 EDR cells decreased. We previously reported that type 2 EDR cells relied on ER-independent and JNK-dependent mechanisms and hypothesized that IGF1R occurs via JNK activation [19]; nevertheless, type 2 EDR cells expressed lower levels of IGF1R and were less sensitive to the IGF1R inhibitor. This was indicated as the reason for differences seen in IGF1R inhibition. We previously used AG1024 as an IGF1R inhibitor, which had different specificities compared with ADW-742. AG1024 has a relatively wide spectrum and also inhibits insulin receptor (IR) to some extent. Moreover, mitogenic effects of IGF occur via the IGF1R and the IR.
Glycometabolism is a well-known function of the IR and is specific to isoform B (IR-B), which is the classical form of IR that binds only insulin (Fig. S1). In contrast, a splice variant of the IR isoform A (IR-A) binds insulin and IGF-II and plays an important role in cell proliferation [25,26,27,28]. Here, we used ADW-742, which had a high specificity to IGF1R. As the JNK pathway plays a significant role in the proliferation of breast cancer cells and increases reliance after estrogen deprivation resistance via ER-independent mechanisms [29], activation of this pathway may be attributed to intracellular phosphorylation pathways, such as those seen in IR-A. Further, the existence of an IR/IGF1R hybrid receptor has been reported [30]. Pharmacological characteristics of hybrid-RA highly depend on proliferation owing to the affinity of IGF. To date, some clinical trials using IGF1R inhibitors have had poorer outcomes than expected [31]. This may be attributed to the existence of IR-A and hybrid-RA.
Considering these results together, IGF signaling is regulated by ER and promotes cell growth and proliferation under normal conditions (Fig. 6a). IGF signaling plays a critical role in proliferation and rescues ER signaling in the temporary absence of ER signaling (Fig. 6b). However, breast cancer cells become reliant on other signaling pathways when the expression and function of ER become diminished by long-term blockade of ER signaling. IGF1R inhibition may be useful after acquired AI resistance as long as ER expression is maintained (Fig. 6c). On the contrary, after fulvestrant resistance is acquired, IGF1R inhibition would not be effective due to loss of IGF signaling. Unfortunately, there is currently no reliable method to specifically inhibit IGF1R, IR-A and hybrid-RA; however, the ability to distinguish between IR-A and IR-B could benefit patients with blockade of IGF signaling agents.
The limitations of this study are that all experiments of this study were conducted in vitro. Further study and investigation especially experiments in animals were needed to apply the IGF1R inhibitor in a clinical setting.
In summary, it is well-established that IGF signaling plays an important role in breast cancer, especially in luminal-type; however, therapies targeting the IGF pathway have not yet been conducted at the bedside. Our study suggests that inhibition of the IGF pathway may be a useful strategy for breast cancer with ER expressing cases, even with acquired AI resistance. These results offer new insights into the selection of patients suitable for effective treatment with these drugs.
References
Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–717.
Hammond MEH, Hayes DF, Wolff AC, Mangu PB, Temin S. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Oncol Pract. 2010;6:195–7.
Senkus E, Kyriakides S, Penault-Llorca F, Poortmans P, Thompson A, Zackrisson S, et al. Primary breast cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24:vi7–23.
Burstein HJ, Temin S, Anderson H, Buchholz TA, Davidson NE, Gelmon KE, et al. Adjuvant endocrine therapy for women with hormone receptor-positive breast cancer: American Society of Clinical Oncology clinical practice guideline focused update. J Clin Oncol. 2014;32:2255–69.
Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer. 2015;15:261–75.
Thomas C, Gustafsson J-Å. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11:597–608.
Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Treuter E, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–31.
Jordan VC, Brodie AMH. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids. 2007;72:7–25.
Schuur ER, Loktev AV, Sharma M, Sun Z, Roth RA, Weigel RJ. Ligand-dependent Interaction of estrogen receptor-α with members of the forkhead transcription factor family. J Biol Chem. 2001;276:33554–60.
Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor α: a new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–24.
Martin MB, Franke TF, Stoica GE, Chambon P, Katzenellenbogen BS, Stoica BA, et al. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I [in process citation]. Endocrinology. 2000;141:4503–11.
Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–4.
Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–83.
Lee AV, Jackson JG, Gooch JL, Hilsenbeck SG, Coronado-Heinsohn E, Osborne CK. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol Balt Md. 1999;13:787–96.
Huynh H, Nickerson T, Pollak M, Yang X. Regulation of insulin-like growth factor 1 receptor expression by pure antiestrogen ICI 182780. Clin Cancer Res. 1996;2:2037–43.
Yu Z, Gao W, Jiang E, Lu F, Zhang L, Shi Z, et al. Interaction between IGF-IR and ER Induced by E2 and IGF-I. PLoS One. 2013;8:1–7.
Hafner F, Holler E, von Angerer ECN-C. Effect of growth factors on estrogen receptor mediated gene expression. J Steroid Biochem Mol Biol. 1996;58:385–93.
Kahlert S, Nuedling S, Van Eickels M, Vetter H, Meyer R, Grohé C. Estrogen receptor a rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;275:18447–53.
Fujiki N, Konno H, Kaneko Y, Gohno T, Hanamura T, Imami K, et al. Estrogen Response element-GFP (ERE-GFP) introduced MCF-7 cells demonstrated the coexistence of multiple estrogen-deprivation resistant mechanisms. J Steroid Biochem Mol Biol. 2014;139:61–72.
Tsuboi K, Kaneko Y, Nagatomo T, Fujii R, Hanamura T, Gohno T, et al. Different epigenetic mechanisms of ERα implicated in the fate of fulvestrant-resistant breast cancer. J Steroid Biochem Mol Biol. 2017;167:115–25.
Hartog H, Wesseling J, Boezen HM, van der Graaf WTA. The insulin-like growth factor 1 receptor in cancer: old focus, new future. Eur J Cancer. 2007;43:1895–904.
Yardley DA, Noguchi S, Pritchard KI, Burris HA, Baselga J, Gnant M, et al. Everolimus plus exemestane in postmenopausal patients with HR+ breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870–84.
Finn RS, Martin M, Rugo HS, Jones S, Im S-A, Gelmon K, et al. Palbociclib and Letrozole in advanced breast cancer. N Engl J Med. 2016;375:1925–36.
Werner H, Sarfstein R. Transcriptional and epigenetic control of IGF1R gene expression: Implications in metabolism and cancer. Growth Horm IGF Res. 2014;24:112–8.
Weroha SJ, Haluska P. IGF-1 receptor inhibitors in clinical trials-early lessons. J Mammary Gland Biol Neoplasia. 2008;13:471–83.
Huang J, Morehouse C, Streicher K, Higgs BW, Gao J, Czapiga M, et al. Altered expression of Insulin receptor isoforms in breast cancer. PLoS One. 2011;6:e26177.
Ulanet DB, Ludwig DL, Kahn CR, Hanahan D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc Natl Acad Sci USA. 2010;107:10791–8.
Pandini G, Medico E, Conte E, Sciacca L, Vigneri R, Belfiore A. Differential gene expression induced by insulin and insulin-like growth factor-II through the insulin receptor isoform A. J Biol Chem. 2003;278:42178–89.
Wang J, Kuiatse I, Lee AV, Pan J, Giuliano A, Cui X. Sustained c-Jun-NH2-kinase activity promotes epithelial-mesenchymal transition, invasion, and survival of breast cancer cells by regulating extracellular signal-regulated kinase activation. Mol Cancer Res. 2010;8:266–77.
Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-yamaguchi Y, et al. Insulin and Insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin / IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Am Assoc Cancer Res J. 1999;5:1935–44.
Robertson JFR, Ferrero JM, Bourgeois H, Kennecke H, de Boer RH, Jacot W, et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. Lancet Oncol. 2013;14:228–35.
Acknowledgements
This study was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, Science and Technology of Japan, a Grant-in-Aid for Cancer Research from the Ministry of Health, Labour and Welfare of Japan, the Program for Promotion of Fundamental Studies in Health Science of the National Institute of Biomedical Innovation (NIBIO), and a grant from the Smoking Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Shin-ichi Hayashi received research grants from Novartis Pharma K.K and Astra Zeneca K.K.
Ethical standard
All experiments complied with current laws of Japan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Iida, M., Tsuboi, K., Niwa, T. et al. Compensatory role of insulin-like growth factor 1 receptor in estrogen receptor signaling pathway and possible therapeutic target for hormone therapy-resistant breast cancer. Breast Cancer 26, 272–281 (2019). https://doi.org/10.1007/s12282-018-0922-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12282-018-0922-0